Publisher’s version / Version de l'éditeur:
Vous avez des questions? Nous pouvons vous aider. Pour communiquer directement avec un auteur, consultez la première page de la revue dans laquelle son article a été publié afin de trouver ses coordonnées. Si vous n’arrivez pas à les repérer, communiquez avec nous à PublicationsArchive-ArchivesPublications@nrc-cnrc.gc.ca.
Questions? Contact the NRC Publications Archive team at
PublicationsArchive-ArchivesPublications@nrc-cnrc.gc.ca. If you wish to email the authors directly, please see the first page of the publication for their contact information.
https://publications-cnrc.canada.ca/fra/droits
L’accès à ce site Web et l’utilisation de son contenu sont assujettis aux conditions présentées dans le site LISEZ CES CONDITIONS ATTENTIVEMENT AVANT D’UTILISER CE SITE WEB.
7th International Conference on Ecomaterials (ICEM7) and 3rd International
Conference on Advanced Materials (ICMAT 2005) [Proceedings], 2, pp. 396-401,
2005-07-01
READ THESE TERMS AND CONDITIONS CAREFULLY BEFORE USING THIS WEBSITE. https://nrc-publications.canada.ca/eng/copyright
NRC Publications Archive Record / Notice des Archives des publications du CNRC :
https://nrc-publications.canada.ca/eng/view/object/?id=8b43f033-5ce3-4910-97c6-f42f0be8ae5a https://publications-cnrc.canada.ca/fra/voir/objet/?id=8b43f033-5ce3-4910-97c6-f42f0be8ae5a
NRC Publications Archive
Archives des publications du CNRC
This publication could be one of several versions: author’s original, accepted manuscript or the publisher’s version. / La version de cette publication peut être l’une des suivantes : la version prépublication de l’auteur, la version acceptée du manuscrit ou la version de l’éditeur.
Access and use of this website and the material on it are subject to the Terms and Conditions set forth at
Geometrical aspects of the crystal chemistry of apatite: an analysis of
calcium-lead fluoro-vanadinites
Mercier, P. H. J.; Whitfield, P. S.; Mitchell, L. D.; Davidson, I. J.; Le Page, Y.;
White, T. J.
Geometrical aspects of the crystal chemistry of
apatite: an analysis of calcium-lead fluoro-vanadinites
Mercier, P.H.J.; Whitfield, P.S.; Mitchell, L.D.
Davidson, I.J.; Le Page, Y.; White, T.J.
NRCC-48350
A version of this document is published in / Une version de ce document se trouve dans : 7th International Conference on Ecomaterials (IECM7) and 3rd International Conference on
Advanced Materials (ICMAT 2005), Singapore, July 3-8, 2005, v. 2, pp. 396-401
Mercier et al. 2005
Geometrical Aspects of the Crystal Chemistry of Apatite: An Analysis of Calcium-Lead Fluoro-Vanadinites.
Patrick H.J. Mercier,a Pamela S. Whitfield,a Lyndon D. Mitchell,b Isobel J. Davidson,a Yvon Le Page,a and Timothy J. Whitec
a
Institute for Chemical Process and Environmental Technology (ICPET), National Research Council Canada, Ottawa, Ontario, Canada K1A 0R6
b
Institute for Research in Construction (IRC), National Research Council Canada, Ottawa, Ontario, Canada K1A 0R6 c
School of Materials Engineering, Nanyang Technological University, Singapore 639798
Background on the crystal chemistry of the apatites and their use for toxic heavy metal immobilization
The incorporation of toxic heavy metals such as Pb, V, As, Zn, Cd, etc. into the apatite crystal structure is a potential solution to the problem of storage of these pollutants. For instance, apatite-type compounds are already applied to the treatment of lead-contaminated soils and waters (Chen et al. 1997; Zhang and Ryan 1999; Ioannidis and Zouboulis 2003) and are under consideration for the co-stabilization and recycling of incinerator ashes with industrial wastes (Taylor et al. 1998; Valsami-Jones 1998; Crannell et al. 2000; Dong et al. 2002; Dong and White 2004a,b; Kim et al. 2005). Therefore, an understanding of the stability relationships of apatites in terms of structure and chemical bonding is of interest. The apatite group of minerals is usually described by the general formula AI4AII6(BO4)6X2 in
correspondence with the contents of a hexagonal unit cell of space group P63/m, i.e. the most common apatite structure
type. The AI and AII distinct crystallographic sites generally accommodate larger divalent (Ca2+, Sr2+, Pb2+, Ba2+, etc.), monovalent (Na+, Li+, etc.), and trivalent (Y3+, La3+, Ce3+, Nd3+, Sm3+, Dy3+, etc.) cations, B cation sites are filled by smaller 3+, 4+, 5+, 6+, and 7+ metals and metalloids (P5+, As5+, V5+, Si4+, etc.), and the X anion site is usually occupied by halides (F-, Cl-, Br-, I-), hydroxyl, or oxygen. The structure is remarkably tolerant to complex chemical substitutions in which charge balance is normally achieved by means of altervalent cation substitutions or through appropriate mixing of monovalent and divalent X-anions and/or X-site vacancies. Cation ordering can also occur over crystallographically distinct AI, AII, and/or B sites, which can promote lower symmetry structure types (e.g. P63, P1121/m, P1121/b, etc.). All
apatite structure types can be conceived as constructed from the following structural units: (1) a hexagonal (or pseudo-hexagonal) network defining the ab-plane which consists of face-connected AIO6 polyhedra stacked along c that share
corners with BO4 tetrahedra; and (2) AIIO6X1,2 polyhedra with irregular coordination located within the tunnels
extending in the c direction that are formed by the (AIO6)-(BO4) polyhedral arrangement. In recent contributions (White et al. 2004; Dong and White 2004a,b; White and Dong 2003), geometrical aspects of the apatite structure were derived from regular triangular anion nets and deviations from this idealization proved useful for tracking systematic crystallographic changes in solid-solution series. Here we present a crystal-chemical analysis of Rietveld refinement results (Dong and White 2004a,b) and ab initio simulations for calcium-lead fluoro-vanadinites based on a geometrical parameterization of P63/m apatite (Mercier et al. 2005) that allows the predictions of unit-cell parameters and all atomic
positions in the crystal structure.
Equivalence of the crystallographic description in P63/m space group with a geometrical parameterization based
on crystal-chemical parameters
Assuming iso-bond-flattened BO4 tetrahedra and cation-centered AIO6 polyhedra, one can show (Mercier et al. 2005)
that the crystallographic description of P63/m apatite (a, c, 12 atom coordinates = 14 crystallographic parameters) is
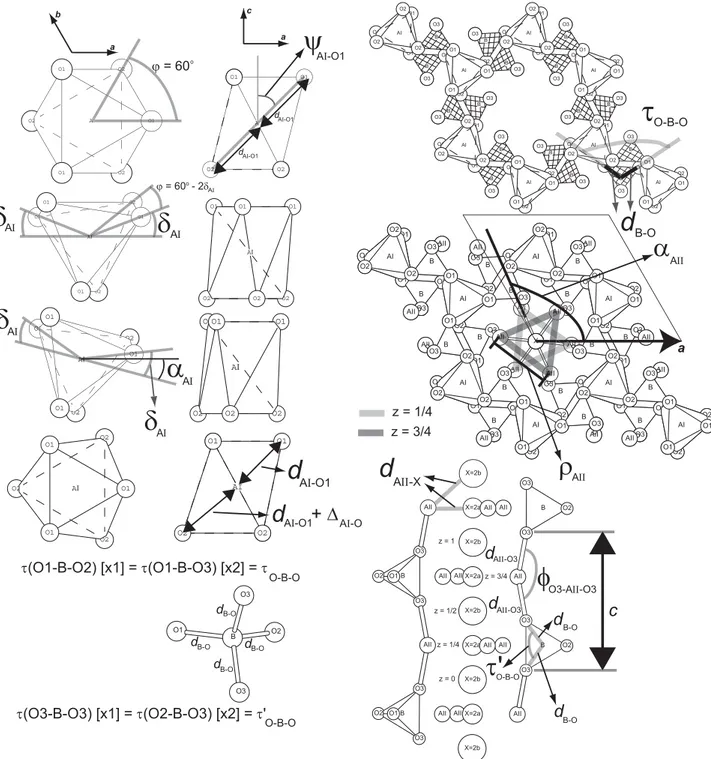
equivalent to a geometrical parameterization based on the following crystal-chemical parameters (Fig. 1): AI-O1 bond length, dAI-O1; difference between AI-O1 and AI-O2 bond lengths, ∆AI-O; angle between an AI-O1 bond and c, ψAI-O1;
counter-rotation angle of AIO6 polyhedra, δAI; orientation of AIO6 polyhedra with respect to a, αAI; tetrahedral BO4 bond
length, dB-O; tetrahedral O-B-O flattening angle, τO-B-O; AII-AII triangular side length, ρAII, or AII-X bond length, dAII-X;
orientation of AII-AII-AII triangles with respect to a, αAII; AII-O3 bond length, dAII-O3; and O3-AII-O3 bond angle, φO3- AII-O3. This geometrical parameterization allows the prediction of all atomic positions for an apatite having a unit cell of P63/m space-group symmetry, i.e. gives the unit-cell parameters and the fractional atomic coordinates for each atom in
the asymmetric unit as functions of 10 algebraically-independent crystal-chemical parameters (dAI-O1, ∆AI-O, δAI, αAI, d B-O, τO-B-O, ρAII or dAII-X, αAII, dAII-O3, and φO3-AII-O3). The difference in the number of independent parameters (14 for the
crystallographic description vs. 10 for the geometrical parameterization) is due to 4 geometric constraints, three of them arising from the assumption of flattened BO4 tetrahedra with four equal B-O bond lengths and the fourth constraining to
zero the z coordinate of the AI cations so as to have cation-centered AIO6 polyhedra. The geometrical parameterization
provides a 1st set of equations for the production a crystallographic description given the crystal-chemical parameters. The decomposition of the crystallographic description of P63/m apatite into its crystal-chemical parameters produces a
2nd set of equations. The two sets of equations are not entirely equivalent because of the reduction from 14 crystallographic parameters to 10 crystal-chemical parameters and 4 geometric constraints. A given set of cell parameters and atomic coordinates then produces an equivalent set of crystal-chemical parameters through use of the 2nd set of equations. Processing those crystal-chemical parameters through equations of the 1st set produces unit-cell parameters and atomic coordinates that are marginally different from the initial ones because the structure has been regularized by the constraints. But after this first incompletely reversible transformation, the transformation is then fully reversible, i.e. the atomic coordinates and lattice parameters of the regularized structure that produced a given crystal chemistry through the equations of the 2nd set are reproduced to numerical accuracy by the equations of the 1st set.
O2 O2 O1 O1 O1 O2 O1 O2 a b a c AI 2 O1
ψ
AI-O1 d AI-O1 d AI-O1 AI O1 O2 AI AI ϕ = 60o O1 O2 O2 O1 AI O1 O2 O2 O1 O1 AII O1 AII O2 A A δδ
AΙ AΙ O2 IIII ϕ = 60 - 2δ
AΙ o O2 O2 O1 O1 O1 O2 O2 O1 AI O1 O2 AII AII O1 AII O2 A A Aα
AΙ AΙ AΙδ
δ
O2 O2 O2 AI O1 O1 O1 O2 O1 AI O2 O1 O1 O2d
AI-O1d
AI-O1+
∆
AI-O O3 O3 O3 O3 O3 O3 O3 O3 B O1 B O2 O1 O1 O1 O2 O1 B O1 O1 O2 O2 O2 O2 B O2 B B O2 B O2 O2 O1 B O1 O1 O2 O3 O3 O3 O3 O3 O3 O3 O3 AI AI AI AI AI AI AI O3 O3 O3 O3 O3 O3 O3 O3 O1 B O1 B B O1 O2 O1 O2 O1 O2 O1 O2 O2 O2 B B B B O2 O2 O2 O2 O2 O2 O1 B O1 O1 O1 O1 O3 O3 O3 O3 O3 O3 O3 O3 O2 O1 B O3 O3 τ(O1-B-O2) [x1] = τ(O1-B-O3) [x2] = τ τ(O3-B-O3) [x1] = τ(O2-B-O3) [x2] = τ' O-B-O d B-O d d d B-O B-O B-Od
B-Od
O2 O1 O O1 AAI O-B-Oτ
O3 O3 O3 O3 O3 O3 O3 O3 O1 O2 O1 O1 O2 O2 B B B B B X B O2 O2 O1 I O1 O2 AII O1 AII O2 O1 O1 O1 AII O1 O2 AII O1 O2 O2 B AII O2 O2 B O1 O3 O3 O3 O3 O3 O3 O3 O3 AI AI AI AI AI AI AI AI O3 O3 O3 O3 O3 O3 O3 O3 O1 O1 O1 O2 O2 O1 O1 O2 AII O2 O1 O2 AII AII O2 B O1 O2 B O1 B B O2 O2 O2 X O1 AII O1 B O1 AII B O1 B O2 O2 B O3 O3 O3 O3 O3 O3 O3 O3 AI AI AI AI AI AI AI AI O3 O3 O3 O3 O3 O3 O3 O3 O2 O1 B O2 O1 O1 AII AII I O2 B O2 O2 B O1 O1 O1 O2 O1 O1 O2 O1 O2 O2 O1 B B O1 B O2 O1 AII X O2 B O2 AII B AII O3 O3 O3 O3 O3 O3 O3 O3 AI AI AI AI AI AI AI AI O3 O3 O3 O3 O3 O3 O3 O3 O1 B O1 X B O2 O1 B O1 B O1 AII O2 O2 AII O2 O2 O1 AII O1 O1 B B O1 O2 O2 B O2 O2 B O2 O1 O1 O1 O2 AII O2 AII O3 O3 O3 O3 O3 O3 O3 O3 aα
AΙΙ AI A I II A A III A A A A AIII A A AII AI AI AII A AI A z = 1/4 z = 3/4ρ
AΙΙ AII AII O2 O2 AII AII O3 O3 O3 O3 B B O1 O1 AII AII X=2b X=2a X=2b X=2a X=2b X=2a X=2b X=2a X=2b AII AII O1 O1 B O3 O3 O3 AII AII O2 O2 AII AIIc
φ
O3-AΙΙ-O3 AΙΙ-O3d
d
AΙΙ-O3 B B B Bd
B-Od
B-Oτ'
O-B-Od
AΙΙ-X z = 1/4 z = 0 z = 1/2 z = 3/4 z = 1 O-B-OFig. 1. Diagrams illustrating the crystal-chemical parameters used in the geometrical parameterization of P63/m apatite.
Ab initio modeling
The modeling and ab initio interface software environment Materials Toolkit (Le Page et al. 2002; Le Page and Rodgers 2005) was used to prepare input files for ab initio optimizations of cell and coordinates for the materials Ca10(VO4)6F2, Ca4Pb6(VO4)6F2 and Pb10(VO4)6F2 with VASP (Kresse 1993; Kresse and Hafner 1993, 1994). The
following execution parameters were used: GGA PAW potentials (Kresse and Joubert 1999), electronic convergence at 1x10-7 eV, convergence for forces of 1x10-4 eV/Å, Davidson-blocked iterative optimization of the wave functions in combination with reciprocal-space projectors (Davidson 1983), a 2x2x2 k-mesh for the reciprocal space integration with a Monkhorst-Pack scheme (Monkhorst and Pack 1976), and a Methfessel-Paxton smearing scheme with order 1 and width 0.2 eV for energy corrections (Methfessel and Paxton 1989). Spin polarization corrections were not used. Thirty single-point minimization iterations took about 20 hours per structure on a single 3-gigahertz Athlon-64 PC running serial VASP.4.6.3 under Windows XP.
Mercier et al. 2005
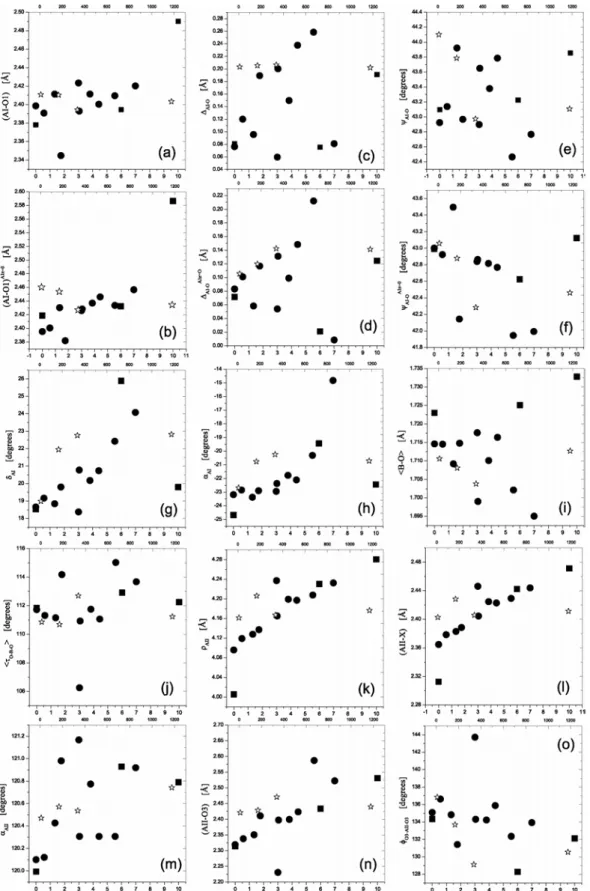
Fig. 2. Crystal-chemical parameters extracted for calcium-lead fluoro-vanadate apatites plotted as function of the lead
fraction x (PbxCa10-x where x = 0 to 10) or annealing time t in hours (t = 0 to 1200). Filled circles = Dong and White
(2004b); open star = Dong and White (2004a); filled squares = ab initio cell-and-coordinate optimizations.
Crystal-chemical analysis of calcium-lead fluoro-vanadate apatites
The crystal-chemical parameters of calcium-lead fluoro-vanadate apatites (Fig. 2) were extracted from the crystallographic parameters of Rietveld and ab initio optimized structures using the 2nd set of equations given by Mercier et al. (2005). The resulting crystal-chemical parameters plotted in Figure 2 are as follows: (a) (AI-O1) is the length of the three AI-O1 bonds in a given AIO6 polyhedra; (b) (AI-O1)AIz=0 is the length that the three AI-O1 bonds
would have if fractional atomic coordinate z(AI) = 0; (c) ∆AI-O = (AI-O1) - (AI-O2), where (AI-O2) is the length of the
three AI-O2 bonds in a given AIO6 polyhedra; (d) ∆AI-OAIz=0 = (AI-O1)AIz=0 - (AI-O2)AIz=0, where (AI-O2)AIz=0 is the
length that the three AI-O2 bonds would have if fractional atomic coordinate z(AI) = 0; (e) ψAI-O1 is the angle that the AI
-O1 bonds make with respect to c; (f) ψAI-O1AIz=0 is the angle that the AI-O1 bonds would make with respect to c if
fractional atomic coordinate z(AI) = 0; (g) δAI is the counter-rotation angle of AIO6 polyhedra; (h) αAI is the orientation
of AIO6 polyhedra with respect to a; (i) <B-O> is the average of the four bond lengths in any given BO4 tetrahedron
(<B-O> = 1/4·[(B-O1)+(B-O2)+ 2·(B-O3)]); (j) <τO-B-O> = 1/3·[τ(O1-B-O2) + 2·τ(O1-B-O3)]; (k) ρAII is the AII-AII
triangular side length; (l) (AII-X) is the length of the AII-X bond in the AIIO6X polyhedra; (m) αAII is the orientation of AII-AII-AII triangles with respect to a; (n) (AII-O3) is the length of the AII-O3 bonds within a given chain of AII -O3-B-O3-AII-etc. atoms in a given tunnel of the apatite structure; and (o) φO3-AII-O3 is the value of the O3-AII-O3 bond angle
within a given chain of AII-O3-B-O3-AII-etc. atoms in a given tunnel. Mercier et al. (2005) compare experimental results of 18 end-member apatite materials studied with single-crystal and Rietveld analysis with computed results of ab initio simulations. The scatter of experimental values for the same materials is observed to be large. Good agreement of ab initio crystal-chemical parameters is observed with only those from single-crystal experiments with the best residuals. When interpreted in this light, Figs. 2 a-o indicate a general agreement of trends between Rietveld experimental results and modeling, but suggest that accurate single-crystal work may still be needed to locate accurately oxygen atoms close to metal atoms as heavy as calcium and lead. Alternatively, one could perform constrained Rietveld-like analyses where the crystal-chemical parameters are refined instead of the usual crystallographic parameters (a, c, atom coordinates). This may reduce parameter trade-off in the least-squares minimization process and result in more accurate Rietveld crystal-structure determinations. Analysis of structure types in terms of crystal-chemical parameters provides a more intuitive framework for the discussion of polyhedral distortions than the independent crystallographic parameters. Ab initio methods currently emerge as an important and relatively inexpensive approach complementary to experiment.
Acknowledgements
This work was supported through the NRC-A*STAR Joint Research Programme: “Advanced Ceramic Methods for the Co-stabilisation and Recycling of Incinerator Fly Ash with Industrial Wastes”.
References
Chen X, Wright JV, Conca JL, Peurrung LM. (1997) Evaluation of heavy metal remediation using mineral apatite. Water, Air, and Soil Pollution 98: 57-78.
Crannell BS, Eighmy TT, Krzanowski JE, Eusden Jr. JD, Shaw EL, Francis CA. (2000) Heavy metal stabilization in municipal solid waste combustion bottom ash using soluble phosphate. Waste Management 20: 135-148.
Davidson, E.R. (1983) Methods in Computational Molecular Physics edited by G.H.F. Diercksen and S. Wilson Vol. 113 NATO
Advanced Study Institute, Series C (Plenum, New York), p. 95.
Dong ZL, White TJ, Wei B, Laursen K. (2002) Model Apatite Systems for the Stabilization of Toxic Metals: I, Calcium Lead Vanadate. J. Am. Ceramic Soc. 85: 2515-2522.
Dong ZL, White TJ. (2004a) Calcium-lead fluoro-vanadinite apatites. I. Disequilibrium structures. Acta Crystallogr. B60: 138-145. Dong ZL, White TJ. (2004b) Calcium-lead fluoro-vanadinite apatites. II. Equilibrium structures. Acta Crystallogr. B60: 146-154. Eighmy TT, Crannell BS, Krzanowski JE, Butler LG, Cartledge FK, Emery EF, Eusden Jr. JD, Shaw EL, Francis CA. (1998)
Characterization and phosphate stabilization of dusts from the vitrification of MSW combustion residues. Waste Management 18: 513-524.
Ioannidis TA, Zouboulis AI. (2003) Detoxification of toxic lead-loaded industrial solid waste by stabilization using apatites. J. Hazardous Materials B97: 173-191.
Kim JY, Dong ZL, White, TJ. (2005) Model Apatite Systems for the Stabilization of Toxic Metals: II, Cation and Metalloid Substitutions in Chlorapatites. J. Am. Ceramic Soc., in press.
Kresse G. (1993) PhD thesis, Technische Universität Wien, Austria.
Kresse G, Hafner J. (1993) Ab initio molecular dynamics for open-shell transition metals. Phys. Rev. B, 48: 13115-13118.
Kresse G, Hafner J. (1994) Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B, 49, 14251-14269.
Kresse G, Joubert J. (1999) From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B, 59, 1758-1775. Le Page Y, Rodgers J. (2005) Quantum software interfaced with crystal-structure databases: tools, results and perspectives. J. Appl.
Cryst., in review.
Le Page Y, Saxe P, Rodgers J. (2002) Symmetry-general ab initio computation of physical properties using quantum software integrated with crystal structure databases : results and perspectives. Acta Crystallogr. B58: 349-357.
Mercier PHJ, Le Page Y, Whitfield PS, Mitchell LD, White TJ, Davidson IJ. (2005) Geometrical parameterization of the crystal chemistry of P63/m apatites: Comparison with experimental data and ab initio results. In preparation.
Methfessel M, Paxton AT. (1989) High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B, 40: 3616-3621. Monkhorst HJ, Pack JD. (1976) Special points for Brillouin-zone integrations. Phys. Rev. B, 13: 5188-5192.
Valsami-Jones E, Ragnarsdottir KV, Putnis A, Bosbach D, Kemp AJ, Gressey G. (1998) The dissolution of apatite in the presence of aqueous metal cations at pH 2-7. Chemical Geology, 151: 215-233.
White TJ, Ferraris C, Kim JY, Madhavi S. (2004) “Apatite–An adaptive structural framework” (invited contribution), Micro-and mesoporous mineral phases, December 6-7, Rome, Italy.
White TJ, Dong Z. (2003) Structural derivation and crystal chemistry of apatites. Acta Crystallographica B59: 1-16.
Zhang P, Ryan JA. (1999) Transformation of Pb(II) from Cerrusite to Chloropyromorphite in the Presence of Hydroxylapatite under varying conditions of pH. J. Environ. Sci. Technol. 33: 625-630.