HAL Id: tel-02169205
https://tel.archives-ouvertes.fr/tel-02169205
Submitted on 1 Jul 2019
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
C-H bond activation catalyzed by Ruthenium
nanoparticles
Longhui Gao
To cite this version:
Longhui Gao. C-H bond activation catalyzed by Ruthenium nanoparticles. Organic chemistry. Uni-versité Paris-Saclay, 2017. English. �NNT : 2017SACLS348�. �tel-02169205�
C-H BOND ACTIVATION
CATALYZED BY RUTHENIUM
NANOPARTICLES
Thèse de doctorat de l'Université Paris-Saclay
préparée à l’Université Paris-Sud
École doctorale n°571 2MIB
Spécialité de doctorat: ChimieThèse présentée et soutenue à CEA Saclay, le 06/11/2017, par
M. Longhui Gao
Composition du Jury :M. Damien Prim
Professeur, Université Paris-Saclay (Université de Versailles Saint-Quentin)
Président
Mme. Montserrat Gomez
Professeur, Université Paul Sabatier Rapporteur
Mme. Françoise Colobert
Professeur, CNRS, Université de Strasbourg Rapporteur
M. Sébastien Roy
Docteur, Sanofi Examinateur
M. Jérôme Hannedouche
Docteur, Université Paris-Saclay (Université Paris Sud) Examinateur
M. Bernard Rousseau
Docteur, CEA Saclay Directeur de thèse
M. Grégory Pieters
Docteur, CEA Saclay Invité
NNT
:
2
0
1
7
S
A
CL
S
3
4
8
Acknowledgments
I would like to thank Dr. Bernard Rousseau and Prof. Peiqiang Huang (Xiamen University) first of all, who offered me the opportunity to do my thesis in CEA Saclay. During these three years, Dr. Bernard Rousseau gave me a lot of useful advices in my works, which have led to the accomplishment of my thesis. Besides, he also gave me a lot of help in my daily life, such as renting and registration.
I would like to thank Dr. Grégory Pieters, who gave me a lot of help in both professional and personal life during these three years. He is always patient with me and is just like a good friend with a great sense of humor. He also gave me very useful advices during the preparation of my thesis.
I would like to thank Dr. Sophie Feuillastre, who helped me a lot during the preparation of my thesis and gave me many useful advices in organic chemistry.
I would like to thank Sébastien Garcia-Argote, who helped me a lot in the operation of tritium labeling and many things in the lab.
I would like to thank Dr. Serge Perato and Dr. Céline Taglang, who taught me the operations of deuterium labeling. Besides, their preliminary works on deuterium labeling inspired me a lot.
I would like to thank the members in analysis group: David Buisson, Céline Chollet and Elodie Marcon. They helped me a lot in the characterization of isotopically labeled molecules and the HPLC analyses.
I would like to thank Prof. Bruno Chaudret (INSA Toulouse), who provided us various well-characterized ruthenium nanoparticles in the exploration of new C-H functionalization reactions. He also gave me some useful advices in my works.
I would like to thank Dr. Gilles Clavier (ENS Cachan), who helped us to detect the photophysical properties of the synthesized compounds.
I would like to thank Dr. Bo Gao, who is a good friend of me and helped me a lot in my daily life. He was my “private translator”, and he was always in good mood and humorous.
I would like to thank Dr. Eric Doris and Dr. Edmond Gravel for their useful advices during these three years.
I would like to thank all the other members in the tritium lab: Florence Pillon for her kindly concern, Dr. Alaric Desmarchelier for his advices and helps, Anaëlle Doerflinger for her help in the lab, and
also Dr. Christophe Dugave, Dr. Emilie Nehlig, Dr. Dinh Vu Nguyen, Dr. Praveen Prakash, Dr. Arun Kumar Ramar, Dr. Gopi Elumalai, Dr. Marielle Tamigney, Victor Pfeifer, Alberto Palazzolo, Minh Duc Hoang, Olivia Carvalho, Leonid Lavnevich, Boris Solda, Mathilde Pauton, Paul Chassagne for their encouragements and kindness during these three years.
Also, I would like to thank Chantal Faux for her patience and responsibility during these three years. In addition, I would like to thank Dr. Frédéric Taran for his responsibility, Dr. Davide Audisio, Dr. Jean-Christophe Cintrat, Dr. Karen Hinsinger and all the other members of SCBM for their kindness.
At the same time, I would like to thank my parents and my grandmother. Their unconditional supports and love offer me the endless power to overcome all the difficulties in my life.
I also thank all my Chinese friends in France, especially Zong, Haiyan, Yongpeng, Chenge, Bo, for their companionship. They brought a lot of joys and laughs to me during these three years.
Finally, I sincerely thank Xiudan, my dear wife, who is always at my side for all these three years. Her endless love makes me strong and offers me endless power. I also thank her for bringing Yiwei, my lovely daughter, into my life. The little girl is my angel, and all the other things become unimportant when I am with her.
Résumé de thèse
Les molécules marquées par des isotopes de l’hydrogène possèdent de nombreuses applications dans divers domaines tels que la chimie, la biologie ou en science des matériaux. Dans le domaine de la recherche de nouveaux médicaments, les études liées à la pharmacocinétique nécessitent un accès rapide à des molécules marquées afin de ne pas impacter les coûts et les délais de développement. Le développement de la métabolomique a aussi entrainé une augmentation du besoin en molécules marquées isotopiquement. En effet, les molécules deuterées peuvent être utilisées en tant qu’étalons internes pout la quantification rapide des métabolites présents dans des tissus ou des fluides biologiques. La première partie de cette thèse concerne le développement d’une méthode générale de marquage de motifs de type thioéther dans des molécules complexes à l’aide d’une nouvelle réaction d’échange isotopique (catalysée par des nanoparticules de Ruthénium).
D’un point de vue fondamental cette transformation représente le premier exemple de C(sp3)-H activation dirigée par un atome de soufre. En termes d’application, cette nouvelle réaction permet la synthèse rapide d’étalons internes pour la quantification LC-MS/MS et le marquage tritium de molécules complexes. La robustesse du système catalytique, et notamment sa capacité à maintenir sa réactivité dans différents solvants protiques deutérés ou aprotiques, a permis de marquer une large gamme de produits contenant un motif thioéther. Ainsi en fonction de la solubilité et de la stabilité des composés d’intérêt, différents milieux réactionnels (solvants : THF, MeOD, DMF, DMA, D2O) ont été utilisés pour marquer des principes actifs et des peptides contenant des sous-structures de type thioethers avec un grande régioselectivité. Par la suite, il a été démontré que cette méthode permettait d’accéder rapidement à molécules deutérées pouvant être utilisé en tant que standards internes pour la quantification LC/MS. En effet, en réalisant plusieurs cycles de catalyse (3) plus de 4 atomes de deutérium ont pu être incorporés sur des molécules complexes. Cette réaction a aussi été optimisée dans le cadre du marquage tritium et le pergolide radiomarqué (activité spécifique : 15 Ci/mmol) a été obtenu en une étape dans des conditions douces (P(T2) = 0.9 bar).
La seconde partie de cette thèse relate le développement d’une nouvelle méthode d’homocouplage de phénylpyridines catalysée par un catalyseur de ruthénium hétérogène (Ru/C) et utilisant le chlorure de
fer (III) comme oxydant. Différents substrats comportant des substituants diverses ont été dimérisés avec de bons rendements (voir figure ci-dessous).
Des études de cinétique et de mécanistique ont été conduites afin de déterminer la nature de l’espèce catalytique active. Ces dimères ont ensuite été utilisés pour synthétiser de nouveaux complexes de bore dont les propriétés photophysiques ont été étudiées. Ces nouvelles espèces possèdent des rendements quantiques élevés et des brillances supérieures aux espèces monomériques. L’utilisation de ces nouveaux complexes de bores en tant qu’émetteurs dans des diodes électroluminescentes sera prochainement étudiée.
Dans une troisième partie, la mise au point d’une réaction palladocatalysée permettant d’obtenir des molécules polycycliques contenant un motif de type pyridine est développée (voir schéma ci-dessous).
Les résultats préliminaires ont montré que la réaction développée permettait d’obtenir de manière sélective le produit résultant de de l’activation de la liaison C-H en para de l’atome d’azote de la pyridine. Le champ d’application de cette réaction a été étudié et un mécanisme réactionnel expliquant la regiosélectivité observée proposé.
I
Table of Contents
ABBREVIATION LIST ... 1
Chapter 1 Deuterium and Tritium Labeling of Bioactive Thioethers ... 5
1 Introduction and background ... 5
1.1 Deuterium and applications of deuterated compounds ... 5
1.2 Tritium and applications of tritiated compounds ... 11
1.3 Synthesis of deuterium or tritium labeled compounds ... 14
1.4 H/D exchange reactions catalyzed by iridium complexes ... 17
1.4.1 Iridium catalyzed deuteration involving oxygen atom as directing groups ... 18
1.4.2 Iridium catalyzed deuteration involving nitrogen atom as directing group ... 21
1.4.3 Iridium catalyzed deuteration of various compounds with distinct functional groups .. 24
1.4.4 Iridium catalyzed deuteration of alkenes ... 26
1.4.5 Iridium catalyzed deuteration of aromatic compounds ... 27
1.5 H/D exchange reactions catalyzed by ruthenium catalysts... 29
1.5.1 Ruthenium catalyzed deuteration involving oxygen atom as directing groups ... 29
1.5.2 Ruthenium catalyzed deuteration involving nitrogen atom as directing groups ... 33
1.5.3 Ruthenium catalyzed deuteration of various compounds with distinct functional groups 42 1.5.4 Ruthenium catalyzed deuteration of other types of organic molecules ... 44
1.6 H/D exchange reactions catalyzed by other metals ... 45
1.6.1 Palladium catalyzed H/D exchange reactions ... 46
1.6.2 Rhodium catalyzed H/D exchange reactions ... 48
1.6.3 Platinum catalyzed H/D exchange reactions ... 50
1.6.4 Iron catalyzed H/D exchange reactions ... 51
1.6.5 H/D exchange reactions catalyzed by mixed metal catalysts ... 52
II
2.1 The investigation of the H/D exchange of thioethers ... 56
2.2 Attempts to apply our method in quantitative LC-MS analysis ... 68
2.3 Application of our method in tritium labeling ... 69
2.4 Mechanistic studies of the H/D exchange of thioethers ... 72
2.5 Summary and perspective... 75
2.6 Exploring the catalytic activities of different kinds of ruthenium catalysts ... 78
3 Experimental section ... 85
3.1 Reagents and General Procedures ... 85
3.2 Experimental details and characterization for compounds 2.1 to 2.15’ ... 87
3.3 Data of mass analyses for compounds 2.1 to 2.15’ ... 107
Chapter 2 Ru/C catalyzed homocoupling of 2-arylpyridines ... 109
1 Introduction and background ... 109
1.1 C-C bond formation through heterogeneously catalyzed C-H functionalization ... 110
1.1.1 C-C bond formation based on heterogeneous palladium catalysts ... 110
1.1.2 C-C bond formation based on heterogeneous ruthenium catalysts ... 117
1.1.3 C-C bond formation based on heterogeneous platinum catalysts ... 121
1.2 C-C bond formation through heterogeneously catalyzed CDC reactions ... 123
1.2.1 C-C bond formation through heterogeneously catalyzed dehydrogenative alkylation 123 1.2.2 C-C bond formation through heterogeneously catalyzed dehydrogenative alkenylation 125 1.2.3 C-C bond formation through heterogeneously catalyzed dehydrogenative arylation . 126 1.2.4 C-C bond formation through heterogeneously catalyzed dehydrogenative alkynylation 129 1.2.5 C-C bond formation through heterogeneously catalyzed dehydrogenative homocoupling ... 131
1.3 Summary and perspectives of CDC reactions through heterogeneous catalysis ... 132
2 The discovery of new C-H activation reactions ... 133
2.1 Exploring new C-H activation reactions catalyzed by ruthenium heterogeneous catalyst .. 133
2.2 Literature review for the homocoupling of 2-arylpyridines ... 136
III
3.1 Optimization of reaction conditions ... 144
3.2 Substrate scope of the homocoupling reactions ... 147
3.3 Experiments for mechanism studies ... 152
3.4 Applications of the homocoupling products ... 153
3.4.1 Perspectives ... 157
3.4.2 Conclusion ... 159
4 Experimental section ... 161
4.1 Reagents and General Procedures ... 161
4.2 Experimental details and characterization for synthesized compounds ... 163
Chapter 3 Pd-catalyzed C-H activation intramolecular arylation via concerted metalation-deprotonation ... 179
1 Introduction and background ... 179
1.1 Synthesis of polycyclic biaryls through intramolecular arylation involving CMD process 182 1.2 Synthesis of polycyclic biaryls through intramolecular arylation involving pyridine derivatives ... 191
2 Results and discussions ... 199
3 Experimental section ... 208
3.1 Reagents and General Procedures ... 208
1
ABBREVIATION LIST
Ac: acetyl group Ad: adamantyl group
ADME: absorption, distribution, metabolism, and excretion AIBN: azobisisobutyronitrile
Ar: aromatic group
BArF: tetrakis[3,5-bis(trifluoromethyl)phenyl]borate BINIQ: 2,2’-binaphthyl-1,1’-biisoquinoline
bMepi: 1,3-(6’-methyl-2’-pyridylimino)isoindolate Bn: benzyl group
Bu: butyl group
CDA: cross-dehydrogenative arylation CDC: cross-dehydrogenative coupling CMD: concerted metalation-deprotonation cod: 1,5-cyclooctadiene
cot: 1,3,5-cyclooctatriene Cy: cyclohexyl group CYP: cytochrome P450s
DCC: N,N'-dicyclohexyl-carbodiimide DCE: dichloroethane
DDQ: 2,3-dichloro-5,6-dicyanobenzoquinone DFT: Density Functional Theory
DIAD: diisopropyl azodicarboxylate DMA: N,N-dimethylacetamide DMAP: 4-dimethylaminopyridine
2 DME: dimethoxyethane
DMF: N,N-dimethylformamide DMSO: dimethyl sulfoxide
dppb: 1,4-bis(diphenylphosphino)butane Et: ethyl group
FTIR: Fourier transform infrared spectroscopy hcp: hexagonal close packed
Hex: hexyl group
HIE: hydrogen isotope exchange
HPLC: high-performance liquid chromatography HREM: high resolution electron microscopy HRMS: high-resolution mass spectrometry
HSCIE: high-temperature solid-state catalytic isotope exchange HTBZ: dihydrotetrabenazine
ICP-AES: inductively coupled plasma-atomic emission spectrometry ICy: N,N-dicyclohexylimidazol-2-ylidene
IDC: intramolecular-dehydrogenative-coupling IR: infrared
KIE: kinetic isotope effect LAE: ligand acceleration effect
LC-MS: liquid chromatography-mass spectrometry LSC: Liquid Scintillation Counter
Me: methyl group
Mes: 2,4,6-trimethylphenyl group MOF: metal-organic framework Ms: mesyl group
3 NBS: N-bromosuccinimide
NHC: N-heterocyclic carbene NMP: N-methyl-2-pyrrolidone NMR: nuclear magnetic resonance
NOBIN: 2-amino-2’-hydroxy-1,1’-binaphthyl OLED: organic light-emitting diodes
PET: positron emission tomography Ph: phenyl group
Piv: pivaloyl group
PMB: 4-methoxybenzyl group PPy: polypyrrole
Pr: propyl group
p-TSA: p-toluenesulfonic acid
PVP: polyvinylpyrrolidone Py: pyridine
SEAr: electrophilic aromatic substitution SEM: scanning electron microscopy SET: single electron transfer SM: starting material
TBAA: tetrabutylammonium acetate TBAB: tetrabutylammonium bromide TBHP: tert-butylhydroperoxyde
TEM: transmission electron microscopy TFA: trifluoroacetic acid
TFAA: trifluoroacetic anhydride THF: tetrahydrofuran
Tf: trifluoromethanesulfonyl group TM: target molecule
4 TOF: turnover frequency
TON: turnover number
TRIP: 2,4,6-triisopropylphenyl Ts: p-toluenesulfonyl group
UHPLC: Ultra High Pressure Liquid Chromatography UV-Vis: ultraviolet-visible
VMAT2: vesicular monoamine transporter 2 WAXS: wide-angle X-ray scattering
XPS: X-ray photoelectron spectroscopy XRD: X-ray diffraction
XRF: X-ray fluorescence spectroscopy ZPE: zero-point energy
5
Chapter 1 Deuterium and Tritium Labeling of
Bioactive Thioethers
1 Introduction and background
1.1 Deuterium and applications of deuterated compounds
A slight difference might cause huge effect, just as the case of hydrogen isotopes (Figure 1.1). Deuterium is one of two natural isotopes of hydrogen, with the symbol D or 2H, which also known as heavy hydrogen. As an isotope of hydrogen, deuterium differs from hydrogen simply by possessing a single neutron, and it is the extra neutron that may make a massive difference in the reactivity of deuterium versus its isotope protium.
Figure 1.1 Hydrogen isotopes
Deuterium was discovered and named in 1931 by Harold C. Urey.1 It has a natural abundance in Earth's oceans of about one atom over 6420 of hydrogen. The most common isotope (1H) occupies more than 99.98% of all the naturally occurring hydrogen, while deuterium has a number of approximately 0.0156% (or on a mass basis 0.0312%). Notably, natural water from different sources or places has a slightly different abundance of deuterium.2 For example, the deuterium content of surface water is about 5% lower than that of marine water, due to the repeated vaporization and condensation.
Multiple applications have been found for deuterated compounds in chemistry, biology and material science.3 One of the most important applications in chemistry is due to the kinetic isotope effect
(Figure 1.2). Since D is heavier than H, the vibrational frequency of a C-D bond is lower than the one
1 a) H. C. Urey, F. G. Brickwedde, G. M. Murphy, Phys. Rev. 1932, 39, 164; b) H. C. Urey, F. G. Brickwedde, G. M. Murphy, Phys. Rev. 1932, 40,
1; c) F. G. Brickwedde, Phys. Today 1982, 35, 34.
2 Y. Horibe, M. Kobayakawa, Geochim. Cosmochim. Acta 1960, 20, 273.
3 a) J. Atzrodt, V. Derdau, T. Fey, J. Zimmermann, Angew. Chem. Int. Ed. 2007, 46, 7744; b) T. Junk, W. J. Catallo, Chem. Soc. Rev. 1997, 26,
6 of a H bond, which leads to a lower zero-point energy (ZPE) of the D bond. The lower ZPE of C-D bond results in higher activation energy for the C-C-D bond cleavage than that of C-H bond one: more energy is needed for a C-D bond to reach its transition state for bond cleavage. As a result, if the rate-determining step of a reaction involves the cleavage of a C-H/D bond, the deuterated analogues will have slower reaction rate than the unlabeled molecules. This effect is called the primary kinetic isotope effect (KIE). Normal KIE means that the reaction rate for a C-H bond cleavage is faster than the one for a D bond cleavage. There is also inverse KIE, which means that the reaction rate for a C-D bond cleavage is faster than the one for a C-H bond cleavage. The secondary isotope effect differs from the primary isotope effect by the fact that no C-D bond is formed or broken in the rate determining step.4
Figure 1.2 Origins of the deuterium kinetic isotope effect5
An important application of deuterated compounds is their use in mechanistic studies.6 For example, deuterated toluenes were employed as substrates for aromatic hydroxylation catalyzed by the natural and G103L isoforms of the diiron enzyme toluene 4-monooxygenase to understand the reaction mechanism by C. E. Rogge and co-workers.6d By observing the magnitudes of isotope effects and patterns of deuterium retention of the hydroxylation, the authors concluded that an active site-directed opening of position-specific transient epoxide intermediates might contribute to the chemical mechanism and the regiospecificity of the reaction (Figure 1.3). Generally, by comparing the reaction rate of deuterated substrates with the hydrogenated ones, the researchers might know whether the rate
4C. L. Perrin, Y. Dong, J. Am. Chem. Soc. 2007, 129, 2759.
5 T. G. Gant, J. Med. Chem. 2014, 57, 3595.
6 a) T. Furuta, H. Takahashi, Y. Kasuya, J. Am. Chem. Soc. 1990, 112, 3633; b) C. H. Oh, H. H. Jung, K. S. Kim, N. Kim, Angew. Chem. Int. Ed.
2003, 42, 805; c) S. D. Nelson, W. F. Trager, Drug Metab. Dispos. 2003, 31, 1481; d) K. H. Mitchell, C. E. Rogge, T. Gierahn, B. G. Fox, Proc.
Natl. Acad. Sci. U.S.A. 2003, 100, 3784; e) D. M. Marcus, M. J. Hayman, Y. M. Blau, D. R. Guenther, J. O. Ehresmann, P. W. Kletnieks, J. F. Haw, Angew. Chem. Int. Ed. 2006, 45, 1933; f) D. M. Marcus, K. A. McLachlan, M. A. Wildman, J. O. Ehresmann, P. W. Kletnieks, J. F. Haw, Angew. Chem. Int. Ed. 2006, 45, 3133; g) T. Zhang, Q. Peng, C. Quan, H. Nie, Y. Niu, Y. Xie, Z. Zhao, B. Z. Tang, Z. Shuai, Chem. Sci. 2016, 7, 5573.
7 determining step is the cleavage of the C-H/D bond or not;7 by observing the final fate of the deuterium atom in substrate, one could suggest the possible reaction pathway.8
Figure 1.3 Binding modes of epoxide intermediates relative to the (Fe2) center
Apart from mechanistic studies, isotopically labeled molecules have also found a major role in the quantitative liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS) analysis as internal standards.9 An internal standard is a compound that is very similar, but not identical to the target analyte. It should have the same effect of sample preparation as the target analyte, but the analytical method should be able to identify the analyte from the internal standard in some factors such as molecular mass. Deuterated compounds can act as internal standards due to several factors: they can be extracted from biological samples at the same content as their corresponding unlabeled analogues; they have the same retention times as the unlabeled analogues in chromatographic methods; the ionization behavior of deuterated compounds is the same as the unlabeled analogues in LC/MS analyses, but their molecular masses are different. Consequently, the quantitative determination of an analyte is possible when the mass difference is large enough between the analyte and its deuterated internal standard. In fact, in order to reduce the cross signal overlap between the analyte and the internal standard, the latter should be deuterated with a low content (less than 0.5%) of unlabeled material (D0) and a representative mass peak (Figure 1.4).9a Thus, it should incorporate 3-5 deuterium atoms in order to be clearly identified from its natural isotope distribution.
7a) E. M. Simmons, J. F. Hartwig, Angew. Chem. Int. Ed. 2012, 51, 3066; b) H. Shi, P. Wang, S. Suzuki, M. E. Farmer, J.-Q. Yu, J. Am. Chem.
Soc. 2016, 138, 14876.
8 a) Z.-X. Wang, X.-Y. Bai, H.-C. Yao, B.-J. Li, J. Am. Chem. Soc. 2016, 138, 14872; b) L. Zhang, G. J. Lovinger, E. K. Edelstein, A. A. Szymaniak,
M. P. Chierchia, J. P. Morken, Science, 2016, 351, 70.
9 a) J. Atzrodt, V. Derdau, J. Label. Compd. Radiopharm. 2010, 53, 674; b) X. Diao, Z. Ma, H. Wang, D. Zhonga, Y. Zhang, J. Jin, Y. Fan, X. Chen,
J. Pharm. Biomed. Anal. 2013, 78-79, 19; c) P. Bruheim, H. F. N. Kvitvang, S. G. Villas-Boas, J. Chromatogr. A 2013, 1296, 196; d) T. Yarita, Y. Aoyagi, T. Otake, J. Chromatogr. A 2015, 1396, 109; e) N. Bakaraki, D. S. Chormey, S. Bakirdere, G. O. Engin, Anal. Methods 2016, 8, 2660; f) H. B. Swan, E. S. M. Deschaseaux, G. B. Jones, B. D. Eyre, Anal. Bioanal. Chem. 2017, 409, 1929.
8
Figure 1.4 MS distribution patterns from different compounds: (a) natural isotope; (b) unselective
H/D exchange; (c) moderately broad isotope cluster with a representative mass peak; (d) highly selective H/D exchange.
For example, 3-n-butylphthalide (Figure 1.5, compound 1.1.1a) is a cardiovascular drug widely used in China for the treatment of cerebral ischemic stroke. It is of great importance to have a better understanding of the in vivo fate of 1.1.1a and its four major metabolites (Figure 1.5, compounds
1.1.2a-1.1.5a) for safety evaluation, good clinic practice and discovery of new antistroke drugs. In
2013, X. Chen et al. reported a method allowing the simultaneous quantitation of 1.1.1a and its four major metabolites in human plasma by LC-MS/MS using the corresponding deuterated 1.1.1b-1.1.5b as internal standards.9b Using this analytical method, they were able to quantify the five compounds with the lowest concentration of 3.00 ng/ml. Moreover, by following a single oral administration of 200 mg 1.1.1a to healthy volunteers, the pharmacokinetic profiles of 1.1.1a-1.1.5a were characterized successfully by this method.
9 More recently, the first deuterated drug, deutetrabenazine (Figure 1.6), was approved by FDA.10 Tetrabenazine is a vesicular monoamine transporter 2 (VMAT2) inhibitor, and it is used for the treatment of chorea associated with Huntington disease. It has been proved that its deuterated analogue, deutetrabenazine, has improved dosing and safety profiles, owing to the stronger bond energy of the C-D bond and thus better withstanding drug-metabolizing enzymes such as the cytochrome P450s (CYP), a class of monooxygenase for the oxidation of C-H bond. According to the in vitro experiments in human liver microsomes, deutetrabenazine is rapidly and extensively metabolized by carbonyl reductase to its major active metabolites, α-dihydrotetrabenazine (α-HTBZ, Figure 1.6) and
β-dihydrotetrabenazine (β-HTBZ, Figure 1.6), which potently inhibit VMAT2 in the central nervous
system. The active metabolites are subsequently metabolized majorly by cytochrome P450 2D6 (CYP2D6) through the oxidation of the methyl group. Since the cleavage of C-H bond is the rate-determining step in CYP catalyzed oxidation,11 as a consequence, the deuterated analogues are more able to withstand the oxidation by CYP2D6 than the hydrogenated ones, owing to the stronger bond energy of C-D bonds than C-H bonds (primary KIE). As a result, the active metabolites of deutetrabenazine have a longer lifetime than the ones of tetrabenazine, hence improving the dosing and safety profiles.
Figure 1.6 Deutetrabenazine and its major active metabolites
Indeed, the selective incorporation of deuterium into drugs can increase significantly their biological half-life, taking advantage of the primary kinetic isotope effect. Moreover, it has been reported that the formation of undesired or toxic metabolites could be reduced through selective deuteration of a drug, together with an enhancement of the formation of desired metabolites.12 In fact, there are more and more deuterated drugs under preclinical and clinical investigations.13
10A. Mullard, Nat. Rev. Drug Discov. 2017, 16, 305.
11 F. P. Guengerich, Biol. Chem. 2002, 383, 1553. 12 S. L. Harbeson, R. D. Tung, MedChem News 2014, 24, 8.
13 a) T. G. Gant, J. Med. Chem. 2014, 57, 3595; b) B. Halford, Chem. Eng. News 2016, 94, 32; c) A. Mullard, Nat. Rev. Drug Discov. 2016, 15,
10 In addition, isotopically labeled compounds also facilitate the rapid development of material science, especially the development of organic light-emitting diodes (OLEDs) and photovoltaics.14 Generally, devices having deuterated molecules do not significantly change the physiochemical properties compared to the ones having hydrogenated compounds,15 but different behaviors of devices having deuterated molecules might be observed due to the mass difference between hydrogen and deuterium. For example, it was reported that by using the devices made with deuterated compounds, the external quantum efficiency of electroluminescent devices could be increased significantly; besides, lower turn-on voltages and lower deterioration rates in the electroluminescence brightness were observed; a better highvoltage stability could happen.14a In another case, the isotopic effects of deuteration on optoelectronic properties of conducting polymers were studied by K. Xiao and co-workers.14d Different photovoltaic characteristics were found between the main-chain deuterated polymer (Figure
1.7, 1.1.6-DM) and side-chain deuterated polymer (Figure 1.7, 1.1.6-DS). For the former deuterated analogue, the short-circuit current was reduced due to the change of film crystallinity and morphology of the active layer resulted by the weak non-covalent intermolecular interactions of deuterated thiophene main-chain; for the latter deuterated analogue, the film morphology remained not changed, while the electronic coupling was decreased together with the formation of a charge transfer state and increased electron-phonon coupling, which has reduced significantly the open circuit voltage.
Figure 1.7 Polymers studied by K. Xiao et al.
There are many other applications for deuterated molecules. For example, in metabolomics, deuterated molecules were used to provide accurate metabolite identification, absolute quantification and flux measurement.16 As shown in Figure 1.8, metabolomic analysis by coupling H/D exchange and LC/MS was conducted by J. F. Banfield and co-workers.16a By comparing the mass spectrum of metabolites obtained from protonated solvents and the one obtained from deuterated solvents, the exchangeable hydrogens of the analytes could be figured out, which could help to identify the structure of the analytes. Through the analyses of the sample metabolites, the authors discovered that natural
14a) C. C. Tong, K. C. Hwang, J. Phys. Chem. C 2007, 111, 3490; b) T. D. Nguyen, G. Hukic-Markosian, F. Wang, L. Wojcik, X.-G. Li, E.
Ehrenfreund, Z. V. Vardeny, Nat. Mater. 2010, 9, 345; c) T. A. Darwish, A. R. G. Smith, I. R. Gentle, P. L. Burn, E. Luks, G. Moraes, M. Gillon, P. J. Holden, M. James, Tetrahedron Lett. 2012, 53, 931; d) M. Shao, J. Keum, J. Chen, Y. He, W. Chen, J. F. Browning, J. Jakowshi, B. G. Sumpter, I. N. Ivanov, Y.-Z. Ma, C. M. Rouleau, S. C. Smith, D. B. Geohegan, K. Hong, K. Xiao, Nat. Commun. 2014, 5, 1; e) H. Tsuji, C. Mitsui, E. Nakamura, Chem. Commun. 2014, 50, 14870; f) A. Danos, R. W. MacQueen, Y. Y. Cheng, M. Dvořák, T. A. Darwish, D. R. McCamey, T. W. Schmidt, J. Phys. Chem. Lett. 2015, 6, 3061; g) A. T. Wagner, R. Zhou, K. S. Quinn, T. A. White, J. Wang, K. J. Brewer, J. Phys. Chem. A 2015, 119, 6781.
15 T A Baillie, Pharmacol. Rev. 1981, 33, 81.
16 a) C. R. Fischer, P. Wilmes, B. P. Bowen, T. R. Northen, J. F. Banfield, Metabolomics, 2012, 8, 566; b) A. Chokkathukalam, D.-H. Kim, M. P.
11 acidophilic microbial communities contained N-methyl lyso phosphatidylethanolamines (PE) as abundant lipids.
Figure 1.8 Sample handling workflow for deuterium exchange metabolomics studies16a
In pharmacokinetic and pharmacological absorption, distribution, metabolism, and excretion studies (ADME), deuterated molecules were used to slow down the metabolic rate and modulate pharmacokinetics.17 In some special cases, deuteration was reported to protect amino acids against racemization,18 enhance catalyst lifetime,19 probe C-H activation process,20 enable structure analysis of protein,21 control the diastereoselectivity of ligand synthesis,22 and facilitate the formation of desired product in total synthesis.23 More importantly, various deuterated solvents are widely used in NMR analyses to understand the structure information of organic compounds.
1.2 Tritium and applications of tritiated compounds
Tritium is a radioactive isotope of hydrogen with a symbol of T or 3H, which contains one proton and two neutrons. It was first produced from deuterium by Rutherford, M. L. Oliphant, and P. Harteck in 1934.24 However, it was L. W. Alvarez and R. Cornog who isolated tritium for the first time and
17 a) R. Sharma, T. J. Strelevitz, H. Gao, A. J. Clark, K. Schildknegt, R. S. Obach, S. L. Ripp, D. K. Spracklin, L. M. Tremaine, A. D. N. Vaz, Drug
Metab. Dispos. 2012, 40, 625; b) J. Schofield, D. Brasseur, B. de Bruin, T. Vassal, S. Klieber, C. Arabeyre, M. Bourrié, F. Sadoun, G. Fabre, J. Label. Compd. Radiopharm. 2013, 56, 504; c) V. Uttamsingh, R. Gallegos, J. F. Liu, S. L. Harbeson, G. W. Bridson, C. Cheng, D. S. Wells, P. B. Graham, R. Zelle, R. Tung, J. Pharmacol. Exp. Ther. 2015, 354, 43; d) J. Jiang, X. Pang, L. Li, X. Dai, X. Diao, X. Chen, D. Zhong, Y. Wang, Y. Chen, Drug Des. Dev. Ther. 2016, 10, 2181.
18 J. D. Lowenson, V. V. Shmanai, D. Shklyaruck, S. G. Clarke, M. S. Shchepinov, Amino Acids 2016, 48, 2189.
19 N. Armenise, N. Tahiri, N. N. H. M. Eisink, M. Denis, M. Jäger, J. G. De Vries, M. D. Witte, A. J. Minnaard, Chem. Commun. 2016, 52, 2189. 20 F. M. Chadwick, T. Krämer, T. Gutmann, N. H. Rees, A. L. Thompson, A. J. Edwards, G. Buntkowsky, S. A. Macgregor, A. S. Weller, J. Am.
Chem. Soc. 2016, 138, 13369. 21
A. Kita, Y. Morimoto, Mol. Biotechnol. 2016, 58, 130.
22 R. A. Arthurs, C. J. Richards, Org. Lett. 2017, 19, 702.
23 a) M. Miyashita, M. Sasaki, I. Hattori, M. Sakai, K. Tanino, Science 2004, 305, 495; b) K. W. Quasdorf, A. D. Huters, M. W. Lodewyk, D. J.
Tantillo, N. K. Garg, J. Am. Chem.Soc. 2012, 134, 1396.
24 a) M. L. Oliphant, P. Harteck, Rutherford, Nature 1934, 133, 413; b) M. L. E. Oliphant, P. Harteck, Lord Rutherford, Proc. R. Soc. A 1934, 144,
12 discovered tritium's radioactivity.25 According to the radionuclide safety data sheet (RSDS) developed by Radiation Safety Services (RSS), tritium has a physical half-life of 12.3 years, a high specific activity of 9650 Ci/g (28.8 Ci/mmol), and it emits beta particle with a range of 5 mm in air and average about 0.56 μm in water or tissue. [Specific activity is defined as the activity per quantity of atoms of a particular radionuclide. The unit of specific activity is Ci/g (Ci = Curie) or Bq/g (Bq = Becquerel), and 1 Ci/g = 3.7×1010 Bq/g.] The high specific activity together with insignificant penetrability through human skin makes tritium widely used as a radioactive tracer element in biological research, especially for the labeling of biomolecules such as peptides, proteins, oligonucleotides, and antibodies.26
Tritium gas is usually stored in the form of a solid metal tritide and the stored tritium will not be released until it is heated. For tritium labeling, the most convenient isotopic source is the tritium gas because it can be easily produced by heating the metal tritide, and it can be reabsorbed upon cooling down the metal tritide. Tritiated water is sometimes used as the isotopic source for tritiation. However, pure tritiated water is corrosive due to self-radiolysis. Hence, it is used in the dilute form, which mainly contains H2O plus some HTO. Since tritium gas is easier and safer to handle, it has been mostly used for tritium labeling. For instance, recently, our group reported a method allowing the tritium labeling of DPA-714 (Figure 1.9), a fluorinated ligand of the translocator protein 18 kDa (TSPO), which could be used as a probe for positron emission tomography (PET) imaging, by using Pd/C as catalyst and T2 gas as the tritium source.26e The tritiated molecule could help to better understand how it binds to the TSPO, and it also gave a combination of in vitro and in vivo studies performed with this tracer.
Figure 1.9 Structure of DPA-714
25 a) L. W. Alvarez, R. Cornog, Phys. Rev. 1939, 56, 613; b) L. W. Alvarez, W. P. Trower, Discovering Alvarez: Selected works of Luis W. Alvarez,
with commentary by his students and colleagues, University of Chicago Press, Chicago and London, 1987, 26-29.
26 a) M. Saljoughian, P. G. Williams, Curr. Pharm. Des. 2000, 6, 1029; b) U. S. Larsen, H. B. Hansen, A.-M. Dahl, L. Sørensen, J. B. Kristensen, J.
Label. Compd. Radiopharm. 2007, 50, 549; c) G. Tóth, J. R. Mallareddy, F. Tóth, A. W. Lipkowski, D. Tourwé, ARKIVOC 2012 (v), 163; d) G. Schwarzmann, C. Arenz, K. Sandhoff, Biochim. Biophys. Acta 2014, 1841, 1161; e) A. Damont, S. Garcia-Argote, D.-A. Buisson, B. Rousseau, F. Dollé, J. Label. Compd. Radiopharm. 2015, 58, 1.
13 In addition, tritiated molecules were applied to ADME studies.27 Tritium labeled compounds are relatively simple and fast to synthesize compared with compounds labeled with other radioactive elements such as 14C and 18F, which are obtained mostly by multi-step synthesis. They can be obtained with high specific activity, which is useful for studying the highly potent compounds at a low dose. Tritium labeling facilitates the procurement of quantitative data of metabolites by avoiding the interference of complex biological matrices and metabolic changes to chemical structure. Through the analysis of radioactive metabolic profiles in excreta, it becomes possible to determine clearance mechanisms of drug candidates. In this context, the use of radioactive labeling is crucial in the safety assessment of novel pharmaceuticals. Therefore, the development of efficient late stage processes enabling the selective deuterium and tritium incorporation into complex molecules is of paramount importance.
For example, F. Lozac’h et al. used tritium labeled 2’-O-methyluridine to study its ADME properties in mice in order to understand the biological impact of this nucleoside in a siRNA sequence and to support preclinical safety studies and clinical development of siRNA therapeutics.27h The concentration of radiolabeled components were determined by Liquid Scintillation Counter (LSC). Metabolites were separated by Ultra High Pressure Liquid Chromatography (UHPLC) and subsequently identified using radiodetection and high-resolution mass spectrometry (HRMS). Through detecting the radioactivity by an intravenous administration of tritium labeled 2’-O-methyluridine after 48 hours, it was observed that tritiated 2’-O-methyluridine represented a minor component of the radioactivity (5.89%) in plasma; three metabolites (Figure 1.10) namely uridine (M1), cytidine (M2), and uracil (M3) were the major circulating components representing 32.8%, 8.11%, and 23.6% of radioactivity area under the curve, respectively.
Figure 1.10 Tritium labeled 2’-O-methyluridine and its metabolites
27 a) D. Dalvie, Curr. Pharm. Des. 2000, 6, 1009; b) F. P. Guengerich, Chem. Res. Toxicol. 2012, 25, 511; c) N. Penner, L. Xu, C. Prakash, Chem.
Res. Toxicol. 2012, 25, 513; d) R. E. White, D. C. Evans, C. E. C. A. Hop, D. J. Moore, C. Prakash, S. Surapaneni, F. L. S. Tse, Xenobiotica 2013, 43, 219; e) R. S. Obach, A. N. Nedderman, D. A. Smith, Xenobiotica 2013, 43, 226; f) M. Pellegatti, Expert. Opin. Drug Metab. 2014, 10, 1615; g) A. Beattie, S. Madden, C. Lowrie, D. MacPherson, Bioanalysis 2015, 7, 507; h) F. Lozac’h, J. Christensen, T. Faller, E. van de Kerkhof, J. Krauser, M. Garnier, K. Litherland, A. Catoire, F. Natt, J. Hunziker, P. Swart, Pharma Res Per 2016, 4, e00209.
14 1.3 Synthesis of deuterium or tritium labeled compounds
There are two possible ways to achieve isotopically labeled molecules: (1) by a conventional multistep synthesis; (2) by direct hydrogen isotope exchange (HIE).
Starting from appropriate commercially available labeled precursors, more complex molecules could be synthesized in several steps. For example, in 2015, V. Uttamsingh and co-workers reported the synthesis of deuterated paroxetine hydrochloride salt 1.3.6 in 5 steps, with a total yield of 20% (Scheme 1.1).28 Notably, deuterated sesamol 1.3.3 was synthesized separately in 3 steps, with a total yield of 53%. The advantage of this method was that it allowed the exclusive introduction of deuterium atoms with high isotopic enrichment (full deuterated starting material); however, it suffered the drawback of time and resources consuming and produced a lot of isotopically labeled wastes. Such drawback would be more underlined when dealing with tritium labeling.
Scheme 1.1 Synthesis of deuterium labeled paroxetine hydrochloride salt
By contrast, the HIE process enables a direct isotopic labeling of the desired molecule in one single step, thus saving time and costs. Different types of catalysts have been applied in the HIE reactions. For metal catalysts, they could be divided into two types according to the active catalytic species: homogeneous catalysts and heterogeneous catalysts. Recently, our group developed a method allowing the regioselective and stereospecific deuteration of nitrogen containing compounds catalyzed by ruthenium nanoparticles,29 and the deuteration of paroxetine has been achieved in one step with high
28 V. Uttamsingh, R. Gallegos, J. F. Liu, S. L. Harbeson, G. W. Bridson, C. Cheng, D. S. Wells, P. B. Graham, R. Zelle, R. Tung, J. Pharmacol.
Exp. Ther. 2015, 354, 43.
29 G. Pieters, C. Taglang, E. Bonnefille, T. Gutmann, C. Puente, J.-C. Berthet, C. Dugave, B. Chaudret, B. Rousseau, Angew. Chem. Int. Ed.
15 deuterium incorporation and high yield (Scheme 1.2). Moreover, stereochemistry was retained over the deuteration process.
Scheme 1.2 H/D exchange of paroxetine catalyzed by RuNp@PVP
Obviously, the HIE process is preferred to obtain deuterated molecules. Achieving isotopically labeled molecules with high isotopic enrichment or high specific activity under mild reaction conditions is the main goal of HIE reactions. The main challenge of HIE process is the development of regio- and stereoselective procedures allowing the labeling of various functional group containing molecules. A remarkable evolution was realized in the field of H/D exchange process since the pioneering work of J. L. Garnett half a century ago.30 Using a homogeneous Pt-catalyst, Garnett succeeded to label a variety of aromatic compounds, despite the poor selectivity and low deuterium incorporation. Since then, rapid development has been achieved in the field of homogeneous catalytic H/D exchange reactions. Moreover, HIE reactions can be accomplished by either acid/base mediated H/D exchange or metal catalyzed H/D exchange. Remarkably, in the early 1980s, W. J. S. Lockley discovered that aromatic carboxylic acids and anilines could be labeled regioselectively using RhCl3.31 These distinguished works opened the door for a series of notable advances in regioselective H/D exchange reactions.
In the last 30 years, many research groups have made great contributions to the selective deuteration of various organic compounds.32 One of the most remarkable advances may be the development and application of the Crabtree's catalyst [(cod)Ir(PPh3)(py)]PF6 (Figure 1.11),33 which was first known as a hydrogenation catalyst in the beginning. Crabtree's catalyst is an air stable homogeneous catalyst developed by R. H. Crabtree in 1979, and it is now widely used for various catalytic reactions because
30 a) J. L. Garnett, R. J. Hodges, J. Am. Chem. Soc. 1967, 89, 4546; b) M. R. Blake, J. L. Garnett, I. K. Gregor, W. Hannan, K. Hoa, M. A. Long, J.
Chem. Soc., Chem. Commun. 1975, 930.
31 a) W. J. S. Lockley, Tetrahedron Lett. 1982, 23, 3819; b) W. J. S. Lockley, J. Label. Compd. Radiopharm. 1984, 21, 45; c) W. J. S. Lockley, J.
Label. Compd. Radiopharm. 1985, 22, 623.
32 For selected reviews, see ref [2], ref [8a] and: a) R. Salter, J. Label. Compd. Radiopharm. 2010, 53, 645; b) W. J. S. Lockley, D. Hesk, J. Label.
Compd. Radiopharm. 2010, 53, 704.
16 of its robust catalytic activity and commercial availability.34 Regarding the H/D exchange reaction mechanism catalyzed by Crabtree’s catalyst, the key processes involve the coordination of substrate to the metal center, the activation of C-H bond adjacent to the functional group to form a five- or six-membered ring intermediate (Figure 1.12), ligand exchange and finally reductive elimination to yield the deuterated product.35 It can be used either as a co-catalyst36 or as an independent catalyst35,37 in different deuteration reactions with high regioselectivity.
Figure 1.11 Structure of Crabtree's catalyst
Figure 1.12 Key intermediates in H/D exchange reactions catalyzed by Crabtree’s catalyst
The substrate scope of Crabtree’s catalyst in H/D exchange is broad, ranging from aromatic esters, amides, ketones to heterocycles such as aryl substituted furan, thiophene, oxazole, thiazole, pyridine, pyrazole and so on.37b In some rare cases, Crabtree’s catalyst is also used for C(sp3)-H bond activation. For example, it has been applied to the selective H/T exchange of methapyrilene at C(sp3)-H bond with high specific activity (Figure 1.13).38
34 R. Salter, M. Chappelle, A. Morgan, T. Moenius, P. Ackermann, M. Studer, F. Spindler, Synthesis and Applications of Isotopically Labelled
Compounds, John Wiley and Sons Ltd, Chichester, 2001, 7, 63-67.
35 a) A. Y. L. Shu, W. Chen, J. R. Heys, J. Organomet. Chem. 1996, 524, 87; b) J. S. Valsborg, L. Sørensen, C. Foged, J. Label. Compd.
Radiopharm. 2001, 44, 209.
36 S. C. Schou, J. Label. Compd. Radiopharm. 2009, 52, 376.
37 a) D. Hesk, P.R. Das, B. Evans, J. Label. Compd. Radiopharm. 1995, 36, 497; b) G. J. Ellames, J. S. Gibson, J. M. Herbert, A. H. McNeill,
Tetrahedron 2001, 57, 9487.
38 a) N. Bushby, D. A. Killick, J. Label. Compd. Radiopharm. 2007, 50, 519; b) R. P. Yu, D. Hesk, N. Rivera, I. Pelczer, P. J. Chirik, Nature 2016,
17
Figure 1.13 H/T exchange of bioactive compounds catalyzed by Crabtree’s catalyst
However, despite the broad applications of the Crabtree’s catalyst, a general drawback is its sensitivity to extraneous proton-bearing impurities.39 Generally, dichloromethane is the only effective solvent used in the reaction. Besides, a significant loss of activity could happen when dealing with substrates with a functional group of alcohols or amines.33 A general study of Crabtree’s catalyst concerning the scope and limitations of deuteration has been done by J. M. Herbert and co-workers.37b They indicated that the Crabtree’s catalyst was regularly required in stoichiometric amounts or even more, and often led to low isotope incorporation levels and undesirably lengthy reaction times. Moreover, the application of this catalyst to the tritium labeling leads to high amounts of (tritiated) wastes (labeled cyclooctane and/or cyclooctene from the ligand) because of the high catalyst loading.40
Numerous works have been recently dedicated to the development of new methods allowing selective and efficient labeling of biologically relevant compounds. Among them, several chemo- and regio-selective metal catalyzed H/D exchange procedures allowing the labeling of oxygen and nitrogen containing molecules have been described.41 Compared to multistep syntheses, the H/D exchange process is a more steps and time saving way to achieve deuterated molecules. We review herein majorly the works published during the past ten years concerning the progress of metal-catalyzed HIE reactions through C-H activation processes.
1.4 H/D exchange reactions catalyzed by iridium complexes
Among all the metal catalysts used for the H/D exchange procedures, iridium complexes are the most wildly studied.42 Many research groups have been working on iridium catalyzed H/D exchange reactions and great achievements have been obtained in this field.32a In recent years, numerous novel
39 Y. Xu, D. M. P. Mingos, J. M. Brown, Chem. Commun. 2008, 199.
40a) J. A. Brown, S. Irvine, A. R. Kennedy, W. J. Kerr, S. Anderssonb, G. N. Nilsson, Chem. Commun. 2008, 1115; b) G. N. Nilsson, W. J. Kerr, J.
Label. Compd. Radiopharm. 2010, 53, 662.
41 For recent examples see ref [35b] and: a) L. V. A. Hale, N. K. Szymczak, J. Am. Chem. Soc. 2016, 138, 13489; b) B. Chatterjee, V.
Krishnakumar, C. Gunanathan, Org. Lett. 2016, 18, 5892; c) B. Chatterjee and C. Gunanathan, Org. Lett. 2015, 17, 4794; d) W. J. Kerr, M. Reid, T. Tuttle, ACS Catal. 2015, 5, 402; e) W. J. Kerr, D. M. Lindsay, M. Reid, J. Atzrodt, V. Derdau, P. Rojahn, R. Weck, Chem. Commun. 2016, 52, 6669; f) L. Neubert, D. Michalik, S. Bähn, S. Imm, H. Neumann, J. Atzrodt, V. Derdau, W. Holla, M. Beller, J. Am. Chem. Soc. 2012, 134, 12239.
18 methods based on HIE have been developed for the selective deuterium/tritium labeling of various organic compounds catalyzed by iridium complexes.43
1.4.1 Iridium catalyzed deuteration involving oxygen atom as directing groups
Oxygen is a common coordinating atom to many metals. It can coordinate in the form of different functional groups, such as alcohols, ketones, esters, carboxylic acids, amides or nitro group. Therefore, many works on H/D exchange have been achieved involving oxygen atom as directing groups catalyzed by iridium complexes.
Among the research groups working on iridium catalyzed H/D exchange reactions, W. J. Kerr’s group has made huge contributions.40a, 41d-e, 42, 43d-k In 2008, Kerr reported40a the synthesis of a series of iridium complexes stabilized by bulky N-heterocyclic carbene (NHC) and their applications in the regioselective H/D exchange reactions (Figure 1.14). Excellent levels of labeling had been achieved over short reaction times and at low catalyst loadings. Moreover, these complexes were all air- and moisture-stable solids, which could be stored under air for more than 8 months without loss of catalytic activity.
Figure 1.14 Structure of Kerr’s catalysts
Since then, Kerr’s group has been concentrating on the core structure of the iridium complex, by modifying the coordinating group or by changing the counterion, they have succeeded to deuterate/tritiate a variety of organic compounds. For example, through replacing PF6 counterion with BArF (tetrakis[3,5-bis(trifluoromethyl)phenyl]borate), the catalytic activity could be improved at lower catalyst loadings, together with an improvement of solubility profile and applicable solvent
43For recent examples of iridium catalyzed H/D exchange reactions, see: a) V. M. Iluc, A. Fedorov, R. H. Grubbs, Organometallics 2012, 31, 39;
b) M. C. Lehman, J. B. Gary, P. D. Boyle, M. S. Sanford, E. A. Ison, ACS Catal. 2013, 3, 2304; c) M. Parmentier, T. Hartung, A. Pfaltz, D. Muri, Chem. Eur. J. 2014, 20, 11496; d) J. A. Brown, A. R. Cochrane, S. Irvine, W. J. Kerr, B. Mondal, J. A. Parkinson, L. C. Paterson, M. Reid, T. Tuttle, S. Andersson, G. N. Nilsson, Adv. Synth. Catal. 2014, 356, 3551; e) A. R. Cochrane, C. Idziak, W. J. Kerr, B. Mondal, L. C. Paterson, T. Tuttle, S. Anderssonb, G. N. Nilsson, Org. Biomol. Chem. 2014, 12, 3598; f) A. R. Kennedy, W. J. Kerr, R. Moir, M. Reid, Org. Biomol. Chem. 2014, 12, 7927; g) W. J. Kerr, R. J. Mudd, L. C. Paterson, J. A. Brown, Chem. Eur. J. 2014, 20, 14604; h) J. Atzrodt, V. Derdau, W. J. Kerr, M. Reid, P. Rojahn, R. Weck, Tetrahedron 2015, 71, 1924; i) J. Devlin, W. J. Kerr, D. M. Lindsay, T. J. D. McCabe, M. Reid, T. Tuttle, Molecules 2015, 20, 11676; j) P. W. C. Cross, J. M. Herbert, W. J. Kerr, A. H. McNeill, L. C. Paterson, Synlett 2016, 27, 111; k) W. J. Kerr, R. J. Mudd, P. K. Owens, M. Reid, J. A. Brown, S. Campos, J. Label. Compd. Radiopharm 2016, 59, 601; l) A. Marek, M. H. F. Pedersen, S. B. Vogensen, R. P. Clausen, B. Frølund, T. Elbert, J. Label. Compd. Radiopharm 2016, 59, 476; m) I. Romanenko, S. Norsic, L. Veyre, R. Sayah, F. D’Agosto, J. Raynaud, C. Boisson, E. Lacôte, C. Thieuleux, Adv. Synth. Catal. 2016, 358, 2317; n) M. Hatano, T. Nishimura, H. Yorimitsu, Org. Lett. 2016, 18, 3674; o) S. Ibañez, M. Poyatos, E. Peris, Dalton Trans. 2016, 45, 14154; p) K. Jess, V. Derdau, R. Weck, J. Atzrodt, M. Freytag, P. G. Jones, M. Tamm, Adv. Synth. Catal. 2017, 359, 629; q) A. Burhop, R. Weck, J. Atzrodt, V. Derdau, Eur. J. Org. Chem. 2017, 1418.
19 scope.43f, k Using the new catalyst 1.4.2, they succeeded to label a drug molecule (niclosamide,
Scheme 1.3) with an improved deuterium incorporation.
Scheme 1.3 Deuteration of niclosamide by iridium complex
Later, Kerr reported the first regioselective C-H deuteration of unsaturated organic compounds at the
β-position with iridium catalyst 1.4.1a.43g Under mild reaction conditions with low levels of catalyst
loading, high levels of deuterium incorporation have been obtained with good selectivity over the potentially competing reduction process, across a series of α,β-unsaturated substrates (Scheme 1.4).
Scheme 1.4 Deuteration of α,β-unsaturated compounds with Kerr’s catalyst
Besides, as an overview of Kerr’s catalysts, they also examined their catalytic activities toward the labeling of benzoate ester derivatives comparing with Crabtree’s catalyst.43i As a result, Kerr’s catalyst
1.4.1a showed more efficiency than Crabtree’s catalyst in the deuteration of benzoate esters.
Additionally, with a slightly raise of reaction temperature (from room temperature to 40 °C), great improvements of deuterium incorporation were observed across all substrates examined. Thus, the application of Kerr’s catalysts became wider by two ways: increasing the reaction temperature or switching the counterion.
Apart from Kerr’s works, A. Marek and co-workers reported the labeling of unsaturated γ-hydroxybutyric acid (1.4.3, a potent ligand for high affinity γ-γ-hydroxybutyric acid binding sites in the central nervous system) by comparison between H/D exchange reaction mediated by iridium complex and reduction by boro-deuterides (Scheme 1.5).43l Through changing the solvent, Crabtree’s catalyst was able to control the selective deuteration of the substrate without over reduction. However, the
20 deuterium incorporation was low (< 10%). Kerr’s catalyst was found to be more efficient than Crabtree’s catalyst for the deuteration. However, over reduction always occurred together with high deuterium incorporation. By contrast, labeling through reduction by boro-deuterides gave α-deuterated alcohol with high yield. Using LiB(OMe)3D as reductant, the conversion of the reaction could reach 99% with 91% of yield, and it was even applicable to tritium labeling by using LiB(OMe)3T.
Scheme 1.5 The strategies of the deuterium labeling of 1.4.3
Overall, Marek’s group has provided an alternative way to the synthesis of deuterated γ-hydroxybutyric acid: the H/D exchange pathway using Kerr’s catalysts 1.4.1a and 1.4.1c, even if the synthesis was accompanied by the formation of over reduced byproduct. Anyway, the different labeling sites of the molecule may help to better understand the in vivo activity of the molecule. In 2016, C. Boisson and co-workers reported the synthesis of a polyethylene-supported iridium
(III)-N-heterocyclic carbene catalyst (Figure 1.15) and its application for the H/D exchange reactions.43m
According to the authors, the catalyst acted as a homogeneous catalyst at 100 °C, but it could be easily recovered by filtration as a precipitate at room temperature due to the unique thermomorphic polyethylene properties.
Figure 1.15 Polyethylene-supported iridium (III)-N-heterocyclic carbene catalyst
The advantages of the polyethylene-supported iridium catalyst were the low catalyst loading, short reaction time and its recyclable ability. When using 2 mol% of catalyst, the deuteration of
21 acetophenone was still efficient after the third cycle of the catalyst in just 5 min reaction time. However, the multistep synthesis of the catalyst and the narrow substrate scope (only 3 examples were given in the publication) have limited the application of the catalyst.
1.4.2 Iridium catalyzed deuteration involving nitrogen atom as directing group
Compared with oxygen, nitrogen atom displays stronger coordinating ability to transition metal catalysts because N-ligands are generally stronger Lewis bases than O-ligands, thus the coordination bond between transition metal and nitrogen is stronger than the one between transition metal and oxygen. Nitrogen can participate as coordinating atom in various functional groups, such as amines, sulfonamides and a series of N-heterocycles. Numerous works have been published recently based on the nitrogen directing groups.
Apart from the works of the deuteration involving oxygen atom, Kerr also applied his catalysts in the labeling of N-heterocycles41e, 43h and primary sulfonamides. 41d For the labeling of N-heterocycles such as pyrimidine, imidazole, oxazole, oxazoline, isoxazole, thiazole, benzimidazole, benzoxazole and benzothiazole, catalyst 1.4.1a proved to be the most efficient (Scheme 1.6, a);43h while for the labeling of unprotected tetrazoles, catalyst 1.4.2 worked best with the addition of Cs2CO3 as base since the reaction was a concerted metalation-deprotonation (CMD) pathway (Scheme 1.6, b).41e Moreover, through the optimization of reaction conditions, deuteration of complex molecules 1.4.7-1.4.9 and deuteration/tritiation of valsartan 1.4.10, an angiotensin receptor blocker, were also achieved using the two catalysts separately (Figure 1.16).
22
Figure 1.16 Labeling of complex molecules by Kerr’s catalyst
In 2014, Kerr reported the first general method allowing the selective deuterium labeling of primary sulfonamides using a modified Kerr’s catalyst 1.4.11 (Scheme 1.7).41d This catalyst had a sterically less hindered and more electron-rich chloride ligand, which could enhance the efficiency of the sulfonamide coordination and subsequent ortho-deuteration. A series of sulfonamides were selectively deuterated with various substituents on aryl rings, including two pharmaceuticals with good chemoselectivities (Scheme 1.7). However, a notable limitation of this method lies in the poor deuterium incorporation of some substrates due to their poor solubility in dichloromethane. A competition study was done to assess the chemoselectivity of the catalyst system, which revealed that the deuteration of primary sulfonamides could proceed smoothly with the presence of ketone, ester, nitro, and various amide directing groups; while the presence of N-heterocycles could change the chemoselectivity of the deuteration process to the labeling of these substrates.
23 More recently, V. Derdau and co-workers reported the selective deuteration of secondary sulfonamides and the first selective aromatic deuteration of tertiary sulfonamides as well as sulfonylureas based on Kerr’s catalyst 1.4.12 and the commercially available Burgess catalyst 1.4.13 (Figure 1.17).43q The Burgess catalyst 1.4.13, which was developed as a catalyst for the asymmetric hydrogenation of olefins,44 has never been applied in H/D exchange reactions before their publication.
Figure 1.17 Structure of Kerr’s catalyst 1.4.12 and Burgess catalyst 1.4.13
By comparison of the catalytic activity of the two iridium complexes, the authors discovered that the Burgess catalyst 1.4.13 was more effective when conducting the deuteration reactions on tertiary sulfonamides, while the Kerr’s catalyst 1.4.12 was more suitable for the labeling of secondary sulfonamides and ureas (Scheme 1.8). They also succeeded to adapt their method to the deuterium labeling of several drug molecules. Finally, the tritium labeling of glibenclamide was achieved after the optimization of reaction conditions (Scheme 1.8). Using 4 μmol of substrate, with 10 mol% of catalyst 1.4.12 and a reduced pressure of tritium gas (160-278 mbar), after heating at 100 °C in PhCl for 3 h, they succeeded to tritiate glibenclamide with a specific activity of 9 Ci/mmol.
Scheme 1.8 Iridium complexes catalyzed isotopic labeling of sulfonamides
24 1.4.3 Iridium catalyzed deuteration of various compounds with distinct functional groups
For the works described above, the catalysts could catalyze the H/D exchange of compounds with comparable functional groups. However, there are some more general catalysts that are able to catalyze H/D exchange of various compounds with different types of functional groups, such as Kerr’s catalysts.42, 43e, f, j, 45
For catalysts containing an NHC and a chlorine ligands, after screening catalysts with different substituents on carbene ligand (Figure 1.18), Kerr discovered that catalyst 1.4.12 was able to deuterate various aryl ketones efficiently.42 The catalyst could also deuterate amides with moderate deuterium incorporation, and N-heterocycles with high deuterium incorporation.
For catalysts containing an NHC and a phosphine ligands, Kerr showed that all the catalysts (Figure
1.18) were active toward the labeling of aryl ketones, aryl amides and N-heterocycles, despite a little
difference of activities was observed among different substrates.43e, f One could choose different catalyst according to the functional groups of substrates. Kerr even succeeded to tritiate various molecules with different functional groups using catalysts 1.4.1a-1.4.1c respectively. Replacing the phosphine ligand with pyridine (Figure 1.18, 1.4.1f) also had similar catalytic activities towards various compounds with different functional groups.43j
Figure 1.18 Chloro-carbene type and phosphine ligand type Kerr’s catalysts
Despite the broad application of Kerr’s catalysts, some other complexes based on iridium have also been designed for H/D exchange reactions. In 2014, D. Muri and co-workers selected a series of reported highly active iridium catalysts for asymmetric double bond reductions with different N, P-ligands to test their catalytic activities toward H/D exchange reactions.43c In this publication, through the scanning of the iridium catalysts with different ligands for the deuterium labeling of 2-phenylpyridine, the authors discovered that the catalysts containing electron-rich ligands (dicyclohexylphosphines or phosphinites) were excellent catalysts for efficient deuterium labeling of various compounds (Scheme 1.9). With these catalysts that contained electron-rich ligands, substrates with strong directing groups such as pyridines, ketones, and amides, as well as weak ligating units
45 a) R. Simonsson, G. Stenhagen, C. Ericsson, C. S. Elmore, J. Label. Compd. Radiopharm. 2013, 56, 334; b) A. Modvig, T. L. Andersen, R. H.
25 such as nitro, sulfones, and sulfonamides, could be labeled efficiently using D2 gas as the isotopic source.
Scheme 1.9 H/D exchange catalyzed by iridium catalysts with N, P-ligands
Interestingly, by adding tris(pentafluorophenyl)borane to the reaction mixture, the authors were able to deuterate acetylbenzonitrile efficiently, which contains a strongly coordinating substituent, through the coordination of borane to nitrile group. Finally, they also developed an iridium catalyst that contained an achiral ligand (Scheme 1.9, 1.4.18). After testing the reactivity of the new catalyst, they claimed that it was very efficient toward a variety of substrates in the H/D exchange process.
Later in 2016, E. Peris reported the synthesis of three iridium(III) complexes with pyrene-containing
N-heterocyclic carbenes (Figure 1.19) and their catalytic activities toward H/D exchange reactions.43o
Several substrates with different functional groups were tested with the three synthesized catalysts. Among them, the chloride complex 1.4.19 worked best for the deuteration of styrene and trans-stilbene with the addition of AgOTf as additive, while the carbonate complexes 1.4.20 and 1.4.21 deuterated THF efficiently without any additive; the latter was the best catalyst for the mono-deuteration of Benzo[h]quinoline. Besides, the catalysts were also tested for their catalytic activities for the C-C coupling of alcohols. Moderate to good yields were obtained for all catalysts, while complex 1.4.21 was the best catalyst for this transformation.
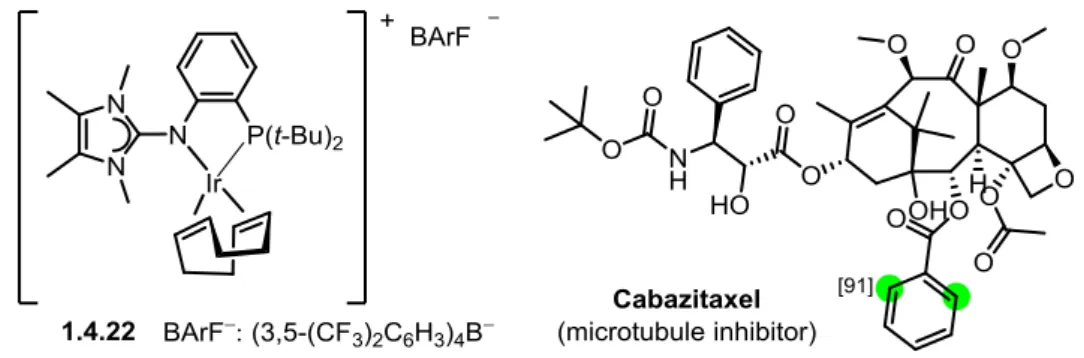
26 Recently, V. Derdau and co-workers reported the synthesis of iridium (I) complexes supported by phosphine-imidazolin-2-imine P, N ligands and its application in the H/D exchange reactions of aryl substrates.43p A total of 12 iridium complexes were synthesized, one of those displayed good activity toward the H/D exchange process (compound 1.4.22, Figure 1.20). Various directing groups such as acetyl, N-heterocycles, amide, sulfone, ester and nitro group were tested with good deuterium incorporation. Moreover, through the optimization of the reaction conditions, substrates with the weakly coordinating methoxy group showed a sufficient degree of deuteration. A total of 23 substrates were deuterated using their method, which was highlighted by the deuteration of the structurally complex molecule Cabazitaxel (Figure 1.20).
Figure 1.20 Iridium complex for H/D exchange reactions and example of substrate: Cabazitaxel
Overall, the reaction conditions of Derdau’s method were mild (5 mol% of catalyst, 1 atm D2 gas pressure and reacted for 1h in CH2Cl2 at ambient temperature), despite the fact that in order to achieve good deuterium incorporation for methoxy derivatives, higher temperature (50 °C), longer reaction time (5 h) and higher catalyst loading (25 mol%) were necessary. The major limitation of Derdau’s method was the multi-step synthesis of the iridium catalysts, and most of the catalysts had poor catalytic activity toward most substrates they used.
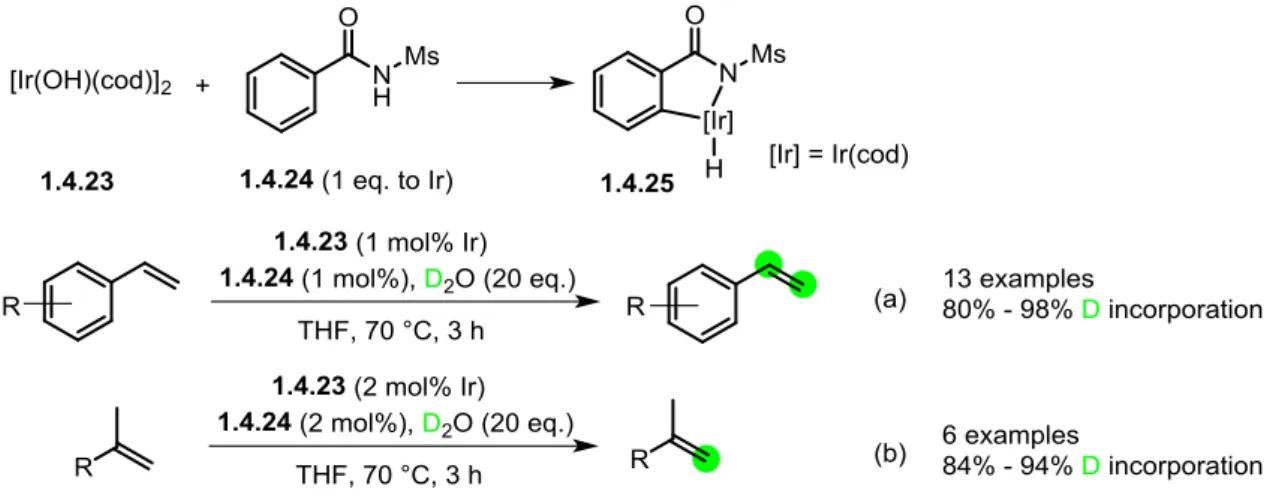
1.4.4 Iridium catalyzed deuteration of alkenes
In 2016, T. Nishimura reported a selective H/D exchange of vinyl and methylidene groups promoted by an iridium catalyst generated in situ from a hydroxoiridium complex and N-mesylbenzamide (Scheme 1.10).43n Various styrene derivatives were tested with good deuterium incorporations selectively at the vinyl group (Scheme 1.10, equation a). Moreover, the catalyst loading could be lowered down to 0.1 mol% with a prolonged reaction time (20 h) to achieve high level of deuterium incorporation, and the isotopic enrichment could also be improved by using a large excess of D2O (200 eq.). Besides, the in situ generated iridium catalyst could also catalyze the deuteration of 1,1-disubstituded alkenes efficiently (Scheme 1.10, equation b), with a slightly increased amount of catalyst. The major disadvantage of this method was that it could not be applied to the H/D exchange