Dicer loss induces an oncogenic epigenetic switch in
mesenchymal stem cells
by
Courtney K. JnBaptiste
B.S. Biology, Bethune Cookman University (2010) Submitted to the Department of Biology
in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
February 2016
© 2016 Massachusetts Institute of Technology All rights reserved
Signature of Author: ______________________________________________________ Courtney K. JnBaptiste Department of Biology January 31, 2016 Certified by: _____________________________________________________________ Phillip A. Sharp Institute Professor of Biology Thesis Supervisor
Accepted by: ____________________________________________________________ Michael T. Hemann Associate Professor of Biology Co-Chair, Biology Graduate Committee
Dicer loss induces an oncogenic epigenetic switch in mesenchymal stem cells
by
Courtney K. JnBaptiste
Submitted to the Department of Biology on January 31, 2016 in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Biology
Abstract
MicroRNAs (miRNAs) are post-transcriptional regulators that tune gene expression. Despite the modest 2-fold repression that miRNA activity generally confers on a target, miRNAs are critical for many biological processes including development and differentiation. Due to this mild repression directly conferred by miRNA activity, miRNAs coordinate with other regulators such as transcription factors to shape the gene expression landscape and phenotypes of a cell.
To understand the function of global miRNA activity in regulating the specification of the somatic state, we deleted Dicer in a murine mesenchymal stem cell model. Upon exploring the consequences of Dicer deletion, we identify a specific let-7 regulated mid-embryonic program within the global de-repression of miRNA targets accompanying Dicer loss. We further observe within the landscape of let-7 regulated targets, an activation of greater than 50-fold of known oncofetal (Igf2bp1/2/3) genes, an effect much greater than that typically reported for miRNA-mRNA interactions. This suggests a requirement of let-7 for the continual suppression of mid-embryonic programs in adult cells.
To investigate the regulation of these oncofetal genes, we restored miRNAs through re-expression of Dicer. Despite complete reconstitution of the post- transcriptional activity of miRNAs, the activated oncofetal genes are incompletely suppressed. Igf2bp1-3 are components of a larger set of irreversible oncogenes whose chromatin signature indicate that they are transcriptionally activated upon Dicer deletion. This transcriptional activation is maintained, despite miRNA restoration in Dicer rescued cells. Consistent with this expression pattern, Dicer rescued cells are able to form tumors in mice, a phenotype absent in the parental wild-type and Dicer knockout cells. Moreover, the irreversible gene set is amplified in human cancers and is predictive of patient survival indicating that our observations are relevant to human disease.
Finally, we develop a computational method to decipher the indirect, transcription factor mediated effects of miRNAs on gene expression. Through comprehensive analysis of ChIP-Seq, CLIP-Seq and RNA-Seq datasets, we quantitatively assess the relative contributions of direct posttranscriptional miRNA activity and transcriptional activity on gene expression changes resulting from Dicer deletion. We find that transcriptional changes contribute significantly to perturbations in gene expression resulting from global miRNA loss upon Dicer deletion.
In summary, our work expands the current knowledge of fundamental roles for miRNAs in differentiated mammalian cells. As further work builds on our observations, the increased understanding of miRNA-mediated regulation will inform therapeutic strategies for human disease.
Acknowledgements
Support from countless individuals over the years has made the work described herein possible. To Dr. Davis, I appreciate you for your commitment to give back to underserved communities. If you had not come to teach at BCU, I wouldn’t be at MIT
today. A million thank-yous for your contagious enthusiasm for science, for seeing potential in me, for supporting my educational pursuits, and for being a mentor and
friend.
To Dr. Sassanfar, affectionately known as Mandana, you took a chance on a student you didn’t know, from a college you didn’t know, when you accepted me into the summer program. Thank you for that vote of confidence and for overriding my preferred lab, to
place me in the Sharp lab. You changed my life!
To Phil, when I was a teenager, feeding my passion for science through the Discovery Channel, I would have never dared to dream that I would get the chance to be at the
forefront of science. I cannot thank you enough! Thank you for accepting me as an undergraduate with minimal research experience, thank you for your enthusiasm to have
me join your lab as a grad student, thank you for the confidence you placed in me by allowing so much intellectual freedom, thank you for the gentle but timely nudges to keep me on the right track, thank you for the culture of open access, thank you for supporting my non-scientific pursuits, and thank you for this amazing experience. To my committee members Jackie Lees and Dave Bartel, you have been with me from the beginning of my journey as a grad student in the Sharp lab. I appreciate your interest in my work and development as a scientist, your thoughtful advice, your frank criticism,
and your encouragement to take charge/ ownership of my work.
To my fellow Sharpies, you are simply amazing! Thank you for the laughter over the years, the celebrations of success, and the fellowship over commiserations on failed experiments. Tim, Jesse and Kevin, you are the core RNAi department of the lab and I
thank you for the huge resource that you have been. Kevin, my favorite (and only) baymate, I especially thank you for teaching me to have a little fun while doing science.
Sara, thanks for always being excited about my progress and for being a great labmate. Mohini and Paul, you were my first mentors in the Sharp lab. Thank you for that initial mentoring experience that got me hooked, and I appreciate you even more now that I am
your colleague. You both have been a constant support! To Allan Gurtan, I was incredibly fortunate to collaborate with you. Thank you for always being confident in my
ability, for your voracious appetite for scientific discussion, for teaching me the experimental skills that I have acquired, and for your remarkable scientific insights. To
Victoria, I thank you for being an amazing undergrad way beyond your peers, and for being someone I could bounce prelim proposal ideas off of.
To my inner circle: Lynne, Jasmine, Tony (STC) and Brian, thank you guyz for being true friends and always planning something to escape the lab. Whitney, I have been incredibly blessed to have you in Boston. Thanks for the coffee dates and for always
cooking me for me. Aunty Francess, I could always escape to your home in NY when I needed to get some St. Lucian food in my stomach. Thanks for being available at all times. Uncle Richard, you were always eager to support my educational pursuits and
made the start of this journey possible. I appreciate you.
I have been incredibly blessed with a God-given family from my time in Florida: Pastors Mark and Michaell, Luke, Logan, Bekki, Jojo, Emmanuel, Vladimir, Kasongo and Brite.
You are a constant source of pride, love and support, thank you!
To my beloved family, I owe everything. You watched me leave home almost 10 years ago, with 2 suitcases embarking on an unknown educational journey. It is thoughts of you that kept me pressing through the difficulties over the years. Mommy, you taught me how to be a diligent student even from my toddler years and your life has taught me to work hard and persevere. Daddy, thanks for being a constant amidst the oscillations of life, and
teaching me how to calmly handle anything thrown at me. Nio, you have set the greatest example of being the eldest sibling, you are my go to person and friend. I thank you. To all my siblings and your spouses, my nieces and nephews, life would not be the same
without you and I am glad that I have you. I love you all dearly.
To my amazing wife, I love you and I am so glad and blessed to have you here with me! You’re always on 100, even when I’m at 0. Thanks for opening up my eyes to the beauty
of the simple things in life, for teaching me to be excited about things, and for being so confident in me, when I need it most. My life is all the more exciting because of you, and
even more so, now that you carry our package of ‘Joy’. To my unborn, one day we will sit together, dust this thesis off the shelf, and talk about all the cool science daddy got to
do. I excitedly await your arrival.
Most importantly, to God who knew and chose me while I was yet in my mother’s womb, I am eternally grateful. Thanks for equipping me with the intellect, skills and gifting to
Table of Contents
Abstract………2
Acknowledgements………..3
Table of Contents.………6
Chapter 1. Introduction………...8
miRNAs: discovery …………..……….9 miRNAs: biogenesis ……….……….………..11miRNAs: mechanisms of action ...14
miRNAs: in gene expression networks...18
miRNAs: in cell state………...22
miRNAs: in cancer……….…..25
Summary & Outline……….…... 34
References………...36
Chapter 2. Let-7 represses Nr6a1 and a mid-gestation developmental
program in adult fibroblasts……….54
Abstract ………...55
Introduction………..55
Results………..59
Discussion………81
Materials & Methods………...86
References………....88
Supplemental Figures..……….94
Supplemental Materials & Methods...………...102
Chapter 3. An epigenetic switch of oncogenes induced by Dicer loss in
mesenchymal stem cells..….……….110
Abstract ……….……….111
Introduction……….………111
Results……….………114
Discussion………...133
Materials & Methods………...137
References………..…….141
Chapter 4. Elucidating microRNA regulatory networks using
transcriptional, post-transcriptional and histone modification
measurements.
…….……….155
Abstract………...156
Introduction………..……….………..156
Results……….………159
Discussion………...173
Materials & Methods………...175
References………...177
Supplemental Figures………..182
Supplementary Materials & Methods………...188
Chapter 5. Conclusions and Future Directions………..195
Conclusions …………..………..196
Future Directions ……….……….………..199
References………...203
Chapter 1
Introduction
Chapter1. Introduction
Cells of multicellular organisms are genetically identical but yet can assume a myriad of functionally distinct cell types. This phenomenon is due to the regulated control of differential gene expression that ultimately defines the catalogue of proteins produced in each cell. Many steps along the path from DNA to RNA to protein allow for the precise tuning of gene expression that is fundamental to a cell’s identity and functional capacity. In this thesis, we particularly focus on microRNA (miRNA) mediated regulation of the gene expression pathway in mammalian systems. In the introduction to follow, we explore miRNA discovery, biogenesis and function, as well as roles in cell identity and cancer.
miRNAs: discovery
One of the most seminal discoveries of recent decades is that RNA is more than just a message to encode protein, but that the majority of the transcribed genome is non-coding RNA (ncRNA). Of this class, miRNAs have risen to prominence for their increasingly pervasive expression across complex organisms and regulation of fundamental physiological processes. It all started with the report of a surprising discovery in 1993. The Ambros group was studying the genetic interaction of lin-4 and lin-14, genes previously known to be important for temporal control of development in C. elegans larvae. In an effort to further elucidate lin-4’s mechanism of action, they cloned the lin-4 gene and surprisingly found that it was not a classical protein coding gene; rather, lin-4 produced a pair of small RNAs (Lee RC, 1993). They found that both RNA products had short blocks of sequence that was repeated multiple times (in an antisense fashion) in the lin-14 3’UTR. About the same time, the Ruvkun group also noticing this antisense complementarity, showed that the lin-4 gene product was required for
post-Chapter1. Introduction
transcriptional down-regulation of lin-14 (Wightman et al., 1993). Together, these studies suggested a model in which antisense RNA-RNA interactions effected repression of a gene product that regulated developmental transitions in worms.
However, there was no evidence to suggest that the lin-4/lin-14 observation was more than an anomaly of worm development for almost another decade. Then, the Ruvkun lab characterized let-7, another gene important for signaling a developmental transition in C. elegans and found it to be a 21nt, untranslated RNA as well (Reinhart et al., 2000). This let-7 RNA promoted the transition from late larval to adult stages through 3’UTR dependent repression of lin-41 and lin-28. Together, these early studies prompted the possibility that the RNA-RNA mediated post-transcriptional repression could be a more widespread mechanism to regulate worm development.
Indeed, shortly thereafter this was confirmed to be the case when Pasquinelli and colleagues reported the conservation of the sequence, as well as temporal expression of the let-7 RNA (Pasquinelli et al., 2000). This marked the beginning of a new world of gene regulation through these miRNAs. Targeted biochemical and computational approaches to identify other members of this class revealed their widespread expression and conservation, confirming that this means of miRNA mediated post-transcriptional regulation was more widespread than previously appreciated (Grad et al., 2003; Lagos-Quintana et al., 2001; Lagos-Lagos-Quintana et al., 2002; Lau et al., 2001; Lee and Ambros, 2001; Reinhart et al., 2002). Today we know that miRNAs are found in viruses, plants, worms, flies, fish, and mammals (Griffiths-Jones et al., 2008), and that their activity is crucial to fundamental biological processes.
Chapter1. Introduction
miRNAs: biogenesis
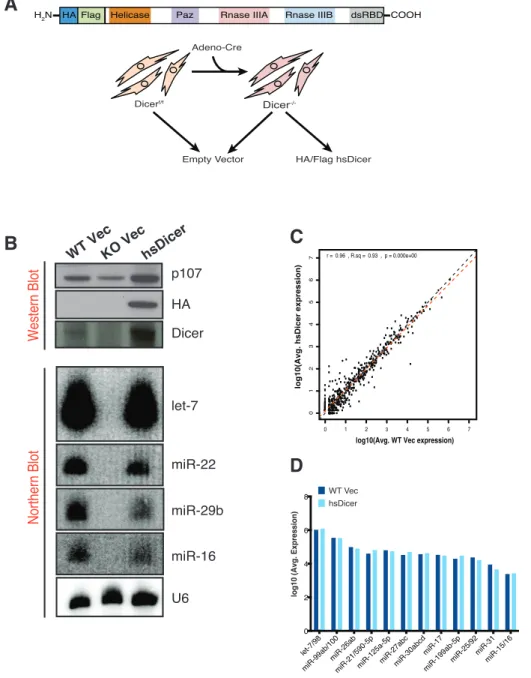
miRNAs are initially transcribed into primary transcripts (pri-miRNA) predominantly by RNA Polymerase II (Pol II), although Polymerase III is thought to transcribe some miRNA genes (Borchert et al., 2006; Lee et al., 2004). Pri-miRNAs can be multi-cistronic (generating many individual miRNAs), come from independent units from introns and exons of protein coding genes, or from intergenic regions (Cai et al., 2004; Rodriguez et al., 2004). Pri-miRNAs generated by Pol II are capped and polyadenylated similar to protein coding genes. The pri-miRNA transcripts folds upon itself to form a hairpin structure with a partially paired stem region and a loop region. From the pri-miRNA, the nuclear microprocessor complex (containing the RNaseIII enzyme Drosha and the double stranded RNA binding protein DGCR8) generates an ~70nt pre-miRNA hairpin (Denli et al., 2004; Gregory et al., 2004; Han et al., 2004; Lee et al., 2003). This pre-miRNA contains a 5’ phosphate and a 3’ 2nt overhang, a characteristic feature of cleavage by an RNaseIII protein. The newly liberated pre-miRNA is then actively exported from the nucleus by Exportin-5 and RanGTP (Figure 1) (Bohnsack et al., 2004; Lund et al., 2004; Yi et al., 2003).
In the cytoplasm, another RNase III endonuclease Dicer, cleaves the pre-miRNA to generate an ~22nt duplex (Bernstein et al., 2001; Grishok et al., 2001; Hutvagner et al., 2001; Ketting et al., 2001). This duplex also bears the 3’ 2nt overhang characteristic feature of RNase III processing. Tar RNA Binding Protein (TRBP) and Protein Activator of PKR (PACT), may associate with Dicer to enhance pre-miRNA cleavage, but are not essential (Chendrimada et al., 2005; Lee et al., 2006). The ~22nt duplex generated post
Chapter1. Introduction
Dicer processing consists of a miRNA guide strand that is essential for mRNA targeting and a miRNA star strand (miRNA*) that is eventually degraded.
Figure 1. Canonical miRNA biogenesis and modes of repression. After transcription by RNA Polymerase II, Drosha/DGCR8 processes the primary transcript into the pre-miRNA. Exportin5 mediates the active export of the pre-miRNA to the cytoplasm where it is recognized by Dicer/TRBP and cleaved into the 22nt duplex. The guide strand is loaded into the Argonaute to form active RISC. Based on sequence complementarity, the miRNA guides RISC to the target mRNA and mediates translational repression or mRNA degradation. Figure adapted from Gurtan, 2013 (Gurtan and Sharp, 2013).
Chapter1. Introduction
Importantly, the mature guide miRNA can be derived from either the 5’arm (5p) or 3’ arm (3p) of the pre-miRNA. Strand selection is largely governed by thermodynamic instability: the strand that is least stable at the 5’end is selected to be the guide (Khvorova et al., 2003; Schwarz et al., 2003). More recent work has revealed that strand selection is more nuanced and involves superimposition of independent rules involving 5’ nucleotide identity and thermodynamic stability (Suzuki et al., 2015). Ultimately, the miRNA guide strand is incorporated into an Argonaute protein (Ago) to form the RNA induced silencing complex (RISC) that effects post-transcriptional repression (Figure 1) (Hutvagner and Zamore, 2002; Liu et al., 2004; Meister et al., 2004).
There is an exception to every rule, and so it is the case with miRNA biogenesis. There are increasing examples of miRNAs that do not follow the canonical processing pathway outlined previously. A class of non-conventional miRNAs termed mirtrons is independent of the nuclear microprocessor (Berezikov et al., 2007; Glazov et al., 2008; Okamura et al., 2007; Ruby et al., 2007). These miRNAs are able to bypass Drosha cleavage because the precursor RNA is generated from mRNA splicing. After debranching, the lariat intermediate in the splicing reaction produces a stem loop structure that resembles a pre-miRNA and can enter the canonical miRNA biogenesis pathway as a Dicer substrate. Consistent with mirtrons bypassing Drosha cleavage, cloning of small RNAs from DGCR8 and Drosha knockout cells revealed no impact on mirtron expression despite complete loss of canonical miRNAs (Babiarz et al., 2008; Chong et al., 2010).
Another class of non-canonical miRNA is the Dicer independent group (Cheloufi et al., 2010; Cifuentes et al., 2010; Yang et al., 2010). To date, only miR-451 constitutes
Chapter1. Introduction
this class. This miRNA is initially processed by Drosha/DGCR8 to generate the pre-miRNA that bypasses Dicer and is loaded directly into Ago2. Ago2 cleaves the 3’ end to generate a 30nt intermediate species, that is trimmed by an unknown nuclease to generate the ~23nt mature miRNA. More recently, a class of transcription start site (TSS) derived miRNAs has been reported (Zamudio et al., 2014). These miRNAs are generated from hairpins that are independent of Drosha and Dgcr8 for processing, and exhibit differential expression across various tissues, thereby indicating that TSS-miRNAs are dynamically regulated. It is worth mentioning that beyond these categories, studies have also documented snoRNA-derived miRNAs (Scott et al., 2009; Taft et al., 2009), and tRNA- derived miRNAs (Cole et al., 2009). Further investigations will determine how pervasive these classes are and the extent of endogenous gene regulation that they confer.
miRNAs: mechanisms of action
Regardless of the pathway that generates miRNAs, the ultimate destination for the mature guide miRNA is the Ago-containing RISC, which effects post-transcriptional repression. Of the thousands of mRNAs expressed in a cell at any single point in time, how does the RISC know which mRNA to target? The specificity for targeting lies in the sequence of the miRNA. At the time of initial miRNA discovery, core elements of complementarity between the target 3’UTR and the 5’ region of the miRNA were observed (Wightman et al., 1993). Soon afterwards, it was experimentally confirmed that principally, pairing between this core seed region of the miRNA (positions 2-7) and complementary sequences in the 3’UTR of the mRNA was crucial for specificity and regulation (Brennecke et al., 2005; Doench and Sharp, 2004; Kloosterman et al., 2004; Lai et al., 2005). Structural studies have shed light on this importance of the seed: upon
Chapter1. Introduction
binding the miRNA, Ago presents the guide in the geometry of an A form helix that preferentially exposes positions 2-6 for nucleating the pairing to the target mRNA (Parker et al., 2005; Wang et al., 2008d). Consistent with their importance for gene regulation, the seed is the most conserved region of miRNAs (Lewis et al., 2003; Lim et al., 2003). Furthermore, target-prediction methods that require perfect pairing to the seed can detect authentic targets above the background of false positives (Lewis et al., 2005; Lewis et al., 2003). Thus, miRNAs can be categorized into families defined by the presence of identical seed sequences and miRNAs of the same family are expected to regulate an overlapping set of genes.
Seed based pairing is the main mode of target recognition, but alternative modes for specifying a target mRNA have been described. Watson-Crick base pairing across positions 13-16 of the miRNA may supplement seed matched sites, and in such cases, are more active, albeit slightly, than seed sites alone (Grimson et al., 2007). In fact, 3’ end pairing may compensate for a mismatch in the seed region. Additionally, continuous pairing centered around positions 4-15 of the miRNA are also effective (Shin et al., 2010). More recently, multiple studies have reported other non-canonical modes of target recognition (Chi et al., 2012; Grosswendt et al., 2014; Helwak et al., 2013; Khorshid et al., 2013; Loeb et al., 2012), suggesting that these types of targeting may be more pervasive than initially thought. However, these non-canonical sites are of relatively lower frequency and it remains controversial whether these sites are truly functional for mediating repression on endogenous targets (Agarwal et al., 2015).
Upon recognizing the target, the RISC can effect endonucleolytic cleavage of the mRNA in rare cases of extensive complementarity to the miRNA (Yekta et al., 2004).
Chapter1. Introduction
More commonly, the modes of RISC-mediated repression are mRNA destabilization and/or translational repression. As there is substantial evidence for both modes of activity, it is still contested whether mRNA destabilization is predominant over translational repression and vice versa.
It was initially reported that 4 caused pronounced repression on 14 and lin-28 exclusively through translational repression, as there was no evidence of change in the levels of the corresponding mRNAs (Lee RC, 1993; Wightman et al., 1993). Later observations that the polysome profile of lin-14 was unchanged from early larval (high lin-14 protein) to late larval stages (low lin-14 protein) suggested that translational repression specifically occurred after initiation of translation (Olsen and Ambros, 1999). This model was consistent with other miRNA target interactions in worms (Seggerson et al., 2002) and later, studies on artificial and endogenous miRNA targets in mammalian cells (Maroney et al., 2006; Nottrott et al., 2006; Petersen et al., 2006). However, other studies suggested that translational repression occurs at the initiation step. Within a short period of time, two groups demonstrated that mRNAs containing an IRES in lieu of the conventional 5’ cap are resistant to miRNA mediated repression, suggesting that miRNA repression requires canonical initiation machinery and hence, interferes with initiation. (Humphreys et al., 2005; Pillai et al., 2005). Furthermore, another report showed that miRNAs inhibited 80S initiation complex formation on an mRNA in cell free extracts (Mathonnet et al., 2007), indicating that the initiation step was repressed by miRNAs. Hence, the precise timing of translational repression is controversial, and it remains possible that miRNAs mediate translational repression both at the point of initiation and afterwards, in a context dependent manner.
Chapter1. Introduction
There is also substantial, if not more, evidence for miRNA induced mRNA destabilization. Interestingly, a study on lin-14 (as well as lin-28 and lin-41), reported their decreased mRNA levels in response to miRNAs (Bagga et al., 2005). This suggested that transcript destabilization was probably a more common mechanism of miRNA activity than previously anticipated. Additionally, in a genome wide study characterizing mRNA expression after transfection of miRNAs, Lim et al. showed that transcripts with seed matches to the transfected miRNA showed reduced levels (Lim et al., 2005). Likewise, there was a pronounced increase in transcript levels for mRNAs with seed matches to experimentally silenced miRNAs (Esau et al., 2006; Krutzfeldt et al., 2005). Together, these studies indicated that miRNAs have broad effects on mRNA stability.
The mechanism underlying target mRNA destabilization is association of Ago (RISC) with GW182. This interaction is crucial to the recruitment of additional factors that induce mRNA degradation. GW182 disrupts the interaction between PABPC (poly(A) binding protein cytoplasmic) and components of the translation machinery (specifically eIF4G) (Moretti et al., 2012), and recruits the deadenylase CAF1-CCR4-NOT complex, ultimately triggering decapping and degradation of the mRNA by the XRN1 exonuclease (Behm-Ansmant et al., 2006; Giraldez et al., 2006; Rehwinkel et al., 2005; Wu et al., 2006; Zekri et al., 2009).
As there is evidence for both modes of miRNA repression, multiple studies have attempted to quantify the exact contribution of translational repression versus target degradation in a genome wide manner. Studies measuring mRNA and protein changes in parallel conclusively show that the effects of miRNAs on protein output are modest and that these changes are ultimately mirrored at the transcript level (Baek et al., 2008;
Chapter1. Introduction
Selbach et al., 2008). Subsequent studies further clarified that in mammalian systems, protein level changes are mainly attributable to mRNA changes (Guo et al., 2010; Hendrickson et al., 2009). The central theme emerging from these and more recent studies is that mRNA degradation is the dominant effect of miRNAs at steady state. (Bazzini et al., 2012; Bethune et al., 2012; Eichhorn et al., 2014; Fabian et al., 2009; Meijer et al., 2013).
miRNAs: in gene expression networks
Since the initial discovery of roles for miRNAs in development, miRNAs have been shown to influence virtually every biological process in some way. These processes include, but are not limited to, cell cycle control (Ivanovska et al., 2008; Wang et al., 2008b), differentiation (Kanellopoulou et al., 2005; Xu et al., 2009a), and apoptosis (Cimmino et al., 2005; Harfe BD, 2005). However, the change of a target in response to a miRNA is typically modest, rarely exceeding 2 fold (Baek et al., 2008). Despite this modest effect, there is a marked selective pressure to conserve miRNA-target interactions (Friedman et al., 2009) indicating that this layer of regulation is of paramount importance. The key to reconciling the apparent contradictory observations of modest effects and universal importance lies in understanding how miRNAs are integrated into gene expression networks. In the following discussion, we detail a few examples that highlight key principles of how the subtle regulation by miRNAs is phenotypically crucial.
miRNAs may amplify the modest effects of single mRNA interactions by disproportionately targeting multiple genes in the same or related pathway. For example,
Chapter1. Introduction
miRNAs of the miR-15/16 seed family negatively regulate the cell cycle by causing an accumulation in G0/G1 (Linsley et al., 2007). In this study, the authors documented the surprising observation that treatment with miR-16 conferred a stronger effect than specific silencing of a miR-16 target via the corresponding siRNA. In fact, the miR-16 effect was best phenocopied by treatment of a pool of siRNAs targeting multiple miR-16 regulated targets (which were also computationally predicted miR-16 targets). Hence, this study demonstrated that for halting of cell cycle progression, simultaneous targeting of genes was more effective than individual gene disruption. Thus the miR-15/16 miRNA family elicits a stronger response on the cell cycle by coordinately regulating targets that act in concert to promote cell cycle progression from G0/G1 to S phase. A similar phenomenon occurs with the miR-17/92 cluster of miRNAs that blunts TGFβ signaling by coordinately repressing several components of the signaling pathway (Dews et al., 2010). Computational approaches for predicting and annotating miRNA targets also indicate that some miRNAs regulate specific biological functions by targeting functionally related genes (Gaidatzis et al., 2007; Grun et al., 2005; Tsang et al., 2010). Hence, coordinate repression of multiple genes in a related pathway is a widespread strategy employed for enhancing the impact of miRNA-mediated regulation.
It has also been recognized that miRNAs are embedded in circuit motifs alongside other regulators of gene expression such as transcription factors (TFs). In fact, miRNAs have the propensity to target transcription factors (Enright et al., 2004; Martinez et al., 2008). For example, miR-134 mediated repression of Nanog and LRH1 is sufficient to promote differentiation of murine embryonic stem cells to ectodermal lineages (Tay et al., 2008). By regulating master transcription factors that dictate the gene expression
Chapter1. Introduction
responsible for a cell state or phenotype, miRNAs centralize their activity at the ‘control center’ and thus confer an amplified effect for a certain phenotype.
Furthermore, miRNAs and TFs may form feed-forward loops (FFL) where a target is regulated by both the miRNA and the TF (Figure 2) (Tsang et al., 2007). The miRNA itself is transcriptionally regulated by the TF. In a subtype of this motif termed a coherent FFL, the transcription factor coordinately represses (or activates) a target both directly and also indirectly through a regulated miRNA. Through this motif, miRNAs repress leaky transcription and reinforce the gene expression dictated by the TF. Because there are two components redundantly acting on the downstream gene, this network structure ensures fidelity of repression in case of a temporary reduction of either the TF or miRNA. A recent study has implicated this type of network in causing an epigenetic switch that leads to cellular transformation in cancer (Iliopoulos et al., 2009). A transient inflammatory signal triggers NF-κB mediated activation of Il6 that ultimately signals through STAT3 to promote cell growth. Simultaneously, NF-κB causes loss of let-7 mediated posttranscriptional repression of IL6 by activating Lin28 to inhibit let-7 (Figure 2). Very high levels of Il6 is required to induce transformation, and hence, both arms of the FFL is essential for this effect (Iliopoulos et al., 2009). By imposing repression of IL6, let-7 effectively minimizes the impact of noise in NF-κB levels and prevents unwanted activation of a network that would otherwise promote cellular transformation and cancer.
Chapter1. Introduction
Figure 2. Network motifs involving miRNAs. Upper panel: to the left is a generalized depiction of a coherent feed-forward motif. To the right is the epigenetic switch outlined in the text. let-7 dampens fluctuations in IL6 levels induced by NF-κB. Only sustained activation of NF-κB is sufficient to de-repress let-7 activity and permit high enough levels of IL6 to cause transformation (Iliopoulos et al., 2009). Lower panel: to the left is a depiction of an incoherent feed-forward motif. To the right is an incoherent motif wherein c-Myc directly activates E2F but also activates miR-17, which negatively regulates E2F (O'Donnell et al., 2005). In both motifs, the direct arm of regulation on the target gene is indicated in orange, while the indirect arm is indicated in purple.
The second type of organization of FFLs is the incoherent FFL, in which the direct effect of the TF on a target is opposed by a miRNA induced by the same transcription factor (Figure 2). This network structure is also important for buffering noise in biological systems. For example, computational studies predict that incoherent FFLs confer stability in expression of a target gene by buffering fluctuations in the upstream TF (Osella et al., 2011). This network structure is an important mechanism for
Chapter1. Introduction
cell cycle control involving c-Myc and the miR-17 cluster. While c-Myc transcriptionally activates E2F (which itself is a master regulator of the cell cycle), it also induces miR-17, which posttranscriptionally represses E2F (Figure 2) (O'Donnell et al., 2005). This allows for the precise, temporal regulation of E2F expression. In this way, miR-17 functions to maintain E2F expression at a threshold that promotes proliferation but does not trigger apoptosis (which occurs when E2F levels are too high). A similar motif is important for the maintenance of pluripotency and self-renewal of embryonic stem cells (Marson et al., 2008). The core embryonic stem cell transcription factor Oct4 activates let-7 pri-miRNA but also concurrently activates lin-28, which subsequently inhibits maturation of let-7 (Marson et al., 2008). In this system, lin-28 functions to buffer let-7 in response to Oct4 fluctuations. By inhibiting mature let-7 expression, this mechanism confers developmental robustness in response to non-uniform expression of Oct4 and prevents premature differentiation.
Thus, despite the mild effects of single miRNA:mRNA interactions, miRNAs reinforce transcriptional programs, confer robustness in the context of environmental perturbations and, dampen fluctuations that are not intended to signal a specific outcome. In total, studies demonstrate that the importance of regulation by miRNAs is not just a mere function of the genes being repressed, but also, that of the underlying network structure in which the miRNAs are embedded.
miRNAs: in cell state
Since the initial discovery of miRNAs, sequencing technology has advanced tremendously and there has been a concomitant increase in the number of annotated
Chapter1. Introduction
mammalian miRNAs. Today, there are over 500 annotated miRNA species in mouse (Chiang et al., 2010). For complex organisms, having such a diverse repertoire of miRNAs increases the power to generate unique combinations of regulatory networks to confer cell type specificity. Indeed, similar to protein-coding genes, miRNAs exhibit a variety of expression patterns: some miRNAs are restricted to specific cell types while others are more ubiquitously expressed. This has given rise to the hypothesis that miRNA expression may specify cell state, a concept that is supported by the observation that miRNA expression can be used to classify human cancers and various tissues, sometimes even more specifically than protein-coding mRNAs (Lu et al., 2005a).
A classic example of a role for miRNAs in specifying and reinforcing cell fate is the developmental switch governed by lin-4 and let-7 in worm development. Before characterization of these gene products as miRNAs, it was known that worms mutant for lin-4 exhibited retarded development: lin-4 mutants reiterated early cell fates at later developmental stages, lacked adult structures and were incapable of egg laying (Lee RC, 1993). Similarly, let-7 loss-of-function mutant worms reiterated larval patterns of cell divisions and failed to develop adult structures (Reinhart et al., 2000). Conversely, overexpression of let-7 triggered precocious differentiation into an adult state. In retrospect, one can more fully appreciate the importance of miRNA regulation underscored by these early studies: miRNAs can act as binary switches to turn off target protein output that ultimately enforces adoption of the differentiated (adult) cell state.
Beyond nematodes, studies also show that miRNAs play crucial roles in early development of mammalian systems. miRNA profiling studies indicate that the total pool of miRNAs in murine embryonic stem cells (mESCs) is dominated by members of the
Chapter1. Introduction
miR-290-295 miRNA cluster (Houbaviy et al., 2003). Normal mESCs exhibit quicker proliferation relative to differentiated cells, as they proliferate in a mitogen independent manner due to a characteristic rapid G1 to S phase transition in the cell cycle (facilitated by constitutively active E2F). However, both DGCR8 and Dicer null mESCs have a pronounced decrease in their proliferative rate due to an accumulation in the G1 phase of the cell cycle (Kanellopoulou et al., 2005; Murchison et al., 2005). In a screen to identify suppressors of this defect, the miR-295 family (among other seed related families) was able to completely rescue this proliferative lag by targeting the cell cycle regulator, p21 (Wang et al., 2008c). Hence, the miR-290-295 cluster facilitates the rapid proliferation that is characteristic of mESCs.
Besides impaired proliferation, miRNA-deficient mESCs are unable to differentiate due to an inability to silence drivers of pluripotency (Kanellopoulou et al., 2005). Absence of the miR-295 family of miRNAs causes an upregulation of Rbl2, a transcriptional repressor of the de novo methyltransferases (Dnmt3a and Dnmt3b), which in turn causes an inability to stably silence the core pluripotency program to facilitate differentiation (Benetti et al., 2008; Sinkkonen et al., 2008). Furthermore, the miR-295 family is embedded in the core pluripotency architecture of mESCs. Oct4, Sox2, Nanog and Tcf3 constitute the drivers of pluripotency in mESCs and they also transcriptionally activate the miR-295 family (Marson et al., 2008). Consistent with such a core role in embryonic stem cell identity, overexpression of miR-295 family members markedly enhances the efficiency of reprogramming fibroblasts to induced pluripotent stem cells (Judson et al., 2009). Similarly, in human embryonic stem cells, miR-145 targets the core pluripotency circuitry involving Oct4, Klf4 and Sox2 to induce differentiation (Xu et
Chapter1. Introduction
al., 2009b). Together, these results show that miRNA-mediated regulation is crucial for maintaining the ability for embryonic stem cells to differentiate.
Other studies further support a role for miRNAs in reinforcing differentiated, somatic cellular states. miR-1’s expression is preferentially confined to the adult heart where it accounts for ~45% of the total miRNA pool in that tissue (Lagos-Quintana et al., 2002). Overepression of miR-1 in Hela cells caused global changes such that the gene expression of Hela cells shifted towards that of heart and skeletal tissue (Lim et al., 2005). This observation is not an anomaly, as a similar experimental approach for the brain specific miR-124, also shifted Hela gene expression toward that of the brain (Lim et al., 2005; Sempere et al., 2004). Notably, genes that are generally more lowly expressed in the brain compared to other tissues, were down-regulated by miR-124 transfection. This latter observation suggests that tissue-specific miRNAs may be actively responsible for the low expression levels of some transcripts in that tissue. Finally, switch-like roles for miRNAs, analogous to that of lin-4 and let-7 in worm differentiation, have been documented in mammalian systems of skin and smooth muscle differentiation (Cordes et al., 2009; Yi et al., 2008). Together, these studies lend support to the concept that transitions from one cell state to another could be driven by miRNAs, and that miRNAs function in defining and maintaining cell type specificities.
miRNAs: in cancer
Back in the early 90s when miRNAs were first reported as regulators of developmental timing in worms, few would have predicted the pervasiveness of this mechanism of controlling gene expression. But as studies began to document miRNA
Chapter1. Introduction
expression in diverse organisms, as well as the importance of the conferred repression on a variety of physiological processes, we began to appreciate the far-reaching implications for miRNA activity. Many of the fundamental processes under miRNA control such as proliferation, differentiation, migration, and apoptosis, become deregulated in diseased states such as cancer. Furthermore, many studies support an emerging paradigm wherein reactivation of embryonic programs promotes tumorigenesis. Given the notable examples of essential roles for miRNAs in early development that we previously discussed, altogether these observation have fueled speculation that altered miRNA activity could be important for tumorigenesis. In the following discussion, we summarize a few studies that strongly support, as well as demonstrate causality, for miRNAs and components of the miRNA biogenesis pathway in cancer.
Initial work from the Croce lab strongly suggested possible contributions for miRNAs in cancer. miRNAs are frequently found at fragile sites in the genome; regions prone to amplification, deletion or translocation in cancer (Calin et al., 2002; Calin et al., 2004). Moreover, cancer is characterized by global alteration of the miRNA profile; as for all tumor types analyzed, the miRNA profile of the tumor differed significantly from that of corresponding normal tissue (Lu et al., 2005b; Volinia et al., 2006). Similar to protein-coding genes, miRNA expression in cancer can be perturbed by chromosomal rearrangements such as translocations, deletions, mutations, promoter methylation and transcriptional repression (Calin et al., 2002; Chang et al., 2008; Davalos et al., 2012; Lujambio et al., 2007). However, other alterations such as mutations in the miRNA biogenesis machinery (Melo et al., 2010; Melo et al., 2009), mutations in the mRNA target site (Chin et al., 2008), and use of alternative 3’UTR isoforms (Mayr and Bartel,
Chapter1. Introduction
2009; Mayr et al., 2007) have also been documented in cancer. Hence, the way a miRNA acts in cancer is context dependent. Depending on the nature of the alteration and function of the target gene, miRNAs can function as tumor suppressors or oncogenes.
Tumor suppressor miRNAs
Tumor suppressor miRNAs are those whose expression limit cell growth or promote apoptosis, and therefore, their loss enhances transformation. The miR-15/16 cluster is located at chromosome 13q14, a region deleted in 68% of patients with chronic lymphocytic leukemia (CLL), as well as in more than 50% of prostate cancers and mantle cell lymphomas (Calin, 2002). In many cases of CLL, the 13q14 deletion is the sole genetic aberration. Thus for an extended time, researchers had been trying to identify the tumor suppressor gene encoded in that region. It was surprising to find that miR-15/16 lies in the minimally deleted region of CLL, thus indicating a selective loss of this miRNA cluster (Calin, 2002). Other circumstantial evidence points to miR-15/16 as a tumor suppressor: some CLL patients carry germline mutations in the pre-miRNA sequence that causes reduced expression of the mature miRNA (Calin et al., 2005). miR-15/16 inhibits the anti-apoptotic protein Bcl-2, whose overexpression is characteristic of CLL (Cimmino et al., 2005), over-expression of miR-15/16 causes cell cycle arrest in non-small lung cancer cell lines (Bandi et al., 2009), and delivery of miR-15/16 in vivo causes regression of prostate cancer xenografts (Bonci et al., 2008).
Perhaps, the miRNA that best fits the role of tumor suppressor miRNA is the let-7 family. Let-7’s expression is temporally restricted so that the mature miRNA is absent in early development but is ubiquitously expressed in differentiated tissues (Lagos-Quintana
Chapter1. Introduction
et al., 2001; Pasquinelli et al., 2000; Reinhart et al., 2000). The let-7 primary transcript is present, but processing to the mature miRNA is hindered by Lin-28 (Thomson et al., 2006; Viswanathan et al., 2008). Hence, let-7 and Lin-28 are inversely correlated in expression across tissues. The let-7 family of miRNAs is globally down-regulated in various tumors (Johnson et al., 2005) in part due to genomic loss of regions encoding let-7 members (Calin et al., 2004). Further evidence for a tumor suppressor role for let-let-7 emerged when it was reported that let-7 is significantly down-regulated in lung cancer (Takamizawa et al., 2004). Additionally, patients with lower let-7 had a poorer prognosis and shortened post-operative survival (Takamizawa et al., 2004). Studies characterizing let-7 targets indicate that let-7 is a master regulator of the cell cycle and self-renewal (Johnson et al., 2007).
Some of the best-characterized targets of let-7 are potent oncogenes. For example, the architectural transcription factor Hmga2 is a target of let-7 that is activated in various tumors. With six conserved seed matches in its 3’UTR, Hmga2 is strongly regulated by let-7 in normal tissues. However, translocations as well as alternative polyadenylation events mediate escape of let-7 inhibition to promote tumorigenesis (Enright et al., 2004; Lee and Dutta, 2007; Mayr et al., 2007). Other oncogenic targets of let-7 are Myc, Ras and the RNA- binding proteins Igf2bp1-3. In some cases, disruption of their let-7 imposed regulation actively promotes cancer development (Boyerinas et al., 2008; Gurtan et al., 2013; Johnson et al., 2005; Kumar et al., 2008; Mayr and Bartel, 2009; Sampson et al., 2007).
Chapter1. Introduction
Let-7 oncofetal targets
A subset of let-7 targets are described as ‘oncofetal’ based on their expression profile: they are highly expressed in early development, off in most adult tissues and their expression is reactivated or amplified in various tumors (Boyerinas et al., 2008) (Figure 3). Thus far, known let-7 regulated oncofetal genes include Hmga2, Igf2bp1-3 and Lin-28, and they all have shared roles in regulating growth and proliferative pathways. This is highlighted in transgenic mouse studies of Hmga2: homozygous loss of Hmga2 causes a dwarf phenotype compared to wild-type mice (Zhou et al., 1995). Conversely, Hmga2 overexpression results in gigantism, and these mice develop various mesenchymal tumors as well as pituitary adenomas (Battista et al., 1999; Fedele et al., 2006; Zaidi et al., 2006). Hence, Hmga2’s causal role in tumorigenesis may be attributed to its regulation of growth and proliferation under normal circumstances. Consistent with this, in humans, polymorphisms in Hmga2 are associated with height variation, Hmga2 amplification is observed in various high-grade tumors, and its overexpression is associated with early metastasis (Hristov et al., 2009; Weedon et al., 2007; Wu et al., 2011).
Igf2bp1-3 constitute a highly conserved family of RNA-binding proteins that also share many of the cancer relevant properties of Hmga2. Mice deficient for Igf2bp1 are dwarfed compared to their wild-type littermates (Hansen et al., 2004), while Igf2bp1 overexpression causes anchorage independent growth in vitro or tumor formation in vivo (Mayr and Bartel, 2009; Tessier et al., 2004). As an RNA-binding protein, Igf2bp1 controls the localization, translation and turnover of target mRNAs such as Igf2, cMyc, K-Ras, and H19, all of which are important in tumorigenesis (Dai et al., 2011; Mongroo et al., 2011; Runge et al., 2000; Weidensdorfer et al., 2009).
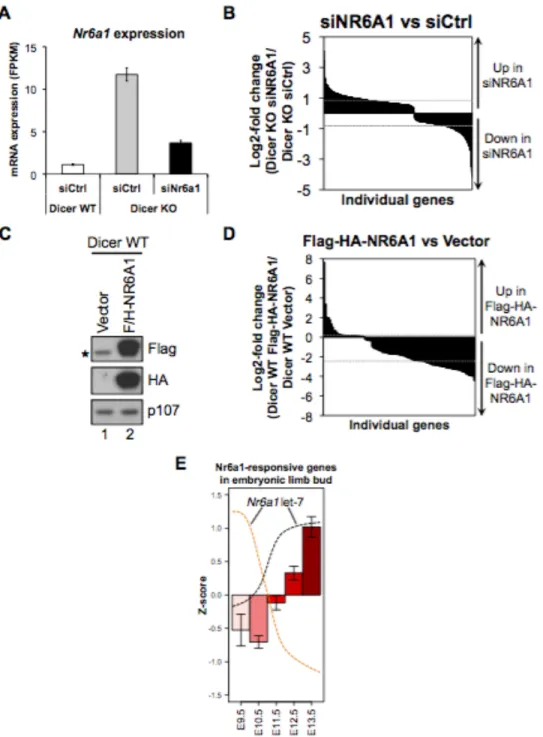
Chapter1. Introduction
Figure 3. Expression of let-7 and oncofetal targets. Schematic depicting anti-correlated expression of let-7 oncofetal targets and let-7 mature miRNA. Let-7 regulated oncofetal genes (Igf2bp1-3, Hmga2, Lin28a/b) peak in expression about E8.5 and their expression subsequently declines, ultimately to undetectable levels in most adult tissues. Contrastingly, mature let-7 becomes expressed about E10.5, its level steadily increases and remains high postnatally. Prior to E10.5, let-7 maturation is inhibited by activity of Lin28. In many cancers Lin28 is reactivated, resulting in suppression of let-7 and upregulation of other oncofetal targets.
Furthermore, high Igf2bp1 expression is associated with advanced ovarian cancer and poor patient survival (Gu et al., 2004). While an oncogenic role is most well defined for Igf2bp1, all members of this family have been implicated in cancer to various extents (Li et al., 2013; Schaeffer et al., 2010; Zhao et al., 2007).
Chapter1. Introduction
Another RNA-binding protein family comprising the paralogues Lin28a and Lin28b also exhibit the oncofetal expression pattern (Figure 3). In mice, overexpression of Lin28 causes increased organismal size (Zhu et al., 2010) and development of tumors in various contexts (Beachy et al., 2012; Nguyen et al., 2014; Tu et al., 2015; Urbach et al., 2014). Genome-wide association studies associate Lin28 polymorphisms with height variations in humans (Lettre et al., 2008). Additionally, Lin28 is activated in many human tumor types and such high Lin28 expression is significantly associated with a poor outcome for hepatocellular carcinoma patients (Viswanathan et al., 2009; Wang et al., 2010). Coordinated repression of multiple let-7 family members partly contributes to Lin28’s oncogenic properties (Chang et al., 2009; Viswanathan et al., 2009). Hence, there is mutual negative feedback between let-7 and Lin28, and disruption of the normal balance of this axis is an important step in cancer development (Molenaar et al., 2012).
The vast interconnection and feedback among the various oncofetal targets of let-7 is increasingly apparent. Igf2bp1-3 are strongly correlated in expression with each other as well as Hmga2 (Boyerinas et al., 2010; Boyerinas et al., 2008). In fact, Hmga2 transcriptionally activates Igf2bp2 (Brants et al., 2004; Cleynen et al., 2007). Moreover, Igf2bp3 granules function as safe houses to protect Hmga2 mRNA from let-7 mediated degradation (Jønson et al., 2014). Evidence also suggests that Igf2bp1 positively regulates Lin28 (Mongroo et al., 2011) and a recent study reported that in mice, Igf2bp3 is required for Lin28b mediated promotion of liver cancer (Nguyen et al., 2014). These observations lend strong support to the idea that cancers recapitulate embryonic features (such as rapid proliferation and de-differentiation) for their initiation and maintenance. Hence, a model (summarized in Figure 3) is emerging in which Lin28, Igf2bp1-3 and
Chapter1. Introduction
Hmga2 are highly expressed during early embryonic development where they function to promote self-renewal, proliferation and growth. As development progresses, differentiation necessitates repression of these oncofetal genes and their expression remains suppressed in most adult tissues. However, tumors reactivate oncofetal genes by co-opting a mechanism crucial for maintenance of self-renewal in embryonic development: re-activation of Lin-28 to globally suppress let-7 miRNAs. Given the feedback and co-regulation among these genes, let-7 suppression can then lead to upregulation of a network of some or all oncofetal genes that in turn promote cellular transformation.
Oncogenic miRNAs
miRNAs that are amplified in cancer and repress tumor suppressor genes are potential oncogenes (oncomiRs). The miR-17/92 cluster is located in the non-protein-coding gene C13orf25 and is frequently amplified in a variety of B-cell lymphomas and some solid tumors such as lung cancer (Hayashita et al., 2005; Ota et al., 2004). Expression of this miRNA cluster is driven by the oncogene Myc (O'Donnell et al., 2005). Mouse studies indicate that miR-17/92 cooperates with Myc activation or Rb loss to promote malignant lymphoma or retinoblastoma respectively (Conkrite et al., 2011; He et al., 2005). Conversely, deletion of this cluster retards Myc induced lymphomas (Mu et al., 2009). A recent study demonstrated that the tumor promoting activity of this miRNA cluster is heavily dependent on miR-19 (Han et al., 2015). Underlying the potent oncogenic activity of the miR-17/92 cluster is repression of tumor suppressor genes such
Chapter1. Introduction
as Pten, Bim, p21 and cyclin D1 (Mu et al., 2009; Olive et al., 2009; Petrocca et al., 2008; Ventura et al., 2008).
Another example of an oncomiR is miR-155, which has low expression in normal lymphoid tissues but becomes highly expressed in lymphomas (Eis et al., 2005; Kluiver et al., 2005), as well as solid tumors including breast, colon (Volinia et al., 2006), cervical (Wang et al., 2008a), and lung cancers (Yanaihara et al., 2006). Perhaps the strongest evidence for any miRNA functioning as an oncogene is for miR-155: transgenic mouse models overexpressing miR-155 in the bone marrow show myeloproliferation and pre-B cell proliferation that ultimately leads full to B-cell leukemia (Costinean et al., 2006; O'Connell et al., 2008).
miRNA biogenesis machinery in cancer
Beyond loss or gain of individual miRNAs, alterations in key proteins involved in miRNA biogenesis also contribute to tumor development by providing the global decrease in miRNA expression that enhances cancer (Calin and Croce, 2006; Kumar et al., 2007). Some tumors show inactivating mutations of XPO5, a protein essential to the nuclear export of miRNAs (Melo et al., 2010). This results in trapping of pre-miRNAs in the nucleus and a subsequent global reduction in mature pre-miRNAs. Furthermore, studies have documented cases of truncating mutations in TRBP2 that lead to destabilization of Dicer, and ultimately, impaired miRNA processing (Melo et al., 2009). Dicer and Drosha transcripts levels are significantly lower in ovarian tumors as compared to normal ovarian tissue (Merritt WM, 2008). Moreover, mouse models show that Dicer functions as a haploinsufficient tumor suppressor (Kumar et al., 2007; Kumar
Chapter1. Introduction
et al., 2009; Lambertz et al., 2010), and in humans, patients with germline mutations in Dicer develop pediatric pleuropulmonary blastoma and cystic nephroma, among other diseases (Bahubeshi et al., 2010; Hill DA, 2009.).
A more recent study reported recurrent mutations within the RNase IIIb domain of human Dicer. In a subset of non-epithelial ovarian tumors, residue D1709 was consistently mutated (Heravi-Moussavi A, 2012.). The significance of this mutation was highlighted by a recent study that reported the first in vivo characterization of various Dicer mutants (Gurtan et al., 2012). Mutation of this residue caused inactivation of Dicer’s RNase IIIb domain. This resulted in a miRNA profile completely deficient in 5p- derived miRNAs, including the ubiquitously expressed let-7 family of tumor suppressor miRNAs, while expression of 3p miRNAs was retained. In total, decreased mature miRNA expression through mutations in Dicer or other miRNA processing proteins is beneficial to tumor development.
Summary and outline
Approximately two decades of research since the initial discovery of miRNAs has tremendously advanced our understanding of the crucial roles that miRNAs play in the processes fundamental to life. A picture has emerged in which miRNAs buffer gene expression to distinguish true signal from noise, promote and reinforce cell fate decisions, and impinge on the various processes that, when deregulated, facilitate cancer development.
Despite this tremendous progress, the field of miRNA research is yet in its infancy as there many important questions that remain to be answered. This thesis
Chapter1. Introduction
represents a small step in that regard. In Chapter 2, we generate a model of Dicer loss to interrogate a role for somatic miRNAs such as let-7, in suppression of embryonic gene expression and the continued maintenance of the differentiated state. Next, we explore the feasibility of global miRNA restoration as a therapeutic modality to suppress oncogene expression networks in cancer. Finally, we develop a computational model for deciphering the indirect effects of miRNAs on gene expression through their control of transcription factor networks.
Chapter1. Introduction
References
Agarwal, V., Bell, G. W., Nam, J. W., and Bartel, D. P. (2015). Predicting effective microRNA target sites in mammalian mRNAs. Elife 4.
Babiarz, J. E., Ruby, J. G., Wang, Y. M., Bartel, D. P., and Blelloch, R. (2008). Mouse ES cells express endogenous shRNAs, siRNAs, and other Microprocessor-independent, Dicer-dependent small RNAs. Genes & Development 22, 2773-2785.
Baek, D., Villen, J., Shin, C., Camargo, F. D., Gygi, S. P., and Bartel, D. P. (2008). The impact of microRNAs on protein output. Nature 455, 64-71.
Bagga, S., Bracht, J., Hunter, S., Massirer, K., Holtz, J., Eachus, R., and Pasquinelli, A. E. (2005). Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell 122, 553-563.
Bahubeshi, A., Bal, N., Rio Frio, T., Hamel, N., Pouchet, C., Yilmaz, A., Bouron-Dal Soglio, D., Williams, G. M., Tischkowitz, M., Priest, J. R., and Foulkes, W. D. (2010). Germline DICER1 mutations and familial cystic nephroma. J Med Genet 47, 863-866. Bandi, N., Zbinden, S., Gugger, M., Arnold, M., Kocher, V., Hasan, L., Kappeler, A., Brunner, T., and Vassella, E. (2009). miR-15a and miR-16 are implicated in cell cycle regulation in a Rb-dependent manner and are frequently deleted or down-regulated in non-small cell lung cancer. Cancer Res 69, 5553-5559.
Battista, S., Fidanza, V., Fedele, M., Klein-Szanto, A. J., Outwater, E., Brunner, H., Santoro, M., Croce, C. M., and Fusco, A. (1999). The expression of a truncated HMGI-C gene induces gigantism associated with lipomatosis. Cancer Res 59, 4793-4797.
Bazzini, A. A., Lee, M. T., and Giraldez, A. J. (2012). Ribosome Profiling Shows That miR-430 Reduces Translation Before Causing mRNA Decay in Zebrafish. Science 336, 233-237.
Beachy, S. H., Onozawa, M., Chung, Y. J., Slape, C., Bilke, S., Francis, P., Pineda, M., Walker, R. L., Meltzer, P., and Aplan, P. D. (2012). Enforced expression of Lin28b leads to impaired T-cell development, release of inflammatory cytokines, and peripheral T-cell lymphoma. Blood 120, 1048-1059.
Behm-Ansmant, I., Rehwinkel, J., Doerks, T., Stark, A., Bork, P., and Izaurralde, E. (2006). MRNA degradation by miRNAs and GW182 requires both CCR4 : NOT deadenylase and DCP1 : DCP2 decapping complexes. Genes & Development 20, 1885-1898.
Benetti, R., Gonzalo, S., Jaco, I., Munoz, P., Gonzalez, S., Schoeftner, S., Murchison, E., Andl, T., Chen, T., Klatt, P., et al. (2008). A mammalian microRNA cluster controls DNA methylation and telomere recombination via Rbl2-dependent regulation of DNA methyltransferases (vol 15, pg 268, 2008). Nature Structural & Molecular Biology 15,
Chapter1. Introduction
Berezikov, E., Chung, W. J., Willis, J., Cuppen, E., and Lai, E. C. (2007). Mammalian mirtron genes. Molecular Cell 28, 328-336.
Bernstein, E., Caudy, A. A., Hammond, S. M., and Hannon, G. J. (2001). Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409, 363-366. Bethune, J., Artus-Revel, C. G., and Filipowicz, W. (2012). Kinetic analysis reveals successive steps leading to miRNA-mediated silencing in mammalian cells. EMBO Rep 13, 716-723.
Bohnsack, M. T., Czaplinski, K., and Gorlich, D. (2004). Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. Rna-a Publication of the Rna Society 10, 185-191.
Bonci, D., Coppola, V., Musumeci, M., Addario, A., Giuffrida, R., Memeo, L., D'Urso, L., Pagliuca, A., Biffoni, M., Labbaye, C., et al. (2008). The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat Med 14, 1271-1277.
Borchert, G. M., Lanier, W., and Davidson, B. L. (2006). RNA polymerase III transcribes human microRNAs. Nat Struct Mol Biol 13, 1097-1101.
Boyerinas, B., Park, S. M., Hau, A., Murmann, A. E., and Peter, M. E. (2010). The role of let-7 in cell differentiation and cancer. Endocr Relat Cancer 17, F19-36.
Boyerinas, B., Park, S. M., Shomron, N., Hedegaard, M. M., Vinther, J., Andersen, J. S., Feig, C., Xu, J., Burge, C. B., and Peter, M. E. (2008). Identification of Let-7-Regulated Oncofetal Genes. Cancer Research 68, 2587-2591.
Brants, J. R., Ayoubi, T. A., Chada, K., Marchal, K., Van de Ven, W. J., and Petit, M. M. (2004). Differential regulation of the insulin-like growth factor II mRNA-binding protein genes by architectural transcription factor HMGA2. FEBS Lett 569, 277-283.
Brennecke, J., Stark, A., Russell, R. B., and Cohen, S. M. (2005). Principles of MicroRNA-target recognition. PLoS Biol 3, 404-418.
Cai, X. Z., Hagedorn, C. H., and Cullen, B. R. (2004). Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. Rna-a Publication of the Rna Society 10, 1957-1966.
Calin, G. A. (2002). Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proceedings of the National Academy of Sciences 99, 15524-15529.
Calin, G. A., and Croce, C. M. (2006). MicroRNA signatures in human cancers. Nature Reviews Cancer 6, 857-866.
Chapter1. Introduction
Calin, G. A., Dumitru, C. D., Shimizu, M., Bichi, R., Zupo, S., Noch, E., Aldler, H., Rattan, S., Keating, M., Rai, K., et al. (2002). Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proceedings of the National Academy of Sciences of the United States of America 99, 15524-15529.
Calin, G. A., Sevignani, C., Dan Dumitru, C., Hyslop, T., Noch, E., Yendamuri, S., Shimizu, M., Rattan, S., Bullrich, F., Negrini, M., and Croce, C. M. (2004). Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proceedings of the National Academy of Sciences of the United States of America 101, 2999-3004.
Chang, T. C., Yu, D. N., Lee, Y. S., Wentzel, E. A., Arking, D. E., West, K. M., Dang, C. V., Thomas-Tikhonenko, A., and Mendell, J. T. (2008). Widespread microRNA repression by Myc contributes to tumorigenesis. Nature Genetics 40, 43-50.
Chang, T. C., Zeitels, L. R., Hwang, H. W., Chivukula, R. R., Wentzel, E. A., Dews, M., Jung, J., Gao, P., Dang, C. V., Beer, M. A., et al. (2009). Lin-28B transactivation is necessary for Myc-mediated let-7 repression and proliferation. Proc Natl Acad Sci U S A 106, 3384-3389.
Cheloufi, S., Dos Santos, C. O., Chong, M. M. W., and Hannon, G. J. (2010). A Dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature 465, 584-U576.
Chendrimada, T. P., Gregory, R. I., Kumaraswamy, E., Norman, J., Cooch, N., Nishikura, K., and Shiekhattar, R. (2005). TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 436, 740-744.
Chi, S. W., Hannon, G. J., and Darnell, R. B. (2012). An alternative mode of microRNA target recognition. Nat Struct Mol Biol 19, 321-327.
Chiang, H. R., Schoenfeld, L. W., Ruby, J. G., Auyeung, V. C., Spies, N., Baek, D., Johnston, W. K., Russ, C., Luo, S., Babiarz, J. E., et al. (2010). Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev 24, 992-1009.
Chin, L. J., Ratner, E., Leng, S. G., Zhai, R. H., Nallur, S., Babar, I., Muller, R. U., Straka, E., Su, L., Burki, E. A., et al. (2008). A SNP in a let-7 microRNA Complementary Site in the KRAS 3 ' Untranslated Region Increases Non-Small Cell Lung Cancer Risk. Cancer Research 68, 8535-8540.
Chong, M. M. W., Zhang, G. A., Cheloufi, S., Neubert, T. A., Hannon, G. J., and Littman, D. R. (2010). Canonical and alternate functions of the microRNA biogenesis machinery (vol 24, pg 1951, 2010). Genes & Development 24, 2228-2228.
Chapter1. Introduction
Pathway Independent of Dicer Requires Argonaute2 Catalytic Activity. Science 328, 1694-1698.
Cimmino, A., Calin, G. A., Fabbri, M., Iorio, M. V., Ferracin, M., Shimizu, M., Wojcik, S. E., Aqeilan, R. I., Zupo, S., Dono, M., et al. (2005). miR-15 and miR-16 induce apoptosis by targeting BCL2. Proceedings of the National Academy of Sciences of the United States of America 102, 13944-13949.
Cleynen, I., Brants, J. R., Peeters, K., Deckers, R., Debiec-Rychter, M., Sciot, R., Van de Ven, W. J., and Petit, M. M. (2007). HMGA2 regulates transcription of the Imp2 gene via an intronic regulatory element in cooperation with nuclear factor-kappaB. Mol Cancer Res 5, 363-372.
Cole, C., Sobala, A., Lu, C., Thatcher, S. R., Bowman, A., Brown, J. W. S., Green, P. J., Barton, G. J., and Hutvagner, G. (2009). Filtering of deep sequencing data reveals the existence of abundant Dicer-dependent small RNAs derived from tRNAs. Rna-a Publication of the Rna Society 15, 2147-2160.
Conkrite, K., Sundby, M., Mukai, S., Thomson, J. M., Mu, D., Hammond, S. M., and MacPherson, D. (2011). miR-17~92 cooperates with RB pathway mutations to promote retinoblastoma. Genes Dev 25, 1734-1745.
Cordes, K. R., Sheehy, N. T., White, M. P., Berry, E. C., Morton, S. U., Muth, A. N., Lee, T. H., Miano, J. M., Ivey, K. N., and Srivastava, D. (2009). miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 460, 705-710.
Costinean, S., Zanesi, N., Pekarsky, Y., Tili, E., Volinia, S., Heerema, N., and Croce, C. M. (2006). Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proceedings of the National Academy of Sciences of the United States of America 103, 7024-7029.
Dai, N., Rapley, J., Angel, M., Yanik, M. F., Blower, M. D., and Avruch, J. (2011). mTOR phosphorylates IMP2 to promote IGF2 mRNA translation by internal ribosomal entry. Genes Dev 25, 1159-1172.
Davalos, V., Moutinho, C., Villanueva, A., Boque, R., Silva, P., Carneiro, F., and Esteller, M. (2012). Dynamic epigenetic regulation of the microRNA-200 family mediates epithelial and mesenchymal transitions in human tumorigenesis. Oncogene 31, 2062-2074.
Denli, A. M., Tops, B. B., Plasterk, R. H., Ketting, R. F., and Hannon, G. J. (2004). Processing of primary microRNAs by the Microprocessor complex. Nature 432, 231-235. Dews, M., Fox, J. L., Hultine, S., Sundaram, P., Wang, W. G., Liu, Y. Q. Y., Furth, E., Enders, G. H., El-Deiry, W., Schelter, J. M., et al. (2010). The Myc-miR-17 similar to 92 Axis Blunts TGF beta Signaling and Production of Multiple TGF beta-Dependent Antiangiogenic Factors. Cancer Research 70, 8233-8246.








![[PDF] Catalogue de formation bureautique PDF | Cours Bureautique](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)