Disassembly of Electron Transport Chain Complexes Drives
Macrophage TLR Responses by Reprogramming Metabolism and
Translation
By Yang Su B.S., Biology Rice UniversitySUBMITTED TO THE DEPARTMENT OF BIOLOGY IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY AT THE
MASSACHUSETTS INSTITUTE OF TECHNOLOGY MAY 2020
© 2020 Massachusetts Institute of Technology. All rights reserved.
Signature of Author: Yang Su Department of Biology May 15, 2020 Certified By: Jianzhu Chen Professor of Biology Thesis Supervisor Accepted By: Amy Keating Professor of Biology Co-Director, Biology Graduate Committee
Disassembly of Electron Transport Chain Complexes Drives Macrophage TLR Responses by Reprogramming Metabolism and Translation
By Yang Su
Submitted to the department on May 15 2020, in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biology
Abstract
Metabolic switch from oxidative phosphorylation (OxPhos) to glycolysis is a key feature of inflammatory response in macrophages, but how this switch occurs in response to inflammatory signals and how it precisely contributes to macrophage function is still obscure. Here we show that stimulation of macrophages through Toll-like receptors (TLR) disrupts the assembly of mitochondrial electron transfer chain (ETC) complexes I-V, leading to the metabolic switch by inhibiting OxPhos and activating HIF-1α-dependent glycolysis. Disassembly of ETC complexes influences the global metabolic status of macrophages not only by inducing glycolysis but also largely by inducing the reprogramming of cellular translational capacity via mTORC1 and ATF4, leading to enhanced global translation rate, cell growth, and production of inflammatory cytokines. Inhibition of OxPhos via myeloid-specific knockout of OPA1, which stimulates ETC complex assembly, exacerbates sepsis in mice while inhibition of mTORC1 reverses this effect. These findings reveal that disassembly of ETC complexes underlies macrophage metabolic switch and inflammatory responses and may be a conserved pathway to reprogram cellular anabolism and function.
Thesis supervisor: Jianzhu Chen Title: Professor of Biology
Table of Contents
Title page 1
Abstract 3
Chapter 1: Introduction 6
Chapter 2: ETC complex disassembly underlies the switch from OxPhos to
glycolysis in macrophage TLR response 17 Chapter 3: ETC complex disassembly promotes macrophage function by
reprogramming translation 51 Chapter 4: Discussion and future directions 101
Chapter 1.
Introduction
This chapter was written by Yang Su, with edits by Dr. Jianzhu Chen, Dr. Teemu Miettinen, Dr. Mary Mu, and Dr. Tracy Anthony.
Macrophages Metabolism and Inflammatory Toll-like Receptor (TLR) responses
Macrophages play a critical role in immunity against bacterial, viral, and fungal pathogens and parasites (Kawai and Akira et al., 2007). Activation of inflammatory responses by macrophage relies on the sensing of pathogen through surface and intracellular pattern recognition receptors, e.g. Toll-like receptors (TLR), which recognize various microbial components including lipopolysaccharide (LPS), unmethylated CpG (CpG), and viral nucleic acids. Various TLR receptors differentially recognize microbial products, with the recognition occurring either intracellularly or extracellularly. For example, the bacterial outermembrane component LPS is recognized extracellularly via TLR4, while CpG and viral nucleic acids are recognized intracellularly through the surface of intracellular compartments by TLR9 and TLR3/7/8, respectively. Upon ligation of TLRs, macrophages activate a signaling cascade through adaptors Myd88/TRIF, protein kinases (e.g. IRAK family), and downstream transcription factors (e.g., NF-κB) responsible for activating the transcription of hundreds to thousands of genes critical for immune responses. In macrophages, such immune responses consist of phagocytosis and degradation of microbial pathogens through the phagosome, increased antigen presentation, as well as mobilization of adaptive immunity and further amplification of immune responses through inflammatory signaling. Inflammatory signaling from macrophages to tissues and other immune cells is enabled by increase surface-surface interaction (e.g., through co-stimulatory molecules such as CD86) as well as through paracrine and exocrine release of cytokines (e.g., TNF), chemokines (e.g., CCL2), and other chemicals and reactive molecules (e.g., reactive oxygen species and nitric oxide). Although such macrophage immune responses are evolutionary adaptations to pathogenic microbes, excessive inflammatory responses are important contributors to tissue damage in disease (Kawai and Akira et al., 2007). For example, excessive cytokine production and signaling in circulation during microbial infections result in lethal systemic inflammatory response known as sepsis. Thus, a deep understanding of TLR responses and its influence on the interaction between pathogen and host is important for designing therapy against microbial infectious disease and other inflammatory conditions.
It is now appreciated that functional changes in TLR-activated macrophages are closely linked to simultaneous metabolic alterations that enable macrophage function by providing necessary cellular conditions for phagocytosis, bacterial killing, and cytokine production. During TLR responses, macrophages dramatically remodel their metabolic pathways such that the synthesis of antimicrobial and inflammatory metabolites are favored. For example, among such molecules are mitochondrial metabolites itaconate and succinate, generated mostly through the extracellularly imported metabolite glutamine following a drastic alteration of the tricarboxylic acid (TCA) cycle that typically serves to maintain cellular energy homeostasis. Itaconate directly inhibits essential bacterial metabolic enzymes, while succinate further serves to amplify host immune responses by stimulating the expression of the inflammatory cytokine IL-1β, which acts in mobilization of innate and adaptive immunity (Tannahill et al., 2013). Such repurposing of essential cellular function from homeostasis to immune response is a now common theme across many metabolic pathways during macrophage TLR response. Among the various metabolic changes in TLR-activated macrophages, the most dramatic but still largely unexplained metabolic alteration in TLR-activated macrophages is the switch from oxidative phosphorylation (OxPhos) to glycolysis, now broadly referred to as Warburg metabolism (Tannahill et al., 2013, Vander Heiden et al., 2009). Although a cellular switch from OxPhos to glycolysis, initially described and popularized by Otto Warburg in 1930s, is a well-documented general phenomenon observed in many cell types in the context of differentiation and modulation of cellular plasticity, how this form of metabolism ultimately enables proper cellular function under specific conditions is still an active area of investigation in diverse fields of biology and medicine. In macrophages, the mechanism by which TLR ligands initially trigger the switch of OxPhos to glycolysis during inflammatory responses is still unknown, and it is unclear how this switch ultimately provides the physiological condition to enable the function of macrophages during immune responses.
Glycolysis, OxPhos, and the Effect of the Metabolic Switch on Cells
To understand how the switch from OxPhos to glycolysis could enable macrophage function during inflammatory response, we studied the mechanism by which
macrophages shut down OxPhos and induce glycolysis in response to TLR stimuli, and how this switch would influence the global transcriptomic and functional state of macrophages to modulate TLR responses.
Glycolysis and OxPhos are two key metabolic pathways by which eukaryotic cells metabolize nutrients to produce ATP. In brief, glycolysis occurs in the cytosol where glucose is phosphorylated and catabolized into pyruvate to produce NADH, H+ and ATP. Mitochondrial metabolism can further catabolize pyruvate generated from glycolysis as well as other carbon sources obtained elsewhere (e.g. glutamine) through the tricarboxylic acid (TCA) cycle to generate ATP and reducing molecules NADH and FADH2, with CO2 and H2O being generated as byproducts. During OxPhos, electron transfer chain (ETC) Complexes I-IV couple the transfer of electrons from NADH and other reducing equivalents in the mitochondria to ATP production. This process is enabled by formation of proton (H+) gradient across the mitochondrial inner membrane, which provides the necessary thermodynamic condition for ATP synthesis via Complex V, also known as ATP synthase. Electrons transfer sequentially from Complex I or II to coenzyme Q (CoQ), then on to Complexes III and IV (Figure 1). Each ETC complex and ATP synthase consists of differing number of subunits, forming high molecular weight complexes. Each ETC complex has also been described to form super- and mega-complexes with other ETC complexes, such as Complex I binding to Complexes III and IV to form what is known as respiratory supercomplex or the respirasome, observed in vitro in mitochondrial fractions of native PAGE. Each respiratory supercomplex can also dimerize to form the respiratory megacomplex (Jiang et al., 2016). Such high molecular weight complex interactions are thought to facilitate the proper transfer of electrons during OxPhos. The function of the ETC in cells is elucidated through pharmacological inhibitors of ETC complexes that include rotenone (inhibiting electron transfer from Complex I to CoQ), antimycin (inhibiting electron transfer from CoQ to Complex III), and oligomycin (inhibiting ATP synthase) (Figure 1). Glycolysis is the less efficient of the two modes in generating ATP, forming only 2 net molecules compared to more than 30 per glucose molecule through TCA cycle and OxPhos.
Despite glycolysis having less capacity of generating ATP from a single glucose molecule compared to OxPhos, many cell types prefer glycolysis as the main mode of energy metabolism. The precise reasons are various and may be cell-type dependent. For example, in cancer cells, glycolytic intermediates generated by breaking down glucose can in turn serve as intermediates in biosynthetic reactions to form cellular building blocks such as nucleic acids and amino acids (e.g., serine). As such building blocks are necessary for cell growth and division, in many cancer cells undergoing Warburg metabolism, OxPhos is thought to be the less favorable of the two metabolic pathways, with one possible reason being that carbon molecules (e.g. pyruvate resulting from glycolysis) are broken down to CO2 through the TCA cycle/OxPhos and thus is prevented from being incorporated into cells (Vander Heiden et al., 2009). Thus, glycolytic metabolism is thought to favor anabolism. To what extent these ideas also apply in macrophages, or whether some alternative mechanisms might exist to support biosynthesis and anabolism during inflammatory responses, remains unclear.
Figure 1: Diagram of electron transfer chain (ETC) and its inhibitors. The electron
transfer chain, with Complexes I-IV, CoQ, cytochrome C, and ATP synthase being shown. The substrates and O2 are also listed. Also shown are the site of electron transfer block/ATP synthesis by rotenone, antimycin, and oligomycin.
Optic Atrophy 1 (OPA1) and the ETC
For many decades, the function of the electron transport chain has been known to be closely linked to the mitochondrial morphology, particularly the shape and surface area
of the mitochondrial inner membrane on which the ETC complexes and ATP synthase are present. The mitochondrial inner membrane forms folded structure protruding into the mitochondrial matrix known as the inner membrane cristae, readily observed by a transmission electron microscope (TEM). It has been observed that different cell types harbor different numbers of the mitochondrial cristae structure with varying shapes and widths of folding, and it is hypothesized that the cristae shape governs the assembly state of ETC complexes and supercomplexes and determine the efficiency of electron transfer through the ETC complexes during OxPhos. The cristae are formed and supported by many structural proteins present at inner membrane, forming junction sites. These structural proteins involve the optic atrophy 1 (OPA1), the ATP synthase, and the multi-protein MICOS complex. In the last decade, it has become increasingly clear these structural proteins required for the formation of the cristae, such as OPA1, also in fact support the assembly of the ETC complexes and supercomplexes (Cogliati et al., 2013). It is now also known that, physiologically, cells use the mechanism of cristae formation and resolution to regulate the assembly status of ETC complexes in cells in response to stress signals, such as apoptotic stimulation, so as to license the release of the ETC component and apoptotic stimulator cytochrome C as well as to switch the role of the ETC from ATP production to reactive oxygen species (ROS) generation (Jiang et al., 2016). Whereas knockout of OPA1 results in partial ETC complex disassembly and sensitizes cells to apoptosis, overexpression of OPA1 enhances ETC complex assembly and protects cells from apoptosis (Cogliati et al., 2013). Whether such mechanism exists in macrophages or whether OPA1/mitochondrial cristae are involved in inflammatory responses is unknown.
Here we examined the role of OPA1 in regulating ETC complex assembly and inflammatory response of macrophages. Besides its role in mitochondrial cristae formation, OPA1 is also involved in mitochondrial fission/fusion. Mechanistically, two isoforms of OPA1, L- and S-OPA1, are thought to oligomerize to promote mitochondrial cristae formation and the fusion of inner membrane. A higher oligomerized state of OPA1 is correlated with increased cristae formation and ETC complex assembly. As the two functions of OPA1 in ETC complex assembly and mitochondrial fusion are separate and
independent, we distinguish the two roles of OPA1 in the context of inflammatory response by also examining the role of dynamin-related protein 1 (DRP1), which is a structural protein present at the mitochondrial outer membrane that is required for mitochondrial fission but not known to be involved in ETC complex assembly.
A Switch from OxPhos to Glycolysis is a General Phenomenon with Poorly Understood Mechanism
TLR-stimulated macrophages have long been known to undergo Warburg-like metabolism, involving both the inhibition of OxPhos and induction of glycolysis as a response to LPS ligation (Hard 1970). Although LPS-treated cells have long been observed to be inhibited for ETC complexes II and III (McGivney and Bradley, 1979), the mechanism is unknown. However, the mechanism of glycolytic induction in inflammatory macrophages is an active area of investigation. In the last two decades, macrophages have been well-characterized to induce glycolysis through the activation of the transcription factor hypoxia-inducible factor 1α (HIF-1α), which stimulates the expression of glycolytic genes and also has pro-inflammatory roles by binding directly to the promoter site of Il1b gene that encodes IL-1β either with or without hypoxia (Tannahill et al., 2013). In turn, HIF-1α protein stability relies on the presence of certain mitochondrial metabolites, such as succinate and fumarate (Cramer et al., 2003; Tannahill et al., 2013). How OxPhos inhibition modulates the accumulation of TCA metabolites or protein levels of HIF-1α in inflammatory macrophages, and thus the link between OxPhos inhibition and glycolysis induction, is still unknown.
The consequence of the metabolic switch in macrophages is also unclear. The switch from OxPhos to glycolysis, and vice versa, is not phenomenon unique to macrophages. During development, cellular differentiation, and physiological stress, many cell types reprogram their metabolism to rewire cell behavior or modulate functional plasticity. Many of these functional changes may not be directly related to the ability of these cells to generate ATP, given that glycolysis is the less efficient in ATP production compared to OxPhos, and in many cases such cells indeed maintain lower levels of ATP. For example, during embryonic development, the balance between glycolysis and OxPhos is also an
important determinant of pluripotency, with glycolysis favoring a more differentiated state (Demarco et al., 2019). As mentioned above, cancer cells prefer to use glycolysis rather than OxPhos, most likely to favor proliferation. The balance between glycolysis and OxPhos is also often altered differently during multiple stages of cellular differentiation. For example, T cells, when activated, initially activate both OxPhos and glycolysis (with glycolysis being the more active of the two) but return to an OxPhos-dependent metabolism to form memory when differentiated to memory T cells (Buck et al., 2015). Many of these changes may reflect evolutionary adaptations to particular microenvironment in which these cells are exposed. For example, glycolytic metabolism in cancer cells as well as inflammatory macrophages and T cells are typically concomitant with increased HIF-1α expression. Such cells are in fact known to be exposed to hypoxia, typical of a crowded tissue environment with excess cells present following uncontrolled proliferation as is the case in cancer, or increased immune cell infiltration as is the case during infection. Nevertheless, more recent studies report the interesting observation that inhibition of OxPhos lone even in the absence of hypoxia can also enhance cellular anabolism, often resulting in increased cellular growth in certain cancer cells and consistent with the Warburg hypothesis that this form of metabolism supports increased cell biomass accumulation (MacVicar et al., 2019; Miettinen et al., 2014). In contrast, by inhibiting glycolysis or HIF-1α expression, activated T cells that typically rely on glycolysis have been observed to differentiate into anti-inflammatory regulatory T cells (Treg) instead of pro-inflammatory T helper 17 (Th17) cells under Th17-polarizing conditions. Thus, a switch from OxPhos to glycolysis has a direct impact on cell physiology under various contexts and is not a phenomenon unique to macrophages. While these phenomena are well-described in various cell types, the mechanisms underlying the metabolic switch still remain obscure.
In this study, we examined such mechanism in macrophages. Given that macrophages appear to undergo a direct inhibition of ETC, as supported by LPS-induced inhibition of ETC complexes II and III (McGivney and Bradley, 1979), and that OxPhos-inhibited cells must rely more heavily on glycolysis to produce ATP, we suspected that OxPhos inhibition may precede glycolytic induction that is now known to involve HIF-1α and correlated to
pro-inflammatory responses. Indeed, here we show that ETC complexes are disassembled in TLR-stimulated inflammatory macrophages, accounting for the metabolic switch. Interestingly, ETC complex disassembly also resulted in a functional switch in macrophages, unexpectedly by enhancing global translation and cell growth via transcriptome rewiring. We further extend our hypothesis to other cell types and speculate that ETC complex disassembly is a general phenomenon enabling metabolic switch and cellular anabolism under various contexts in other cells.
mTORC1/ATF4 are General Regulators of Cell Anabolism and Growth
Functional changes occurring in differentiating cells are often coupled to anabolic changes necessary to meet their increased metabolic demand. A central regulator of protein synthesis and cell growth that senses and integrates nutrient and energy levels to control anabolic processes, often linked with glucose metabolism, is the mammalian target of rapamycin (mTOR) (Laplante and Sabatini, 2012). Various complexes of mTOR serve different and sometimes opposing functions depending on the context or cell type. mTOR forms two distinct complexes mTORC1 and mTORC2, of which mTORC1 stimulates translation by phosphorylating p70 S6 kinase (S6K; ribosomal kinase) and 4E-BP1 (translation initiation factor inhibitor) and is sensitive to the mTOR inhibitor rapamycin. mTORC1 drives inflammatory macrophage function, while mTORC2 drives anti-inflammatory/anti-parasitic macrophage function (Byles et al., 2013). Nevertheless, how mTORC1/2 are linked to energy metabolism, translation, and cell growth in macrophages and thus govern cellular status during immune response is unknown. Recently, the transcription factor, activating transcription factor 4 (ATF4) has emerged as an important downstream effector of global translation acting downstream of mTORC1, promoting the transcript induction of genes driving ribosome biogenesis, tRNA synthesis, translation initiation, and amino acid anabolism (Ben-Sahra et al., 2016; Han et al., 2013). Some ATF4 targets examined in this study are Aars (tRNA synthase), Slc3a2 (glutamine transporter), and Asns (asparagine synthase). Further, based on studies of primary mitochondrial disease and model organism ETC complex knockout tissues, mTORC1 and ATF4 have now been further linked to mitochondrial function. Surprisingly, such cells and tissues remain viable and accumulate both mTORC1 and ATF4 above normal levels,
indicating that anabolic pathways are upregulated in response to decreased OxPhos/OxPhos defect under certain contexts despite severe energy deficiency. These observations strongly suggest the presence of compensatory or alternative anabolic pathways acting specifically in ETC-inhibited cells, perhaps including TLR-stimulated inflammatory macrophages. Thus, inhibition of ETC is linked to alterations in cellular anabolic pathways and may drastically alter cellular function in ways not directly related to cell survival alone. How the mTORC1 and ATF4 pathways are related to mitochondrial metabolism and inflammatory response in macrophages is still unknown.
In this study, we use a transcriptomic network approach to identify mTORC1 and ATF4 as regulators of translation in macrophages during inflammatory response and induced following ETC complex disassembly. We assess the molecular pathways regulated by mTORC1 and ATF4 and examine the phenotype of macrophages knocked out or depleted for each. To measure de novo translation rate in macrophages under various conditions influencing ETC activity and translational activity, we use a well-described fluorescent assay analogous to the traditional 35S methionine assay with the same principle of measuring methionine incorporation following starvation and pulse, known as FlUorescent Non-Canonical Amino acid Tagging (FUNCAT). We confirm the validity of this assay in macrophages and show that ETC complex disassembly alone results in increased translation in macrophages, with mTORC1 and ATF4 being responsible for this effect.
ETC Complex Disassembly Drives Macrophage Inflammatory Response by Reprogramming Cellular Metabolism and Translation
In conclusion, we investigated the molecular mechanisms by which macrophages undergo metabolic switch and how this switch contributes to macrophage TLR responses. We show that a surprisingly simple mechanism mediated by the disassembly of ETC Complexes I-IV and ATP synthase drives both macrophage metabolic switch and inflammatory responses. Disassembly of ETC complexes is triggered by TLR stimulation and involves optic atrophy 1 (OPA1), which regulates mitochondrial fusion and cristae structure (Cogliati et al., 2013). Disruption of ETC complexes inhibits OxPhos and
activates glycolysis through HIF-1α, driving the metabolic switch. Functionally, the disassembly of ETC complexes also unexpectedly triggers various glycolysis-independent translational pathways to enhance macrophage anabolism via mTORC1 and ATF4, driving inflammation. Consistently, inhibition of OxPhos via myeloid-specific knockout of OPA1 exacerbates sepsis in mice, while inhibition of mTORC1 reverses this effect. These findings show that ETC complex disassembly is a common mechanism that not only underlies the TLR-induced metabolic switch but also reprograms cellular translation and growth pathways needed for macrophage inflammatory responses, thus coupling cellular metabolism to acquisition of new function.
Chapter 2.
ETC complex disassembly underlies the switch from OxPhos to
glycolysis in macrophage TLR inflammatory response
This chapter was written by Yang Su, with edits by Dr. Jianzhu Chen, Dr. Teemu Miettinen, Dr. Mary Mu, and Dr. Tracy Anthony.
TLR stimulation results in ETC complex disassembly
It has long been known that ETC Complexes II and III become inactivated as a result of TLR stimulation (McGivney and Bradley 1979), but the precise mechanism is still unknown. More recent studies have revealed mitochondrial morphology to govern mitochondrial function and does so largely by controlling the assembly status of the ETC. Whether TLR signaling modulates the mitochondrial ultrastructure or ETC complex assembly status in macrophages to regulate metabolism is unknown. As the absolute expression levels of ETC complex components (as commonly detected on denaturing PAGE) often do not reflect the assembly status of ETC complexes (Jiang et al., 2016) that requires special techniques to detect, previous reports have largely overlooked the possibility that inhibition of OxPhos in inflammatory macrophages might be caused by the disassembly of ETC complexes. To investigate whether TLR stimulation causes ETC complex disassembly, we used the Blue Native gel electrophoresis (BNGE) technique to detect ETC complexes at their native state. First, we stimulated mouse bone marrow-derived macrophages (BMM) with LPS, separated mitochondrial proteins by BNGE and SDS-PAGE in parallel, and detected subunits of ETC complexes I-V by Western blotting. Indeed, while the levels of specific ETC subunit components remained similar on SDS-PAGE, their presence in high molecular weight ETC complexes dramatically decreased over time after LPS stimulation on BNGE (Figure 2), suggesting that the native state of ETC complexes are altered by LPS but the absolute levels of individual subunits are not. As expected, VDAC (loading control for mitochondrial fraction) level was unaltered by LPS stimulation in mitochondrial fractions (Figure 2). Thus, LPS stimulation results in disassembly of Complexes I-V.
Figure 2: LPS stimulation induces ETC complex disassembly. Mouse BMM was not
treated or treated with LPS (100 ng/ml) for 6, 12, or 24 hours. Protein extracts were prepared from mitochondrial fraction of mouse BMM and separated by BNGE and SDS-PAGE. Western blotting with antibody specific for NDUFA9 indicates total NDUFA9 protein levels in mitochondria on SDS-PAGE and NDUFA9 in complex I on BNGE. Similarly, Western blotting with antibodies specific for SDHA, UQCRC2, and ATP5A indicates their levels in mitochondria on SDS-PAGE and in complex II, III and V on BNGE, respectively. Complex IV was detected with antibody specific for COX IV.
To assess whether ETC complex disassembly is conserved across multiple TLR signaling pathways other than LPS-dependent TLR4 signaling, we stimulated macrophages with CpG, which stimulates TLR9 located in intracellular compartments, as well as poly I:C, which are synthetic oligonucleotides mimicking viral nucleic acids recognized intracellularly by TLR3. CpG or poly I:C stimulation also decreased the levels of ETC complexes in BMM (Figure 3), suggesting that ETC complex disassembly is conserved across multiple TLR pathways that operate under different mechanisms.
Figure 3: Various TLR ligands induce ETC complex disassembly. Mouse BMM was
not treated or treated with LPS (100 ng/ml), CpG (5 μM), or poly I:C (10 μg/ml) for 6 hours. Protein extracts were prepared from mitochondrial fraction of mouse BMM or membrane
fraction of human MDM and separated by BNGE and SDS-PAGE. Western blotting was done as in Figure 2.
Many immune processes are not evolutionarily conserved between mouse and human. To determine whether ETC complex disassembly during TLR response occurs in human macrophages, we derived human monocyte-derived macrophages (MDM) from peripheral blood monocytes. LPS stimulation also decreased the levels of ETC complexes in human MDM, indicating that the pathway is conserved from mouse to human (Figure 4).
Figure 4: TLR-induced ETC complex disassembly is conserved in human. Human
MDM was not treated or treated with LPS (100 ng/ml) for 24 hours. Protein extracts were prepared from P15 membrane fraction of human MDM and separated by BNGE and SDS-PAGE. Western blotting was done as in Figure 2.
Although LPS is well-known to cause a switch from OxPhos to glycolysis in mouse BMM, whether the same metabolic switch occurs in human MDM is unknown. We measured OxPhos and glycolysis using the Seahorse metabolic assay, using MDM derived from multiple human donors. We found that oxygen consumption rate (OCR), a parameter of OxPhos, was significantly decreased whereas extracellular acidification rate (ECAR, a parameter of glycolysis) and ECAR/OCR ratio were increased in human MDM following LPS stimulation (Figure 5), indicating that a switch from OxPhos to glycolysis indeed occurs in human macrophages. Together, these results show that TLR stimulation disrupts ETC complexes in macrophages while switching metabolism from OxPhos to glycolysis in a highly conserved manner across various TLR signaling pathways and across species, providing a mechanistic explanation for the metabolic switch from OxPhos to glycolysis in inflammatory macrophages.
Figure 5: LPS induces a switch from OxPhos to glycolysis in human macrophages.
MDM from human donors (n=3) were treated with LPS (100 ng/ml) for 24 hours and basal OCR and basal ECAR were measured by Seahorse assay.
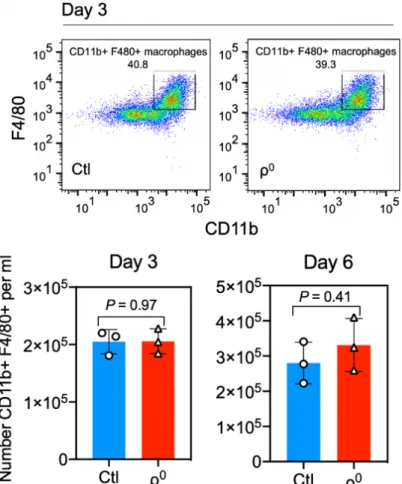
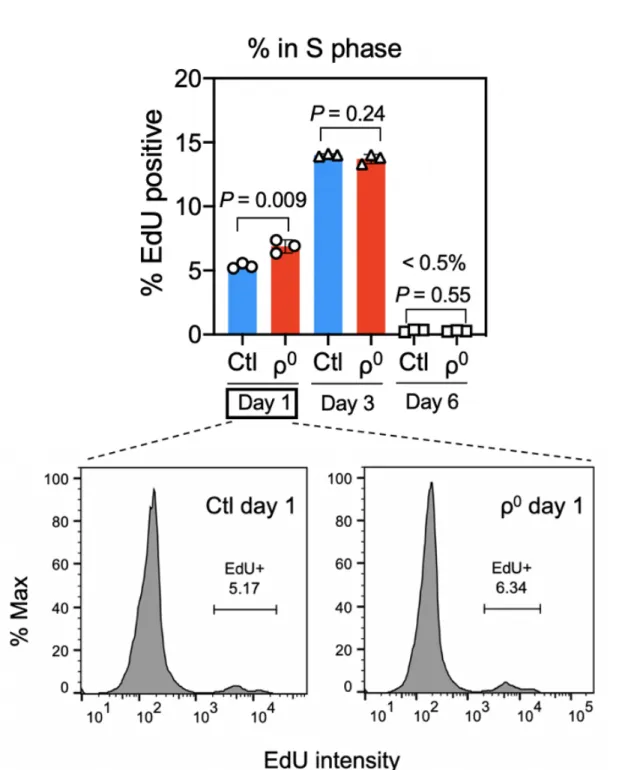
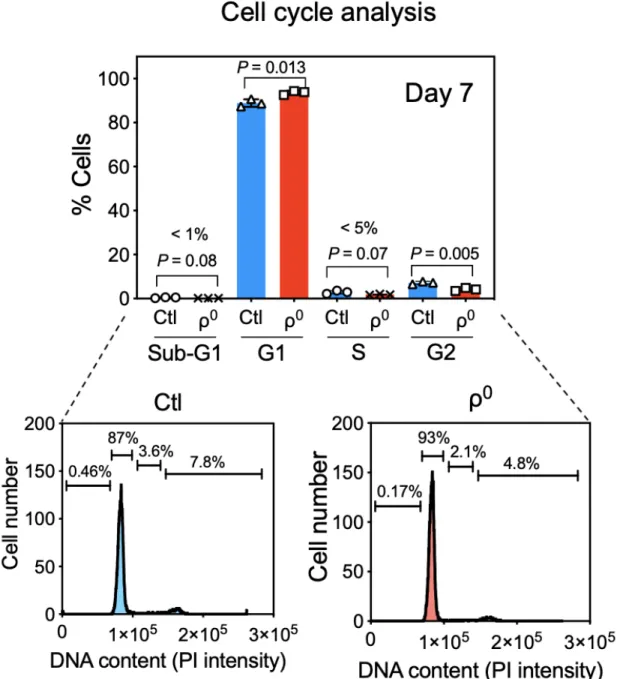
To investigate the consequence of ETC complex disassembly on macrophage phenotype and function, we constructed mouse BMM deficient in the ETC components by culturing bone marrow cells in the presence of ethidium bromide to deplete mtDNA (Chandel and Schumacker, 1999). Interestingly, ethidium bromide treatment did not compromise macrophage differentiation as indicated by normal expression of CD11b and F4/80, normal total cell counts (Figure 6), normal S-phase entry and exit (Figure 7), and normal cell cycle status (Figure 8), suggesting that macrophages have robust cellular mechanisms compensating for the lack of OxPhos, as expected from the fact that
PBS LPS
0
20
40
60
80
Basal OCR (pmol/min)
P = 0.006
PBS LPS
0
10
20
30
40
50
Basal ECAR (pmol/min)
P = 0.078
PBS LPS
0.0
0.5
1.0
1.5
ECAR/OCR ratio
P = 0.006activated macrophages naturally switch off OxPhos during inflammation. The resulting BMM differentiated under ethidium bromide, referred to as “ρ0” cells, were completely deficient in Complexes I, III, IV and ATP synthase (V), but not the nuclear-encoded Complex II (Figure 9).
Figure 6: ETC complex disassembly and ethidium bromide do not influence macrophage differentiation from bone marrow cells. Bone marrow cells (n=3 mice)
were differentiated for 3 (top) or 3 or 6 days (bottom) and stained for CD11b and F4/80 mature macrophage markers and subject to flow cytometry. The number of mature CD11b+ F4/80+ macrophages were counted per ml of culture media.
Figure 7: ETC complex disassembly and ethidium bromide do not influence entry to or exist from S phase during macrophage differentiation from bone marrow cells.
Bone marrow cells (mean +/- SD of n=3 mice) were differentiated for 1, 3, or 6 days and stained DNA synthesis activity using 5’-ethynyl-2’-uridine (EdU) and analyzed on flow cytometry for percent in S phase.
Figure 8: ETC complex disassembly and ethidium bromide do not influence cell cycle status during macrophage differentiation from bone marrow cells. Bone
marrow cells (mean +/- SD of n=3 mice) were differentiated for 7 days, fixed, and stained with propidium iodide nuclear dye and assessed for cell cycle status. Percentage of cells in each cell cycle phase is shown in top panel with representative plots shown below.
Figure 9: Differentiation of macrophages under ethidium bromide results in absence of assembled Complexes I, III, IV, and V. Mouse BMM was not treated or
treated with LPS (100 ng/ml) for 6 hours. Protein extracts were prepared from mitochondrial fraction of mouse BMM and separated by BNGE and SDS-PAGE, and then probed with antibodies specific to subunits of Complexes I-V.
Consistent with the near total absence of Complexes I, III, IV, and V, ρ0 BMM showed higher ECAR and lower OCR compared to normal BMM and responded minimally to oligomycin and FCCP (Figure 10), suggesting a near complete inhibition of OxPhos and a strong activation of glycolysis. We then assessed the inflammatory phenotype of ρ0 BMM. Interestingly, ρ0 BMM expressed higher levels of inflammatory marker CD86 compared to wild-type, either with or without LPS stimulation (Figure 11), and produced significantly higher levels of inflammatory moledules TNF, IL-6, CXCL-2, and NO following LPS stimulation compared to wild-type BMM (Figure 12). Thus, disruption of
ETC complexes through mtDNA depletion enhanced the inflammatory response under LPS, and similar to LPS, depletion of ETC complexes alone was sufficient to switch macrophage metabolism from OxPhos to glycolysis.
Figure 10: Elimination of Complexes I, III, IV, and V switches macrophage metabolism from OxPhos to glycolysis. Control and ρ0 BMM (n=3 per group) were subjected to Seahorse metabolic assay, measuring ECAR as a glycolysis parameter and
OCR as an OxPhos parameter. Oligo: oligomycin, FCCP: uncoupling agent, A/R: antimycin/rotenone. Data are representative of biological replicates.
Figure 11: ρ0 macrophages upregulate the inflammatory marker CD86. Control and
ρ0 BMM (n=3 per group) not treated or treated with LPS for 24 hours were stained for CD86 and analyzed for its expression by flow cytometry.
Figure 12: ρ0 macrophages upregulate inflammatory cytokines and chemokines.
6 or 24 hours and supernatant was analyzed for TNF, IL-6, CXCL-2, and nitric oxide (NO) levels by ELISA.
Loss of OPA1 Disrupts ETC Complex Assembly and Enhances Inflammation
Interestingly, ETC complex disassembly has been observed to occur during apoptosis (Jiang et al., 2016). We suspected that mechanistic parallels with the known apoptosis pathway might exist. As OPA1 oligomerization status is known to modulate ETC assembly and mitochondrial
cristae formation during apoptosis (Cogliati et al., 2016), partly through conversion of L-OPA1 to S-L-OPA1 (Jiang et al., 2016), we investigated whether LPS also influences OPA1 oligomerization and cristae stability in mouse BMM. Indeed, the levels of oligomeric OPA1 decreased over time following LPS stimulation when assessed on BNGE whereas the total levels of OPA1 or the relative levels of L- and S-isoforms did not change when assessed by SDS-PAGE (Figure 13A). A unique mechanism may exist to regulate OPA1 oligomerization state. Morphologically, LPS stimulation decreased the number of mitochondrial cristae and increased the cristae width per given cross section on transmission electron microscopy (TEM) (Figure 13B), thus demonstrating remodeling of the inner membrane cristae. These data suggest that macrophage metabolic switch during inflammation is governed by mitochondrial shape and OPA1, which in turn could regulate ETC complex assembly.
Figure 13: TLR-stimulated macrophages demonstrate OPA1 deoligomerization and fewer and wider cristae. (A) Wild-type macrophages were treated for 0, 6, 12, or 24
hours with LPS and mitochondrial fractions were isolated. Proteins were purified and separated on either BNGE or SDS-PAGE and probed with antibodies specific to OPA1 and VDAC. (B) Wild-type macrophages were either not treated or treated with LPS for 24 hours. Cross-sections of macrophages were observed on transmission electron microscope (TEM) with quantifications from 50 mitochondria shown below. Scale bar: 500 nm. Representative of 3 independent experiments.
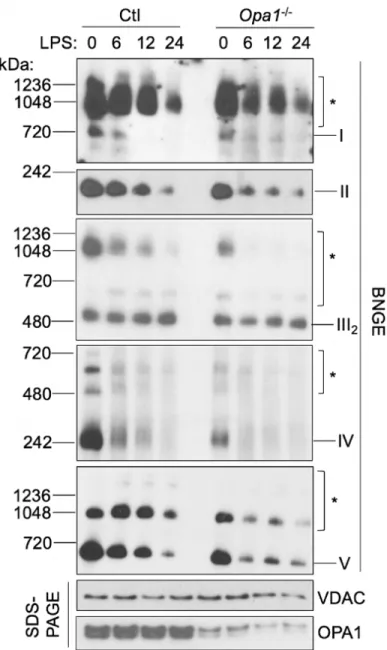
Knocking out OPA1 is known to disrupt the assembly of ETC complexes while
sensitizing cells to apoptosis (Cogliati et al., 2013). We thus tested whether depleting OPA1 would be sufficient to disrupt ETC complex assembly in macrophages and modulate macrophage metabolism and function. We derived BMM from Opa1fl/fl mice that expressed homozygous Cre+/+ under the control of lysozyme promoter (referred to as Opa1-/- BMM or Opa1-/- mice). Without LPS stimulation, Complexes I-V were partially
depleted in Opa1-/- BMM, but their levels still remained higher than those in normal BMM following 6 hours LPS stimulation (Figure 14). After LPS stimulation, the levels of ETC complexes were decreased further in Opa1-/- BMM and were in general slightly lower than those in LPS-treated normal BMM. While the differences in ETC complex
assembly status was only slight, phenotypically the effects were drastic. First, Opa1-/- BMM exhibited increased ECAR and decreased OCR compared to wild-type floxed BMM. The ECAR/OCR ratio of unstimulated Opa1-/- BMM was similar to that of normal BMM stimulated with LPS for 6 hours (Figure 15), suggesting that even a small
reduction in ETC complexes can significantly skew macrophage metabolism toward glycolysis. Consistently, Opa1-/- BMM also accumulated glycolytic intermediates that included glucose-6-phosphate and lactate as measured by LC/MS (Figure 16). Notably, although succinate is a substrate of Complex II and is also known to drive
pro-inflammatory responses in macrophages through HIF-1 (Tannahill et al., 2013), succinate levels as detected by LC/MS were not increased but decreased in Opa1-/- BMM, indicating that the pro-inflammatory response resulting from ETC complex disassembly occurred independently of succinate (Figure 16).
Figure 14: OPA1 knockout results in partial ETC complex disassembly. Wild-type
and Opa1-/- macrophages were treated for 0, 6, 12, or 24 hours with LPS and mitochondrial fractions were isolated. Proteins were purified and separated on either BNGE or SDS-PAGE and probed with antibodies specific to ETC complex subunits and VDAC.
Figure 15: OPA1 knockout results in a switch from OxPhos to glycolysis. Wild-type
macrophages were treated for 0, 6, 12, or 24 hours with LPS, and OCR (left), ECAR (middle), and ECAR/OCR ratio (right) were measured using Seahorse metabolic assay. Data represents n=3 biological replicates.
Figure 16: OPA1 knockout macrophages accumulate glycolytic intermediates but have reduced levels of succinate. Wild-type and Opa1-/- BMM were not treated or treated with LPS for 12 hours and metabolites were extracted and quantified by LC/MS.
We further assessed the physiological effect of OPA1 knockout on TLR response by intraperitoneal injection of Opa1-/- mice with LPS, an in vivo model for sepsis. Myeloid cells contribute the bulk of TNF, IL-6, CCL-2, and CXCL-2 in circulation during septic shock in mice. We found TNF, IL-6, CCL-2, and CXCL-2 to be significantly elevated in
0 6 0 6 0.0 0.1 0.2 0.3 0.4 Relative abundance
Succinate
Ctl Opa1 -/-LPS (h): P < 0.001 P = 0.0050
12
0
12
0.000
0.005
0.010
0.015
Relative abundance
Glucose-1 or 6-phosphate
Ctl
Opa1
-/-LPS (h):
P = 0.26 P = 0.004 P < 0.0010
12
0
12
0.0
0.5
1.0
1.5
2.0
Relative abundance
Lactate
Ctl
Opa1
-/-LPS (h):
P = 0.003 P = 0.044 P = 0.12the sera of Opa1-/- mice compared to control Opa1fl/flmice after LPS injection,
suggesting that OPA1 knockout enhances inflammatory responses in vivo (Figure 17).
Figure 17: Myeloid OPA1 knockout sensitizes mice to septic shock by increasing pro-inflammatory protein production. Comparison of the levels of cytokines and
chemokines by ELISA in Opa1-/- and Opa1fl/fl mice 3 hours after PBS or LPS injection.
As OPA1 is well-known to also be required for mitochondrial fusion, we distinguished the dual roles of OPA1 in ETC complex assembly and mitochondrial fusion by studying the effect of knocking out dynamin-related protein 1 (DRP1), a related protein that acts
in mitochondrial fission but not in ETC complex assembly, on macrophage inflammatory responses. This model thus provides a separation-of-function approach. Using
macrophages deficient in DRP1 (also known as Drp-/- BMM; Wang et al., 2017), we showed that while DRP1 knockout results in constitutively elongated/fused mitochondria (Figure 18), which remained elongated following LPS treatment, the prevalence of fused mitochondria did not correlate with altered ETC complex assembly status (Figure 19) or in altered levels of TNF, IL-6, CCL-2, and IL-12/23p40 (Figure 20), suggesting that mitochondrial fission/fusion status does not affect ETC complex assembly or macrophage TLR responses. Thus, the role of OPA1 in macrophage inflammatory response is likely independent of its role in mitochondrial fusion. Taken together, these results show that, similar to depletion of mtDNA, OPA1 knockout in macrophages disrupts ETC complex assembly, switches metabolism from OxPhos to glycolysis, and enhances LPS-induced production of inflammatory molecules.
Figure 18: DRP1 knockout in macrophages results in elongated mitochondria.
using MitoTracker and nucleus using Hoechst 33342. Shown are representative images from confocal microscopy.
Figure 19: DRP1 knockout in macrophages does not influence ETC complex assembly. BMM were treated with LPS (100 ng/ml) for 6, 12, and 24 hours. Protein
extracts were prepared from the mitochondrial fractions and separated by BNGE and SDS-PAGE, followed by Western blotting with antibodies specific for ETC Complexes I-V on BNGE, and antibodies specific for I-VDAC to detect relative levels of I-VDAC on SDS-PAGE. Representative of 2 experiments.
Figure 20: DRP1 knockout in macrophages does not influence inflammatory responses. Comparison of TNF, IL-6, CCL-2, and IL-12/23p40 by ELISA between
Drp1fl/fl (Ctl) and Cre+/- Drp1fl/fl (Drp1-/-) BMM with or without LPS stimulation for 24 hours. N=3 technical replicates from 2 biological replicates per group.
Discussion: ETC complex disassembly drives metabolic switch from OxPhos to glycolysis during macrophage TLR response
Our data demonstrate that a surprisingly simple mechanism involving ETC complex disassembly underlies the metabolic switch in macrophages from OxPhos to glycolysis during inflammatory response. Based on our analysis of ETC complex assembly status on native PAGE in macrophages stimulated with various TLR ligands, we conclude that ETC complex disassembly is a process that is conserved across multiple different TLR signaling pathways and is sensitive to bacterial or viral ligands, and operates either extracellularly or intracellularly. We also observe conservation from mouse to human,
suggesting high evolutionary importance of its conservation. We modeled ETC complex disassembly using two methods, a complete depletion of Complexes I, III, IV, and V using ρ0 macrophages as well as a partial depletion of Complexes I-V using OPA1 knockout macrophages, and observe that in both scenarios the production of inflammatory markers and proteins by such macrophages is increased during LPS response. This pro-inflammatory phenotype was also correlated with decreased OxPhos and increased glycolysis, mitochondrial cristae remodeling, and OPA1 oligomerization. Indeed, knocking out OPA1 further sensitized macrophages to LPS stimulation and switched macrophages from OxPhos to glycolysis, despite causing only a slight reduction in ETC complex disassembly. We examined the other known role of OPA1, mitochondrial fusion, by using a DRP1 knockout strain, and found that the mitochondrial fission and fusion status did not influence ETC assembly or the inflammatory response. Thus, we reason that a slight reduction in ETC complex assembly, whether caused by cristae remodeling or some other yet uncharacterized mechanism, is sufficient to drive the macrophage metabolic switch during TLR response and has a significant physiological effect. Together, our data suggest that the biological consequence of ETC complex disassembly in macrophages is a heightened potential to maintain an inflammatory response and thus macrophage function. It is unintuitive as to how limiting OxPhos in macrophages would ultimately boost its function, raising the question as to what underlying mechanisms could be responsible, a topic of next chapter.
Notably, our observations made in macrophages during TLR response are remarkably similar to those made in previous studies on apoptosis and may even be a general phenomenon in other cell types in contexts other than cell death or inflammation. During apoptosis, cristae remodeling enables cytochrome C release while the resulting disruption of ETC complexes result in lethal ROS production. Here we observe a similar phenomenon in macrophages, with a unique consequence being a switch from OxPhos to glycolysis rather than cell death. LPS-treated and ETC complex-deficient macrophages in fact produced lower levels of mitochondrial ROS compared to unstimulated wild-type macrophages (data not shown), demonstrating the diverse context-dependent or cell type-specific effects of ETC complex disassembly on cell physiology. Whether ETC
complex assembly status is altered in cancer cells undergoing Warburg metabolism, for example, has not yet been examined. It is not unlikely that many cell types (including cancer cells) may undergo ETC complex disassembly to some extent that is not necessarily as drastic as seen in inflammatory macrophages, so as to still maintain some OxPhos capacity to support ETC-dependent biosynthesis and cell proliferation. Whether ETC complex disassembly is a general phenomenon under other scenarios remains to be examined.
Our study also raises the question of how OPA1 or ETC complex assembly status might be regulated downstream of TLR signaling. Interestingly, as assessed by SDS-PAGE, the absolute expression level of neither Complex I-V subunits nor OPA1 were altered following LPS stimulation, despite their assembly on high molecular weight complexes being drastically reduced. The assembly status of OPA1 also did not depend on the L-OPA1/S-OPA1 ratio as previously reported in apoptosis (Jiang et al., 2016), suggesting the existence of a unique mechanism. Further understanding is required to dissect these processes. Initially, with hopes to investigate how OPA1 stability could be regulated by TLR stimulation, we performed LC/MS on co-immunoprecipitated OPA1 sample in macrophages treated with or without LPS. Although we identified many previously found interactors of OPA1 during this LC/MS run, including subunits of Complex III and ATP synthase, all such interactions were confirmed false positive following controlled Western experiments in which we used IgG and bead-only samples to serve as negative controls (data not shown), suggesting that the previously discovered interactions between OPA1 and ETC components may not be real and that it may be difficult to discover binding partners of OPA1 by this approach. The regulatory mechanism of OPA1 stability and ETC complex assembly is still an active and a new area of investigation, and to have a fuller understanding would require significantly more detailed molecular study using more genetically tractable models. Because little is understood regarding the control mechanism of OPA1 and ETC complexes, more molecular studies would be required before reasonable hypothesis can be formed as to how ETC complex assembly status is regulated downstream of TLR signaling. However, some immediate work can be done to identify the earliest signals acting in the pathway. Our preliminary results using existing
mouse strains suggest that the Myd88 branch of TLR signaling may not be required for LPS-induced ETC complex disassembly (data not shown), suggesting the possibility that it may instead rely on the TRIF-dependent branch. Taken together, our study offers a mechanistic explanation for the long-described phenomenon of OxPhos-to-glycolysis switch in inflammatory macrophages.
Methods
Mouse Strains and Human Subject Studies Approval
Human subject experiments involving monocytes were approved by Institutional Review Board. Wild-type C57BL/6J mice were obtained from The Jackson Laboratory. Animals (6-12 weeks) were maintained under specific pathogen-free conditions. Studies were done in accordance of Institutional Animal Care and Use Committee. Myeloid Opa1 knockout mice were generated by crossing B6.129P2 LysM-Cre mice and Opa1 floxed mice (obtained from H. Sesaki; Johns Hopkins University). Myeloid Hif1a knockout mice were generated by crossing B6.129P2 LysM Cre mice and Hif1a floxed mice (obtained from Jackson laboratory). Cre-/- flox/flox mice of each respective genotype were used as controls in experiments involving Cre-mediated knockout. Leg bone samples from Atf4 knockout mice (12-30 weeks) were obtained from T.G. Anthony (Rutgers University). Leg
bone samples from myeloid Drp1 knockout mice were a gift of I. Tabas (Columbia University). Sex- and age-matched pairs were used for all mouse experiments.
Bone Marrow Macrophage (BMM) Derivation
Mouse euthanasia was performed by CO2 asphyxiation followed by cervical dislocation. Bone marrow cells were isolated from EtOH-sterilized leg bone samples and
differentiated in macrophage growth media. The macrophage growth media consisted of RPMI with no glutamine supplemented with 10% FBS, 1x pen/strep, 2 mM glutamine, and
20% L-929 supernatant which consisted of L-929-derived growth factors in DMEM supplemented with 10% FBS and 1x pen/strep. Where indicated, M-CSF (50 ng/ml) was used instead of L-929 supernatant for differentiating macrophages. On day 6 of
differentiation, mature macrophages were detached by incubating in PBS containing 5 mM EDTA for 5 minutes followed by gentle pipetting, centrifuged at 300 x g and
resuspended in fresh macrophage growth media at 0.5x106 cells/ml and seeded overnight
RC inhibitors, cells were maintained under 5 μg/ml uridine. TLRs were stimulated by 100
ng/ml LPS, 5 μΜ CpG, or 10 μg/ml poly I:C.
Endotoxin-induced Model of Sepsis
For serum cytokine measurements, B6.129P2 LysM Cre-/- Opa1fl/fl or LysM Cre+/+ Opa1fl/fl
mice were injected i.p. with LPS (10 mg/kg), and whole blood samples were collected 3 h later by isoflurane anesthesia followed by cardiac puncture. To monitor survival, mice were assigned into experimental groups and then injected i.p. with either vehicle
(DMSO
dissolved in 5% PEG-400/5% Tween-20) or rapamycin (1.2 mg/ml dissolved in 5% PEG-
400/5% Tween-20). Three daily injections were done, and 1 h after the third rapamycin injection, LPS was injected i.p. (10 mg/ml). Following LPS injection the identity of mice was blinded from the experimenter for further monitoring. Mice were observed for 72 h by
experimenter and facility veterinarians for signs of morbidity. Clinical scores were assessed using published guidelines based on neurological and respiratory symptom (Shrum et al., 2014), where scores of > 21 or sudden change (within 3 hours) in respiration
rate was considered the humane endpoint.
Generation of Respiratory Chain-deficient ρ0 BMM
ρ0 macrophages were generated by culturing bone marrow cells in the continued presence of EtBr (200 ng/ml) during differentiation. For experiments involving ρ0 cells, bone marrow cells including control cells were supplemented with 5 μg/ml uridine throughout differentiation and during experiments.
Growth media lacking glucose was generated using RPMI media containing no glucose with 1x pen/strep, 2 mM L-glutamine, and 10% dialyzed FBS (ThermoFisher). On day 7 of differentiation, normal growth media was replaced with either no glucose media or no glucose media reconstituted with 10 mM glucose. The lack of glucose in these
preparations were confirmed by running glucose concentration measurements on YSI biochemistry analyzer (YSI Life Sciences) using manufacturer’s setting. ECAR under the presence of 0 mM or 10 mM glucose was assessed using Seahorse machine to confirm depletion of glycolysis under 0 mM glucose.
Blue Native Gel Electrophoresis, Western Blot Analysis, and Protein Quantification
Mitochondria were isolated as described previously (Baldanta et al., 2017). 7.5 million mouse macrophages were collected in PBS containing 5 mM EDTA and washed once with PBS. Cell pellets were frozen at -80oC, followed by homogenization in 10 volumes of buffer A (83 mM sucrose, 10 mM MOPS, pH 7.2 containing 1x Roche Complete Mini protease inhibitor cocktail with EDTA), followed by dilution in 1 volume of buffer B (250 mM sucrose, 30 mM
MOPS, pH 7.2 containing 1x Roche Complete Mini protease inhibitor cocktail with EDTA). Nuclei and debris were removed by 1000 x g centrifugation for 5 min.
Mitochondrial pellets were collected by centrifuging at 12,000 x g for 2 min and washed once with buffer C (320 mM sucrose, 1 mM EDTA, 10 mM Tris-HCl, pH 7.4 containing 1x Roche Complete Mini protease inhibitor cocktail with EDTA). Mitochondrial protein was extracted in 1x NativePAGE
sample buffer (ThermoFisher) containing 1% digitonin followed by 20 min spin at 12,000 x g to pellet debris. Protein quantification was done by running a small volume of extract by BCA Protein Assay Kit (Pierce), with protein standard curve being constructed using bovine serum albumin. Extraction efficiency was confirmed by detecting VDAC
expression on SDS-PAGE. SDS-PAGE samples were denatured by resuspended in RIPA buffer (Cold Spring Harbor
protocol) followed by dilution with 2x Laemmli buffer (Sigma Aldrich) and boiling at 100oC for 5 min. Protein extracts were run on Blue Native gel electrophoresis using
Novex NativePAGE system (ThermoFisher) and subsequently transferred to PVDF membrane, fixed, and blotted for native proteins.
Human mitochondrial protein isolation was done using the P15 isolation procedure (Jiang X. et al., 2016). In brief, 1 million macrophages were washed once in cold PBS and scraped and swelled in hypotonic buffer A (20 mM HEPES, 40 mM KCl, 1.5 mM MgCl2, 1 mM EGTA, 1 x Roche Complete Mini protease inhibitor cocktail with EDTA) containing 250 mM sucrose and incubated on ice for 15 min. Cells were then passed through 26G needle 15 times and supernatant cleared by 12,000 x g centrifugation for 10 min. P15 fractions were then extracted using 1% digitonin sample buffer as above.
Analysis of Differentiation and Proliferation by Flow Cytometry
Flow cytometry analysis was done using BD Biosciences flow cytometer LSR II HTS. For analysis of macrophage differentiation, single cells were gated based on forward and side scatter, and then dead cells (positive for propidium iodide) were gated out. Compensation matrices were constructed based on dropout controls. Mature
macrophages were identified based on positive expression for CD11b and F4/80. Total live cells were manually counted based on Trypan exclusion to compare differentiation yields across different strain backgrounds, with total count multiplied with % CD11b+ F4/80+ to obtain final yield. EdU labeling protocol was used to assess proliferation during S phase. In brief, cells were labeled with 10 μM EdU for 1 h, and then EdU was made to fluoresce using click chemistry following the same wash, fixation, and
permeabilization protocol as FUNCAT translation assay detailed below, except that the click cocktail was made using the following components: 1 mM CuSO4, 10 mM ascorbic acid, and 8 μM Azide Fluor 488 in PBS. Cells positive for EdU on flow cytometry were considered proliferating cells. Cell cycle status was analyzed using propidium iodine (PI) staining of DNA. The cells were first washed with ice cold PBS after which the cells in PBS were mixed with ice cold EtOH to a final EtOH concentration of 70%. EtOH fixation was carried out overnight in -20ÆC. The following day the cells were washed with PBS and stained with FxCycle
PI/RNase Staining Solution (Invitrogen) for 30 min. After staining the cells were placed on ice until DNA content was analyzed using BD Biosciences flow cytometer LSR II HTS with 488 nm excitation laser and 585/15 nm emission filter. Flow cytometry data were analyzed and graphed using FlowJo.
Extracellular Flux Assays for ECAR and OCR Measurement
Extracellular flux was measured using the Seahorse XFe96 analyzer (Agilent) with manufacturer’s protocol. In brief, macrophages were seeded at the density of 0.5 million cells (mouse) or 0.25 million cells (human)/ml and incubated overnight, followed by TLR stimulation. Media was then replaced with Seahorse base media and extracellular flux assessed in the presence of appropriate mitochondrial/glycolysis inhibitors according to manufacturer’s protocol. The drugs with final concentrations used were: glucose (10 mM), ETC inhibitor (0.5 μM rotenone and 0.5 μM antimycin A), glycolysis inhibitor (50 mM 2-DG), H+ influx inhibitor (1 μM oligomycin), and uncoupler (1 μM FCCP).
Microscopy for Mitochondrial Morphology Assessment
BMM were treated or not with LPS (100 ng/ml) for 24 h. For transmission electron microscopy (TEM), cells were fixed in fixation buffer (2% glutaraldehyde, 3%
paraformaldehyde, and 5% sucrose in 0.1M sodium cacodylate (pH 7.4), washed once in 0.1 M sodium cacodylate containing 7.5% sucrose, and post-fixed in 1% osmium tetroxide for 1 hour on ice (Palade 1951). Samples were rinsed once and stained overnight in 1% aqueous uranyl acetate. Samples were
rinsed in cold 50% ethanol and dehydrated with graded series of ethanol and embedded in 50% propylene oxide/50% epoxy resin. Samples were sectioned onto grids and
stained with uranyl acetate and lead citrate. Grids were viewed on Technai Spirit
transmission electron microscope (FEI). Quantification of cristae width were done using ImageJ.
For fluorescence imaging, cells were stained with MitoTracker Deep Red (100 nM) for 15 minutes, rinsed 3 times with warm PBS, fixed in 3.7% paraformaldehyde. Cells were rinsed 3 times in PBS and resuspended on VECTASHIELD antifade mounting medium
prior to imaging (Vector Laboratories). Samples were imaged on FV1200 Laser Scanning Confocal Microscope (Olympus Life Sciences), on 35 mm dish (MatTek).
Metabolite Profiling
LC/MS was performed at Whitehead Institute metabolome core facility. Following treatment, cells were washed once in ice-cold 0.9% NaCl and lysates were extracted in 80% methanol solution containing internal standards for LC/MS by scraping on dry ice followed by 10 minute mixing with vortex in 4oC. Following lysate extraction, debris were removed by high speed centrifugation and supernatant was dried using speedvac. Samples were stored at -80oC until
downstream analysis. Samples were analyzed by LC/MS on QExactive Orbitrap instruments (Thermo Scientific) in Whitehead metabolite profiling core facility. Data analysis was performed using the in-house software described previously (Lewis et al., 2014).
References
Kawai, T., and Akira, S. (2007). Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 13, 460–469.
Vander Heiden, M.G., Cantley, L.C., and Thompson, C.B. (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029– 1033.
Hard, G.C. (1970). Some biochemical aspects of the immune macrophage. Br. J. Exp. Pathol. 51, 97–105.
McGivney, A., and Bradley, S.G. (1979). Effects of bacterial endotoxin on lysosomal and mitochondrial enzyme activities of established cell cultures. J. Reticuloendothel. Soc. 26, 307–316.
Cramer, T., Yamanishi, Y., Clausen, B.E., Förster, I., Pawlinski, R., Mackman, N., Haase, V.H., Jaenisch, R., Corr, M., Nizet, V., et al. (2003). HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112, 645–657.
Tannahill, G.M., Curtis, A.M., Adamik, J., Palsson-McDermott, E.M., McGettrick, A.F., Goel, G., Frezza, C., Bernard, N.J., Kelly, B., Foley, N.H., et al. (2013). Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242.
MacVicar, T., Ohba, Y., Nolte, H., Mayer, F.C., Tatsuta, T., Sprenger, H.-G., Lindner, B., Zhao, Y., Li, J., Bruns, C., et al. (2019). Lipid signalling drives proteolytic rewiring of mitochondria by YME1L. Nature 575, 361–365.
Miettinen, T.P., Pessa, H.K.J., Caldez, M.J., Fuhrer, T., Diril, M.K., Sauer, U., Kaldis, P., and Björklund, M. (2014). Identification of transcriptional and metabolic programs
related to mammalian cell size. Curr. Biol. 24, 598–608.
Sênos Demarco, R., Uyemura, B.S., D’Alterio, C., and Jones, D.L. (2019). Mitochondrial fusion regulates lipid homeostasis and stem cell maintenance in the Drosophila testis. Nat. Cell Biol. 21, 710–720.
Laplante, M., and Sabatini, D.M. (2012). mTOR signaling in growth control and disease. Cell 149, 274–293.
Ben-Sahra, I., Hoxhaj, G., Ricoult, S.J.H., Asara, J.M., and Manning, B.D. (2016). mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 351, 728–733.
Han, J., Back, S.H., Hur, J., Lin, Y.-H., Gildersleeve, R., Shan, J., Yuan, C.L., Krokowski, D., Wang, S., Hatzoglou, M., et al. (2013). ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 15, 481–490.
Byles, V., Covarrubias, A.J., Ben-Sahra, I., Lamming, D.W., Sabatini, D.M., Manning, B.D., and Horng, T. (2013). The TSC-mTOR pathway regulates macrophage polarization. Nat. Commun. 4, 2834.
Cogliati, S., Frezza, C., Soriano, M.E., Varanita, T., Quintana-Cabrera, R., Corrado, M., Cipolat, S., Costa, V., Casarin, A., Gomes, L.C., et al. (2013). Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155, 160–171.
Chandel, N.S., and Schumacker, P.T. (1999). Cells depleted of mitochondrial DNA (rho0) yield insight into physiological mechanisms. FEBS Lett. 454, 173–176.
Wang, Y., Subramanian, M., Yurdagul, A., Jr, Barbosa-Lorenzi, V.C., Cai, B., de Juan-Sanz, J., Ryan, T.A., Nomura, M., Maxfield, F.R., and Tabas, I. (2017). Mitochondrial Fission Promotes the Continued Clearance of Apoptotic Cells by Macrophages. Cell 171, 331–345.e22.
Jiang, X., Li, L., Ying, Z., Pan, C., Huang, S., Li, L., Dai, M., Yan, B., Li, M., Jiang, H., et al. (2016). A Small Molecule That Protects the Integrity of the Electron Transfer Chain Blocks the Mitochondrial Apoptotic Pathway. Mol. Cell 63, 229–239.
Palade, G.E. (1952). A study of fixation for electron microscopy. J. Exp. Med. 95, 285– 298.
Lewis, C.A., Parker, S.J., Fiske, B.P., McCloskey, D., Gui, D.Y., Green, C.R., Vokes, N.I., Feist, A.M., Vander Heiden, M.G., and Metallo, C.M. (2014). Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol. Cell 55, 253–263.
Chapter 3.
ETC complex disassembly promotes macrophage function by
reprogramming translation
This chapter was written by Yang Su, with edits by Dr. Jianzhu Chen, Dr. Teemu Miettinen, Dr. Mary Mu, and Dr. Tracy Anthony.
Whole transcriptome analysis to elucidate the mechanism by which metabolic switch governs inflammatory response
How does a switch from OxPhos to glycolysis enable macrophage function? To address this question, we performed whole transcriptome analysis to identify candidate cellular pathways specifically altered following ETC complex disassembly that could potentially drive inflammatory responses. Our ρ0 and OPA1 knockout macrophage models, combined with or not combined with LPS treatment, allow for identification of pathways enriched following ETC complex disassembly in both resting macrophages and inflammatory macrohpages. RNA-seq was performed on control (Opa1fl/fl), Opa1-/-, and ρ0 BMM (with ρ0 BMM being derived from Opa1fl/fl mice), either not treated or treated with LPS for 6 hours. As expected, transcriptomes of the same genetic background clustered together by hierarchical clustering analysis (Figure 21).
Figure 21: Hierarchical clustering of transcriptomes of control Opa1fl/fl, ρ0, and
Opa1-/- BMM not treated or treated with LPS. (Left) Unstimulated BMM with each
sample as shown. (Right) BMM treated with LPS for 6 hours. N=3 biological replicates per group.
Differentially expressed genes (DEGs) were identified between Opa1-/- and Opa1fl/fl BMM as well as between ρ0 and Opa1fl/fl BMM, without or with LPS stimulation (Figure 22). Without LPS stimulation, 198 DEGs were common between Opa1-/- and ρ0 BMM (Figure 23), while with LPS stimulation 248 were common (Figure 23), suggesting that significant overlap exists between DEGs in Opa1-/- and ρ0 BMM. Unexpectedly, gene ontology (GO)
analysis revealed that DEGs upregulated in both Opa1-/- and ρ0 BMM were mostly enriched for amino acid metabolism and translation pathways (Figure 24) rather than inflammatory or glycolytic pathways despite their pro-inflammatory and glycolytic bias. These data suggest that, at the transcript level, ETC complex disassembly induces protein anabolism pathways more significantly than inflammatory and glycolytic pathways.
Figure 22: Differential gene expression analysis between control Opa1fl/fl, ρ0, and
Opa1-/- BMM not treated or treated with LPS. Volcano plots of fold change versus FDR
of each group comparisons as shown, with select genes annotated. N=3 biological replicates per group.
Figure 23: Venn diagrams showing the overlap between genes differentially expressed in ρ0 BMM and those differentially expressed in Opa1-/- BMM. (Left)
Unstimulated macrophages. (Right) BMM treated with LPS for 6 hours. N=3 biological replicates per group.
Figure 24: ETC complex disassembly results in upregulation of translation pathways. DAVID enrichment analysis of DEGs for pathways specifically upregulated
following ETC complex disassembly. (Left) Pathways upregulated in LPS-stimulated Opa1-/- BMM compared to LPS-stimulated wild-type BMM. (Right) Pathways upregulated in LPS-stimulated ρ0 BMM compared to LPS-stimulated wild-type BMM. N=3 biological replicates per group.
These observations were further supported in a whole genome K-means clustering analysis, identifying 4 expression clusters among variable genes across entire transcriptome (maximum Pearson correlation = 0.41) among LPS-stimulated control, Opa1-/-, and ρ0 BMM (Figure 25; Figure 26). Clusters 2 and 4 contained the majority of genes and showed opposite expression patterns, with gene expression progressively increasing (cluster 2) or decreasing (cluster 4) from control to Opa1-/- to ρ0 BMM (Figure 26). Functional enrichment analysis (DAVID) revealed that the biological process most significantly enriched across all clusters was translation in cluster 2 (Figure 25), indicating that genes involved in translation were increased from control to Opa1-/- to ρ0 BMM in a dose-dependent manner roughly correlated to the amount of ETC complexes present.
Figure 25: K-means clustering analysis for pathways altered following ETC complex disassembly in LPS-treated BMM identifies a highly significant translation cluster. (Top) Shown are the heatmaps of the 4 clusters (top) with GO
0 2 4 6 8 0 20 40 60
log
2(enrichment)
-log(FDR)
Cluster 1 Cluster 2 Cluster 3 Cluster 4DAVID annotation of GO processes