Fast tree aggregation for consensus hierarchical clustering
Texte intégral
Figure
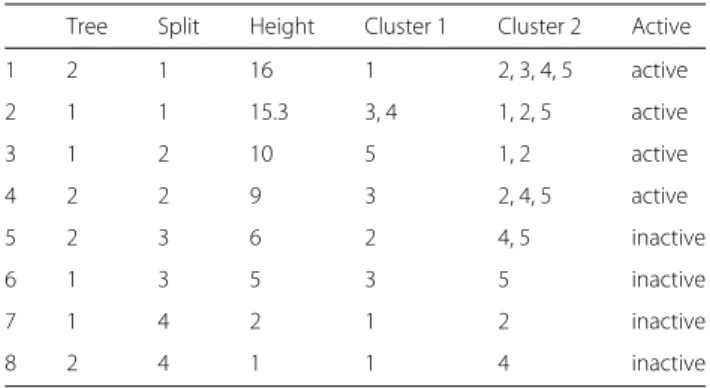
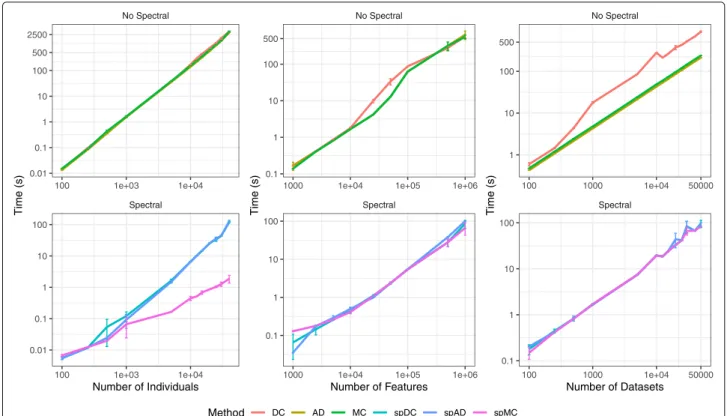
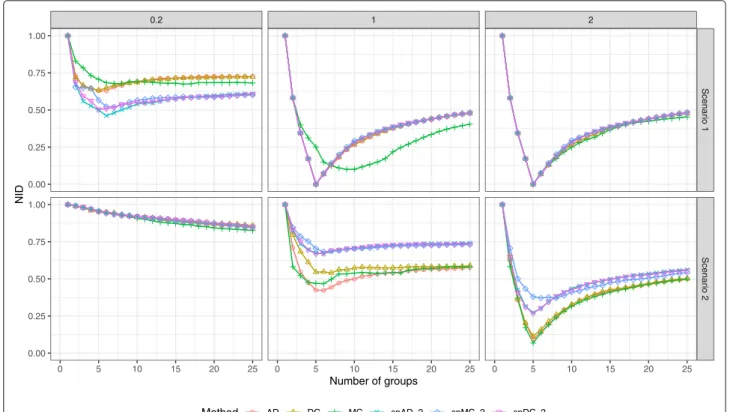
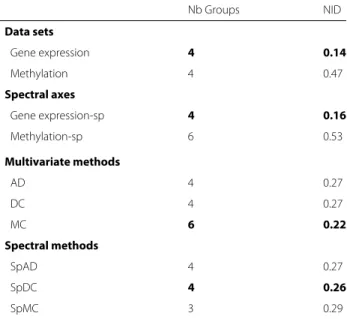
Documents relatifs
Another example of such a statistic is provided with the Denert statistic, “den”, introduced recently (see [Den]) in the study of hereditary orders in central simple algebras1.
In this article, we continue this tradition of tabulating automorphic forms on Shimura varieties by computing extensive tables of Hilbert modular cusp forms of parallel weight 2
The proof of theorem 4.1 actually shows that the heat kernel associated to the Lichnerowicz operator is positivity improving in case (M n , g 0 (t)) t≥0 is an expanding gradient
Then, with this Lyapunov function and some a priori estimates, we prove that the quenching rate is self-similar which is the same as the problem without the nonlocal term, except
First introduced by Faddeev and Kashaev [7, 9], the quantum dilogarithm G b (x) and its variants S b (x) and g b (x) play a crucial role in the study of positive representations
In order to describe the evolution of the PSD in an emulsion polymerization system mathematically, it is necessary to account for the various phenomena capable of effecting the
T is the address of the current subterm (which is not closed, in general) : it is, therefore, an instruction pointer which runs along the term to be performed ; E is the
15 and Claverie and Ogata 4 , claim that viruses (particularly large DNA viruses such as the Mimivirus) can indeed be placed in a TOL on the basis of phylogenetic analysis of
