HAL Id: tel-01950418
https://tel.archives-ouvertes.fr/tel-01950418
Submitted on 10 Dec 2018
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
les processus de différenciation
Anissa Guillemin
To cite this version:
Anissa Guillemin. Rôle de la stochasticité de l’expression des gènes dans les processus de
différencia-tion. Biologie cellulaire. Université de Lyon, 2018. Français. �NNT : 2018LYSE1188�. �tel-01950418�
THÈSE DE DOCTORAT DE L’UNIVERSITÉ DE LYON
opérée au sein de
l’Université Claude Bernard Lyon 1
École Doctorale ED340
Biologie Moléculaire Intégrative et Cellulaire
Spécialité de doctorat
Biologie
Soutenue publiquement le 5/10/2018, par :
Anissa GUILLEMIN
Rôle de la stochasticité de l’expression
des gènes dans les processus de
différenciation
Devant le jury composé de :
Paldi Andràs, DE, EPHE Président
Jaffredo Thierry, DR, CNRS Rapporteur
Peronnet Frédérique, DR, CNRS Rapporteure
Perié Leïla, CR, CNRS Examinatrice
Gonin-Giraud Sandrine, MCU, UCBL Examinatrice
Président de l’Université
Président du Conseil Académique
Vice-président du Conseil d’Administration
Vice-président du Conseil Formation et Vie Universitaire Vice-président de la Commission Recherche
Directrice Générale des Services
M. le Professeur Frédéric FLEURY
M. le Professeur Hamda BEN HADID
M. le Professeur Didier REVEL M. le Professeur Philippe CHEVALIER M. Fabrice VALLÉE
Mme Dominique MARCHAND
COMPOSANTES SANTE
Faculté de Médecine Lyon Est – Claude Bernard
Faculté de Médecine et de Maïeutique Lyon Sud – Charles Mérieux
Faculté d’Odontologie
Institut des Sciences Pharmaceutiques et Biologiques Institut des Sciences et Techniques de la Réadaptation Département de formation et Centre de Recherche en Biologie Humaine
Directeur : M. le Professeur G.RODE
Directeur : Mme la Professeure C. BURILLON Directeur : M. le Professeur D. BOURGEOIS Directeur : Mme la Professeure C. VINCIGUERRA Directeur : M. X. PERROT
Directeur : Mme la Professeure A-M. SCHOTT
COMPOSANTES ET DEPARTEMENTS DE SCIENCES ET TECHNOLOGIE
Faculté des Sciences et Technologies Département Biologie
Département Chimie Biochimie Département GEP
Département Informatique Département Mathématiques Département Mécanique Département Physique
UFR Sciences et Techniques des Activités Physiques et Sportives Observatoire des Sciences de l’Univers de Lyon
Polytech Lyon
Ecole Supérieure de Chimie Physique Electronique Institut Universitaire de Technologie de Lyon 1 Ecole Supérieure du Professorat et de l’Education Institut de Science Financière et d'Assurances
Directeur : M. F. DE MARCHI
Directeur : M. le Professeur F. THEVENARD Directeur : Mme C. FELIX
Directeur : M. Hassan HAMMOURI Directeur : M. le Professeur S. AKKOUCHE Directeur : M. le Professeur
G. TOMANOV
Directeur : M. le Professeur H. BEN HADID Directeur : M. le Professeur J-C PLENET Directeur : M. Y.
VANPOULLE
Directeur : M. B. GUIDERDONI Directeur : M. le Professeur E.PERRIN Directeur : M. G. PIGNAULTDirecteur : M. le Professeur C. VITON
Directeur : M. le Professeur A. MOUGNIOTTE Directeur : M. N. LEBOISNE
Pour commencer, un grand merci aux membres de mon jury qui ont accepté de venir évaluer
mon travail. Merci au Dr. Thierry Jaffredo et au Dr. Frédérique Peronnet d’avoir accepté de
relire et de rapporter mon manuscrit. Merci également au Dr. Leïla Perié, au Pr. Andràs Paldi et
à Sandrine de bien vouloir examiner ma thèse. C’est un plaisir et un honneur que de présenter
devant vous toustes le travail de plus de trois années.
C’est à mes yeux les lignes les plus importantes de ce manuscrit, je tiens à exprimer toute
ma reconnaissance et ma gratitude à Olivier et Sandrine pour avoir accepté de m’intégrer dans
l’équipe et pour avoir été présents pendant toutes ces années. Un grand merci à Olivier qui a été
un excellent formateur, juste et qui a su faire de moi une passionnée de la bonne recherche. J’ai
énormément appris et ces années de thèse ont été les plus enrichissantes de ma vie. Tout ça a
été possible grâce à sa pédagogie, son humanité, son travail et sa patiente. Également un grand
merci à Sandrine pour m’avoir donné la chance de venir dans cette équipe en me choisissant au
début de l’aventure. Merci pour ses conseils, sa confiance et son sourire. Merci également à tous
les deux d’avoir pris le temps de relire mes manuscrits.
Merci au CGPhiMC et au LBMC pour m’avoir accueilli et fourni l’environnement nécessaire
pour travailler.
Merci à l’ED BMIC pour avoir jugé mon travail et m’avoir permis de financer ma thèse. Un
petit mot pour Roxane pour son accessibilité et sa gentillesse.
Je souhaite également remercier le Pr. Andràs Paldi et le Dr. Pierre Sujobert pour avoir fait
partie de mon comité de suivi de thèse et pour leurs discussions très enrichissantes. Merci
égale-ment à toutes les équipes avec lesquelles j’ai discuté et travaillé au sein des différents laboratoires.
Merci à l’équipe du Pr. Andràs Paldi pour nos échanges intéressants. Merci à l’équipe du Dr.
Vincent Gache et à l’équipe du Dr. Véronique Maguer-Satta pour leur collaboration et pour avoir
participé à l’éclectisme de mon travail. Je souhaite également remercier Fabien Crauste pour la
touche de modélisation qu’il a apporté dans ma thèse avec Ronan. Pour ses relectures et ses
précieux conseils.
été à mes côtés pendant ces deux dernières années, je les remercie pour leurs conseils et leur
confiance.
Si j’ai autant aimé mon travail c’est sans compter sur mon équipe qui a su créer l’ambiance
parfaite pour être à l’aise. Encore un grand merci à Olivier et Sandrine pour leur rôle au sein
de l’équipe, la cohésion qu’iels ont créé, ainsi que pour les moments joyeux qu’iels ont su nous
faire profiter. Merci à Olivier d’avoir réuni autant de personnes différentes dans la même équipe,
ça a été beaucoup de schémas au tableau et de discussions sur des sujets originaux qui m’ont
fait adorer la science, et surtout les échanges avec les autres domaines de la recherche. Merci à
Gérard pour son temps, tous ces conseils, ses discussions et son café, toujours à disposition pour
notre plus grand bonheur. Merci à Angélique pour avoir été là depuis le début. J’ai passé des très
bons moments à discuter et rigoler avec elle. Merci pour ses petits services, sa grande aide, son
soutien et son sourire reposant. Merci à mon coéquipier de thèse, Ronan. Le travail en équipe
avec lui a été inoubliable. J’ai beaucoup appris à ses côtés, sur ma façon de travailler. Mais j’ai
surtout appris de lui qu’on pouvais toujours apprendre plus et aller plus loin. Ma thèse a pris une
plus-value quand il est arrivé et je lui en suis extrêmement reconnaissante. Comme précisé plus
haut, l’une des choses que j’ai le plus apprécié dans cette équipe a été sa pluridisciplinarité. Merci
à Ulysse pour avoir été un passionné de mathématiques et pour m’avoir impressionné dans ton
travail. Merci également pour tes conseils et tes discussions. Merci à Arnaud pour m’avoir donné
envie de faire des algorithmes. Ton travail a été une source d’inspiration pour moi et pour ma
future carrière. Un grand merci à Elodie pour son aide précieuse, sa complicité et les moments
de détente à discuter avec elle. Merci également à Catherine pour ses connaissances et sa bonne
humeur dans le bureau. Merci à Geneviève pour ses questionnements intéressants et sa veille
bibliographique. Je n’oublie pas les ancien.e.s comme Rebecca pour sa bonne humeur, merci à
François Briat pour son humour et pour m’avoir fait découvrir le Roller-Derby. Merci également
à toustes les stagiaires ancien.e.s, actuels et actuelle, d’avoir participé à la bonne ambiance de
l’équipe, à sa multidisciplinarité et à sa cohésion.
boulot. Merci aux gestionnaires du LBMC pour leur gentillesse. Merci aux personnes curieux.ses
du travail que j’ai réalisé, pour leurs questions inattendues.
Le travail a été passionnant mais c’est aussi grâce au fait que j’ai su m’entourer des bonnes
personnes à l’extérieur des murs du laboratoire que j’ai autant apprécié mon travail. Merci donc
à mes ami.e.s de promo qui n’ont jamais été très loin, merci à Ommar, Léo, Godart, Kéké ainsi
qu’à tous les autres membres
γβχ. Un petit mot en particulier pour Manon qui a été le pied
de la chaise sur laquelle je me suis assise pendant trois ans. Merci à mes ami.e.s rencontrés à
Lyon qui m’ont écouté parler de mon travail et qui ont su me faire passer de bons moments de
détente. Merci à mes ami.e.s de Chalon qui sont venu me voir. La thèse, c’est aussi apporter son
savoir-faire ailleurs comme à DéMesures où j’ai rencontré des personnes de tous les horizons et
partagé des moments instructifs. Merci pour votre confiance et votre volonté. Merci également à
l’équipe de l’AlternatiBar et à tous les autres membres de l’Alternatiba Rhône pour m’avoir fait
vivre leur passion et pour avoir partagé leurs engagements. Merci à toutes les personnes que j’ai
rencontré à l’ENS. Ça a été un cadre très stimulant avec beaucoup de moments de discussions et
de détente. Merci à Ronan encore une fois pour m’avoir fait découvrir ce cadre. Ça m’a permis de
voir plus loin que le bout de mon nez. Je vais terminer cette partie en remerciant des ami.e.s un
peu particuliers mais surtout mes compagnons du midi et du soir Benji et Gram. Je n’aurai pas
espéré de meilleurs co-thésards, aussi faisans et semi-croustillants soient-ils, dans cette aventure.
Un grand merci à ma famille pour leur soutien et leur patiente. Merci à Fatima pour avoir pris
soin de moi. Merci à Sophia, Nadia et Elies pour m’avoir fait sortir de temps en temps et pour
m’avoir rappelé que la famille n’est jamais bien loin.
Table des matières
7
Table des figures
11
1
Introduction
13
1.1
La stochasticité de l’expression des gènes (SEG) . . . .
14
1.1.1
L’histoire de la stochasticité biologique . . . .
14
1.1.2
Les sources de cette variabilité . . . .
16
1.1.3
Les techniques de mesure et d’analyse de la variabilité . . . .
19
1.1.3.1
La mesure de la variabilité au niveau protéique . . . .
19
1.1.3.2
La mesure de la variabilité au niveau transcriptomique . . . . .
22
1.1.3.3
Les limites et biais de mesure de la variabilité
. . . .
26
1.1.3.4
L’analyse de la variabilité . . . .
28
1.1.4
Les rôles connus dans les processus biologiques . . . .
29
1.2
La SEG et les processus de différenciation . . . .
33
1.2.1
L’hématopoïèse, un processus de différenciation multi-lignage . . . .
33
1.2.2
Des processus de différenciation mono-lignage
. . . .
35
1.2.2.1
La différenciation érythropoïétique . . . .
36
1.2.2.2
La différenciation musculaire . . . .
37
1.2.3
Les pathologies associées au processus de différenciation . . . .
38
1.2.3.1
La cancérogénèse . . . .
38
renciation érythrocytaire
43
2.1
Introduction . . . .
44
2.2
Article . . . .
45
2.3
Données supplémentaires
. . . .
81
2.4
Conclusions . . . .
92
3
Automatisation de la mesure du cycle et de la taille cellulaire pour l’analyse
en cellule unique
95
3.1
Introduction . . . .
96
3.2
Article . . . .
97
3.3
Conclusions . . . .
105
4
Inférence de réseaux dynamiques de régulation de gènes par un algorithme
itératif : WASABI
107
4.1
Introduction . . . .
108
4.2
Article . . . .
109
4.3
Conclusions . . . .
138
5
Calibration, sélection et analyses de l’identifiabilité d’un modèle
mathé-matique de la différenciation érythrocytaire
in vitro dans des conditions
contrôles et perturbées.
139
5.1
Introduction . . . .
140
5.2
Article . . . .
141
5.3
Conclusions . . . .
160
6
Effet des drogues qui modulent la stochasticité de l’expression des gènes sur
le processus de différenciation érythrocytaire
161
6.1
Introduction . . . .
162
7
Effet des drogues qui modulent la stochasticité de l’expression des gènes sur
le processus de différenciation musculaire
177
7.1
Introduction . . . .
178
7.2
Matériel et méthodes
. . . .
178
7.2.1
Culture cellulaire . . . .
178
7.2.2
Extraction d’ARN, rétro-transcription et PCR quantitative en temps-réel
179
7.2.3
Immunofluorescence (IF) . . . .
180
7.3
Principaux résultats . . . .
180
7.3.1
Effet de l’Artemisinin et de l’Indomethacin sur la myogénèse . . . .
180
7.3.2
Effet de MB-3 sur la myogénèse . . . .
182
7.4
Conclusions et perspectives . . . .
182
8
Étude des mécanismes de résistance à l’Imatinib dans le cadre de la leucémie
myéloïde chronique par une approche en cellules uniques
185
8.1
Introduction . . . .
186
8.2
Matériel et méthodes
. . . .
187
8.2.1
Culture cellulaire . . . .
187
8.2.2
RTqPCR haut débit en population et en cellules uniques . . . .
187
8.2.3
Analyses statistiques . . . .
188
8.3
Principaux résultats . . . .
188
8.3.1
Établissement d’une liste de gènes d’intérêts . . . .
188
8.3.2
Identification et caractérisation d’un modèle in vitro de cellules myéloïdes
chroniques . . . .
189
8.3.3
Analyses préliminaires des données transcriptomiques en cellules uniques .
190
8.4
Conclusions et perspectives . . . .
192
9
Discussion et perspectives
193
9.1
La rôle de la SEG dans la différenciation
. . . .
195
9.3
Contrôle de la différenciation dans le cadre des pathologies . . . .
199
1.1
Les processus stochastiques sont les moteurs de la variabilité cellulaire . . . .
18
1.2
Techniques de mesure de la variabilité . . . .
27
1.3
Illustration de l’utilisation de l’entropie par Claude Shannon dans le cadre de la théorie
de l’information . . . .
30
1.4
Modèles d’engagement des CSHs dans les lignages hématopoïétiques . . . .
34
7.1
Effet de l’Artemisinin et de l’Indomethacin sur les C2C12
. . . .
181
7.2
Effet de MB-3 sur les C2C12 . . . .
183
8.1
Un modèle dynamique du comportement des cellules LMC . . . .
187
8.2
Analyses en composantes principales (ACP) des cellules TF1-BA sensibles et résistantes190
8.3
Analyses en t-SNE des cellules TF1-BA sensibles à l’Imatinib
. . . .
191
9.1
Schéma représentant le point de vue dynamique de la différenciation et de la tumorigénèse202
Certains processus cellulaires ont été décris comme des mécanismes prédéfinis, garantissant
une homogénéité phénotypique. Cependant, depuis plusieurs dizaines d’années, il a été montré
l’existence d’un caractère hétérogène au sein d’une population isogénique où chaque cellule
pos-sède un comportement différent et aléatoire. Cette variabilité non génétique, est un phénomène
probabilistique dont l’observation remet en cause le programme génétique. Les récentes études
portant sur ce mécanisme, rendues possible grâce aux nouvelles technologies d’analyse en cellules
uniques, montrent qu’il est désormais nécessaire d’intégrer cette dimension stochastique dans
l’interprétation de tous les processus biologiques [146, 33, 59, 212, 95]. Bien que cette réalité
existe depuis quelques décennies, ce phénomène reste encore mal compris. Dans cette première
partie, je présenterai l’état des connaissances sur la stochasticité de l’expression des gènes de sa
découverte à aujourd’hui, puis je détaillerai ses implications dans différents processus biologiques
dont la différenciation.
1.1.1
L’histoire de la stochasticité biologique
Toutes les découvertes scientifiques, qui ont bâti le socle de notre connaissance actuelle, ont
tout d’abord commencé par de simples observations. Avant même que le terme "génétique" n’ait
été utilisé pour la première fois par William Bateson en 1905, des passionnés, des philosophes, des
chercheur.e.s et souvent même les trois à la fois ont fait avancer pas à pas notre compréhension
des mécanismes biologiques. Dans le but d’obtenir des plantes de couleurs nouvelles, Gregor
Men-del remarqua en 1865 une certaine régularité dans le phénotype à chaque croisement entre deux
variétés différentes. Cette observation donna naissance à une des premières lois sur l’hérédité de
caractères phénotypiques. C’est en 1879 que Walther Flemming décrivit en détail pour la première
fois le mouvement des chromosomes pendant la mitose en colorant des cellules d’embryon de
salamandre avec de l’aniline. De fil en aiguille, les liens se sont formés. La compréhension des lois
de l’hérédité évolue et se confirme pour arriver à la définition de gènes et de phénotypes par
Wil-helm Johannsen en 1909. Moins de 50 ans plus tard, des physiciens et des biologistes découvrent
le support de l’information génétique. En 1970, Francis Crick, l’un des co-découvreurs de cette
dernière avancée, détailla le transfert séquentiel de l’information génétique par la notion de dogme
central. L’idée que le fonctionnement des cellules dans un organisme était un processus précis et
extrêmement bien cadré donna ainsi naissance à la notion de programme génétique. Il paraissait
évident à l’époque que des organismes aussi bien organisés ne pouvaient résulter que de
méca-nismes parfaitement ordonnés. Même dans les années 1950, le développement et l’utilisation de la
cytométrie en flux n’a pas changé la vision des choses. Cette technologie, alors capable de mesurer
des caractéristiques morphologiques à l’échelle de la cellule unique, permettait d’observer de la
variabilité entre cellules génétiquement identiques et cultivées dans un environnement homogène.
Ce n’est que 30 ans plus tard, que l’idée du désordre dans les processus génétiques a commencé à
être envisagée. Nous devons ce nouveau paradigme aux avancées technologiques notamment en
matière de mesures quantitatives du transcriptome/protéome de cellules individualisées telles que
les RTqPCR en cellules uniques ou le suivi de la fluorescence de protéines in vivo. Malgré tout, la
réticence scientifique était encore dominante face à cette nouvelle vision des processus cellulaires,
et ce malgré les observations de nombreux chercheur.e.s [12, 30, 173].
La balance a penché dans les années 2000, où l’évidence ne pouvait plus être ignorée. En
utilisant des technologies plus récentes, des chercheur.e.s ont démontré l’existence du caractère
aléatoire de l’expression des gènes. C’est principalement en observant la variabilité d’expression
d’une protéine fluorescente rapportrice entre chaque cellule d’une population bactérienne
iso-génique que Peter Swain, Michael Elowitz, Ertugrul Ozbudak et bien d’autres ont participé à
l’émergence de ce nouveau domaine de recherche [46, 146]. La pensée biologique a vu son histoire
scientifique s’ébranler pour faire place à un renouveau profond. En effet, les notions de dogme
central et de programme génétique ont dû être revisitées au profit des théories modernes de
l’expression aléatoire des gènes [113].
C’est à ce moment que nous avons intégré la dimension stochastique à l’expression génique.
L’image d’un gène actif dans un type cellulaire et inactif dans un autre apparaissait inadaptée et
laissa place à des modèles à caractère probabilistique. L’expression des gènes est donc soumise à
1.1.2
Les sources de cette variabilité
Du fait du processus moléculaire qui mène à l’expression des gènes, les sources de la
stochas-ticité sont nombreuses. Cette variabilité affecte toutes les étapes de production d’une protéine à
partir d’un gène [102]. L’étape de transcription reste la source majeure de variabilité inter-cellulaire
[129, 200, 98].
Tout d’abord, l’ADN est une molécule dynamique, dont la forme change constamment. Les
modifications épigénétiques associées au remodelage de la chromatine ont été suggérées comme
étant impliquées dans la régulation de la SEG [194, 10, 170, 142, 31]. Par exemple, il a été montré
chez S. cerevisiae que deux complexes de déacétylation des histones répriment le niveau de bruit
dans l’expression génique [216]. Des sites de méthylation des histones ont également été associés
à des modifications de la SEG [223]. De plus, les protéines responsables de la transcription ou
de la régulation de la transcription, s’accrochent à l’ADN de manière transitoire et répétée. La
fréquence d’assemblage et de désassemblage des protéines sur l’ADN se fait plus rapidement que la
demi-vie de l’ARNm [182]. Ces processus dynamiques qui créent cette reconfiguration permanente
rend donc imparfaite l’efficacité des réactions, dans le sens où toutes les réactions commencées
ne se terminent pas forcément. Le processus de transcription lui-même requiert également des
formations de multi-complexes protéiques formant la machinerie transcriptionnelle, qui occupent
un espace considérable. Cet encombrement spatial créé de la compétition topologique entre les
différents complexes protéiques. Par exemple, le nombre de sites de fixation de l’ARN polymérase II
sur l’ADN a été estimé à environ 2500 [82, 70], ce qui réduit considérablement le nombre de gènes
exprimables simultanément et favorise une expression différentielle entre cellules. En conséquence,
même si tous les réactifs sont disponibles pour engager des réactions, l’espace disponible reste une
limite à ne pas négliger. Sans compter le fait que les protéines régulatrices de l’expression génique
1. Ici les termes sont importants : nous ne sommes pas passés d’une vision d’un ordre parfait à un chaos total. Au fil des découvertes, nous avons admis que l’expression d’un gène ne donne pas un phénotype mais donne une probabilité d’avoir ce phénotype, soumis à une certaine contrainte. Cette limitation doit être vu comme une exploitation dynamique et non pas comme une nécessité de réduction de la stochasticité.
un instant donné, 80% des gènes exprimés dans E.Coli produisent moins de 100 protéines par cellule
[161]. Ce faible nombre de copies a deux conséquences majeures sur les réactions nucléaires [9].
D’une part, les réactions sont instables. Comme toutes les protéines nécessaires à une réaction ne
sont pas toutes disponibles au même moment et au même endroit, le fonctionnement réactionnel
varie selon la disponibilité des composants de la chaîne. D’autre part, lors des divisions cellulaires,
vu le faible nombre de copies d’une protéine, celles-ci ne vont pas se partager idéalement en
quantité égale dans chacune des deux cellules filles. Un pourcentage non négligeable de cellules
filles auront donc un nombre de copies de protéines inférieur à celui de sa cellule soeur [128, 106].
A ce stade, nous commençons à entrevoir l’étendue des conséquences de ces différents éléments
sur les variations phénotypiques.
Cette accumulation de phénomènes aléatoires, étape par étape, renforce les effets sur la
sto-chasticité générale. En effet, plus le niveau de variabilité augmente dans les réactions en amont
de cette chaîne de production, plus le niveau de SEG et ses conséquences sont importants à
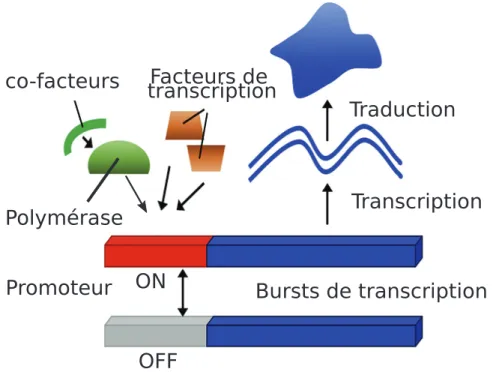
l’arrivée (Figure 1.1) [73]. Il a été montré que la modification de l’activité d’un promoteur chez
la levure entraînait des variations d’amplitude de SEG [15]. Cette phase critique dans l’activité
du promoteur a été étudiée, démontrée et modélisée [46, 67, 163]. Ces modèles sont appelés
des modèles deux-états. Ils représentent le processus de transcription où le promoteur d’un gène
fluctue entre un état ON et un état OFF [169, 152, 98, 151, 183, 83]. Parmi les modèles utilisés
pour simuler ces oscillations stochastiques, nous connaissons le "random telegraph" de 2005 [67],
qui était initialement utilisé en physique pour simuler les états dissipatifs des couches minces
supra-conductrices [226]. Utilisé en biologie, ce modèle assume que le promoteur d’un gène donné
passe d’un état OFF à un état ON avec une probabilité constante. Il assume également qu’une
fois le promoteur dans un état ON, il peut soit permettre la transcription (c’est ce que l’on
ap-pelle un "burst de transcription") soit repasser dans un état OFF, toujours avec une probabilité
constante. Les bursts de transcription sont décris selon deux paramètres : leur taille (caractérisée
par l’intensité ou la durée d’un burst) et leur fréquence (nombre de bursts en un temps donné).
Transcription
Traduction
Polymérase
Promoteur
ON
Bursts de transcription
OFF
Figure 1.1 – Les processus stochastiques sont les moteurs de la variabilité cellulaire.
Le promoteur du gène est soit en état ON soit en état OFF. Dans son état ON,
des ARN sont produits pendant ce que l’on appelle un burst de transcription. A
cette étape, les protéines permettant et régulant la transcription sont sujettes à de
la variabilité résultante par exemple du faible nombre de copies des protéines ou
de l’encombrement stérique des complexes de réaction. L’ARN est ensuite traduit
en protéine, étape où là aussi les protéines permettant et régulant ce processus sont
soumises à de la variabilité. Le niveau de la stochasticité augmente donc en partant du
gène vers la production des protéines, due à l’accumulation des différents processus,
soumis à de la variabilité. Source d’image : modifiée à partir de [73]
favorisés par les avancées technologiques afin de mieux comprendre l’origine de la SEG.
Enfin, à tout cela s’ajoute la topologie. C’est à dire le rôle de la conformation globale de
l’ADN et des territoires chromosomiques [17]. L’accessibilité à l’ADN est une étape déterminante
à plusieurs niveaux. Par son volume encombrant, ses repliements complexes peuvent augmenter
le temps qui séparent un facteur de transcription à son promoteur. Chez la levure, il a été montré
que la stochasticité de l’expression des gènes essentiels était un phénomène très contrôlé
principa-lement par leur position dans les chromosomes [9]. Les gènes essentiels sont concentrés dans les
régions ouvertes de la chromatine, où le bruit est le plus faible [7]. Une trop forte variabilité dans
distance entre un gène et le centromère du chromosome était liée au niveau de stochasticité de
l’expression du gène en question [9, 7].
1.1.3
Les techniques de mesure et d’analyse de la variabilité
Pour rendre compte de cette variabilité dans l’expression des gènes, une nouvelle dimension
quantitative à l’échelle de la cellule unique doit être utilisée. Dans ce paragraphe, j’ai choisi de
vous présenter les différentes techniques que j’ai utilisé durant me thèse ou celles qui existent dans
la littérature et dont je me suis intéressée de près.
1.1.3.1
La mesure de la variabilité au niveau protéique
Les rapporteurs fluorescents
Depuis plusieurs décennies, il est possible de mesurer la quantité
d’une ou plusieurs protéines dans toutes les cellules d’une population. Ces mesures ont été possibles
grâce à la découverte et à l’utilisation de rapporteurs fluorescents (GFP pour Green Fluorescent
Protein ou encore mCHerry). Ces molécules sont capables d’être excitées par une longueur d’onde
donnée (longueur d’onde d’excitation) et de restituer une partie de cette énergie sous l’aspect
d’une longueur d’onde de plus faible énergie (longueur d’onde d’émission). Chaque fluorochrome
est caractérisé par ces deux longueurs d’onde. La première protéine fluorescente a été découverte
par Osamu Shimamura en 1962 mais ce n’est que 30 ans plus tard, que les chercheur.e.s ont
mesuré tout le potentiel de cet outil (prix Nobel de chimie en 2008). Les premières utilisations
de ces rapporteurs dans le but d’observer la variabilité dans le processus d’expression datent des
années 2000 [146, 166, 125].
A partir de constructions génétiques, les chercheur.e.s sont capables de fusionner à une
pro-téine, une molécule fluorescente, sans que la production de la protéine ne soit altérée. Le principe
est le suivant : un gène rapporteur fluorescent produit une protéine fluorescente détectable par des
outils spécifiques (dont certains seront développés plus tard dans cette section) et permet de
me-surer l’expression d’un gène d’intérêt [108]. Ce gène rapporteur va être fusionné au gène d’intérêt
(dont on veut mesurer l’expression) ou mis sous le contrôle de son promoteur. Cet ADN
recombi-Ensuite, une fois purifiée, cette construction va être intégrée dans l’ADN de l’organisme ou dans
la cellule vivante cible par transgénèse (généralement par transduction via un vecteur viral).
La molécule fluorescente va donc être produite dans la cellule de la même manière que la
protéine endogène d’intérêt. De cette façon, la mesure de la fluorescence permet d’avoir accès à
la quantité de protéine d’intérêt. Pour la mesurer, plusieurs techniques sont utilisées. Parmi toutes
ces techniques, je vais vous en présenter trois : la cytométrie en flux, la microscopie à fluorescence
ou encore la vidéo-microscopie.
La cytométrie en flux
La cytométrie en flux est une technique de caractérisation individuelle,
quantitative et qualitative de particules dans une suspension. Cet outil est né d’un besoin
d’auto-matisation de comptage des composants cellulaire du sang. C’est en 1934 que Moldavan conçut
le premier appareil dans le but de compter des cellules qui passaient dans un capillaire fin et où
chacune étaient captées par un récepteur photo-électrique [156].
Un système de centrage hydrodynamique permet la formation d’un flux qui centre les cellules
d’une suspension cellulaire à travers une aiguille. La dimension de ce capillaire ainsi que
l’ac-célération progressive du flux permet d’aligner les cellules les unes derrière les autres [156]. La
cytométrie en flux permet d’analyser plusieurs types de paramètres : la taille et la granularité de
chaque cellule mais aussi la fluorescence de marqueurs spécifiques. Un laser est focalisé sur le flux
de cellules et sa diffusion est captée par des photodiodes. Sa diffusion frontale, captée par un
premier photodiode, renseigne sur la taille des cellules et sa diffusion latérale captée par un second
photodiode, renseigne sur la granularité des cellules. Les cellules éventuellement marquées par
des fluorochromes émettent des signaux de longueurs d’onde différentes. Un jeu de miroirs et de
filtres permet à des photo-multiplicateurs de les capter. Ces signaux optiques sont ensuite traduits
en signaux numériques proportionnels pour pouvoir être analysés avec un ordinateur. La première
démonstration de cette méthode d’analyse d’un fluorochome date de 1942 où Albert Coons utilisa
la cytométrie en flux pour mesurer la fluorescence d’un antigène anti-pneumocoque dans un tissu.
La puissance de cette technique réside dans sa capacité à analyser un grand nombre de cellules
ressante. Cependant, cette technique ne permet d’avoir qu’une image instantanée de l’expression
d’un ou plusieurs gène. Son utilisation pour la mesure de la variabilité dans l’expression a été
décrite des années plus tard, dans les années 2000, avec l’émergence de la nouvelle vision du "non
déterminisme" [14].
La microscopie confocale & la vidéo-microscopie
Parallèlement, l’utilisation de la
micro-scopie confocale dans le but d’observer la SEG s’est également développée [46].
Le microscope confocal est un microscope optique utilisé sur des échantillons biologiques
comme des cellules vivantes qui expriment un ou plusieurs gènes rapporteurs. Il est composé de
deux diaphragmes qui sont placés sur le trajet de la lumière (Figure 1.2 A). Le premier diaphragme
permet la focalisation du faisceau seulement sur une petite partie de l’échantillon. Les fluorophores
alors présents en ce point émettent une lumière qui est ensuite déviée par un miroir dichroïque. Un
deuxième diaphragme est placé au point de focalisation du faisceau et ne laisse passer seulement
la lumière venant du point visé dans l’échantillon. Un balayage horizontal de l’échantillon est
nécessaire pour obtenir une image de fluorescence à haute précision. Ensuite, un déplacement
vertical permet d’avoir accès à différents plans de coupe pour recommencer le balayage horizontal.
Un traitement informatique permet finalement de constituer une image en volume [210].
Cette technique permet de détecter des molécules fluorescentes avec une bonne résolution
spa-tiale à trois dimensions sur chacune des cellules composant l’échantillon. La microscopie confocale
est une variante de la microscopie à fluorescence, dont la détection de la fluorescence ne se
li-mite qu’à la surface des cellules (une seule couche). Ces images obtenues permettent d’avoir
accès à une image instantanée de l’expression d’un ou plusieurs gènes sur un nombre de cellules
relativement limité par rapport au cytomètre de flux [210].
Pour avoir une vision de l’évolution de l’expression génique en cellule unique dans le temps,
la vidéo-microscopie a récemment pris une place importante dans les mesures de la stochasticité.
Cette technique permet de visualiser et de quantifier en temps réel la ou les protéines d’intérêt
(toujours via des rapporteurs fluorescents) produites dans chaque cellule in vitro [141, 35, 36]. Cet
d’une caméra à haute résolution. La plate-forme motorisée permet de balayer la surface de chaque
couche de l’échantillon. La caméra permet d’enregistrer les images en temps réel. La caméra et
traitement informatique permettent de récolter les images et de les transformer en vidéo. Cette
dernière technique ne nous permet plus d’avoir un "snapshot" de l’expression génique mais nous
permet de suivre l’expression génique en temps réel [141].
Grâce à ces différentes techniques, la variabilité de l’expression génique est observée à l’échelle
de la protéine.
1.1.3.2
La mesure de la variabilité au niveau transcriptomique
La technique d’hybridation
in situ D’autres techniques, basées cette fois-ci sur des amorces
d’ARN ou d’ADN, nous donnent accès à la quantité d’ARN dans les cellules. Par exemple, la
technique de FISH pour Fluorescent in situ hybridization a été utilisée pour la première fois pour
hybrider l’ADN [8]. Cette méthode a permis par exemple la cartographie des gènes polytènes chez
les drosophiles [119]. L’hybridation in situ s’est ensuite développée pour l’ARN [189]. Initialement,
cette méthode était utilisée pour localiser la position intra-cellulaire de certains ARN dans un
organisme comme chez la levure [54]. Son utilisation combinée à celle de la microscopie ou de
la vidéo-microscopie permet de quantifier le nombre d’ARN d’un gène ou plusieurs gènes donnés
dans une cellule. Le principe est le suivant : une sonde complémentaire de l’ARNm d’intérêt est
tout d’abord synthétisé. Cette sonde va être préparée de manière à incorporer des marqueurs
fluorescents. Une fois les cellules fixées et perméabilisées, une étape d’hybridation va permettre
aux sondes de se fixer sur les ARNm d’intérêt. Les ARNm vont alors pouvoir être détectés par de la
microscopie à fluorescence. Plus récemment, cette technique a été adaptée pour pouvoir l’utiliser
sur des cellules vivantes [131]. En utilisant de la perméabilisation réversible, de la micro-injection
ou encore de l’électroporation, les sondes marquées peuvent être introduites dans des cellules
vivantes et leur fluorescence peut être suivie par microscopie confocale ou bien vidéo-microscopie
[4].
lules fixées comme dans des cellules vivantes, comme par exemple la technique de marquage MS2.
Le principe est le suivant : par construction génétique (de la même manière que pour introduire
un gène rapporteur dans l’ADN de cellules), une séquence particulière va être introduite dans
les parties non-codantes de l’ADN du gène d’intérêt. Cette séquence va permettre la formation
de structures tige-boucles dans l’ARN transcrit (Figure 1.2 B). Ces structures tige-boucles dans
l’ARN vont être reconnues et être fixées spécifiquement par une protéine appelée MS2 (protéine
découverte chez un bactériophage en 1961 [38, 4]). Préalablement, la protéine MS2 va être
pro-duite, fusionnée à une protéine fluorescente (par construction génétique). C’est la reconnaissance
des structures tige-boucles par la protéine MS2 marquée par un fluorochrome qui va permettre le
suivi de l’ARN d’intérêt dans la cellule vivante [91, 13, 54, 4].
La microfluidique combinée à la RTqPCR
Les nouvelles avancées technologiques ont
per-mis de mesurer directement la quantité d’ARN en cellule unique sans passer par une mesure des
constructions génétiques. Ces techniques utilisent après isolation des cellules dans des chambres
réactionnelles, la combinaison de rétro-transcription (RT), pré-amplification et quantification de
l’ADNc [220]. Durant ma thèse, j’ai utilisé principalement la RTqPCR en cellule unique de
Flui-digm (96 échantillons et 96 paires d’amorces d’intérêt). Les premières étapes de cette technique
(isolation des cellules, RT et pré-amplification de l’ADNc) peuvent être réalisées de plusieurs
manières différentes. Je vais décrire les deux méthodes que j’ai utilisé :
— Le système C1 utilise le principe de pièges hydrodynamiques pour isoler les cellules. Une
micro-puce, composée d’un circuit fluidique principal, de canaux latéraux et de chambres
réactionnelles (dont les dimensions sont préalablement choisies en fonction de la taille des
cellules), va voir passer un flux de cellules. Par un jeu de pression, les cellules vont être
dirigées vers les canaux latéraux et piégées dans des sites de capture. A cette étape, la
micro-puce peut être sortie du C1 et observée au microscope pour identifier les puits avec
une cellule et ceux avec un nombre de cellules différent. C’est également à cette étape que
la micro-puce peut être observée sous différents laser dans le cas où les cellules auraient été
et leur contenu va être conduit par le flux dans différentes chambres réactionnelles pour
subir la RT et la pré-amplification (avec un mélange des 96 paires d’amorces d’intérêt).
Les ADNc de chaque cellule vont être ensuite dilués et être transférés dans une plaque 96
puits où chaque puit reçoit le contenu transcriptomique de chaque cellule piégée. Elle sera
alors utilisée comme support pour la quantification d’ADNc [178].
— La tri cellulaire est une technique du Fluorescence-Activated Cell Sorting (FACS) (cas
par-ticulier de cytométrie en flux dont les caractéristiques basiques ont été décrites plus haut).
Dans une population de cellules hétérogène, des sous-populations peuvent être définies et
séparées physiquement de la population globale [32]. Dans la cadre de ma thèse, les critères
de séparation étaient uniquement morphologiques (cellules vivantes individuelles) choisies
sur des graphiques représentant la granularité versus la taille des cellules. Il est possible de
séparer les cellules par d’autres critères comme par la quantité d’un marquage fluorescent
par exemple. Pour séparer les cellules, le système de centrage hydrodynamique vibre grâce
à un dispositif piezo-électrique et rompt le flux de cellules. Des gouttelettes, contenant au
plus d’une cellule, se forment et passe dans un champs électrostatique. Celles contenant
les cellules de sous-population d’intérêt sont alors chargées et déplacées
perpendiculaire-ment à la direction du flux. Elles sont ensuite récupérées dans un récipient collecteur [32].
Les gouttelettes non chargées sont éliminées (Figure 1.2 C). Dans notre cas, le récipient
collecteur est une plaque 96 puits contenant un mélange réactionnel permettant la lyse de
la cellule dès réception dans le puit. Une fois récolté et lysé, le contenu de chaque puit est
mis en contact avec différents milieux réactionnels. Ils sont ajoutés manuellement dans la
plaque pour poursuivre avec la RT et la pré-amplification de l’ADNc (avec un mélange des
96 paires d’amorces d’intérêt). Après une dernière étape de dilution, les ADNc sont prêt à
être quantifiés.
Le contenu des 96 puits est mis en contact avec un nouveau mélange réactionnel : une sonde
EvaGreen (agent fluorescent lorsqu’il est lié à de l’ADN double brin) et son tampon de
réac-tion. Tout ceci est ensuite chargé dans une nouvelle micro-puce avec les 96 paires d’amorces.
rescence de l’EvaGreen à chaque cycle d’amplification [61]. Pour chaque méthode d’isolement
des cellules (C1 ou FACS
2), la traçabilité entre la cellule de départ (condition pour le FACS,
condition/morphologie/marqueurs pour le C1) et l’expression de ses 96 gènes d’intérêt est
conser-vée. Cette information permet de comparer l’expression de plusieurs gènes en cellule unique entre
différentes conditions ou de mesurer des corrélation entre morphologie cellulaire et expression
génique.
Le séquençage de l’ARN
Plus récemment, la technologie du séquençage de l’ARN permet
d’avoir accès au transcriptome entier à l’échelle de la cellule unique [66]. Cette technique, de la
même manière que la RTqPCR en cellule unique, utilise l’isolation de cellules, la rétro-transcription
et l’amplification de l’ADNc comme support pour ensuite passer au séquençage (technologie
Illumina majoritairement). L’isolation des cellules en haut débit peut se faire par FACS ou par
microfluidique dont le détail des techniques est expliqué plus haut. Les méthodes de séquençage
de l’ARN en cellule unique diffèrent par leur manière de préparer les échantillons notamment
pendant les étapes de rétro-transcription et d’amplification de l’ADNc. Ces techniques ont donc des
sensibilités variables pour la détection des gènes faiblement exprimés. Une fois les ARN convertis
en ADNc, ils sont fragmentés, lus et alignés sur la base du génome de l’espèce étudiée, donnant
un nombre de fragments d’ADNc pour chaque gène, proportionnel au niveau d’expression de ce
gène.
Une technique récente utilise une combinaison de plusieurs méthodes visant à améliorer
l’ef-ficacité du séquençage et à limiter les biais [228]. Elle consistent à isoler des cellules dans des
gouttelettes d’huile par microfluidique. Chaque gouttelette contient les réactifs nécessaires, de
la lyse à la fragmentation des ADNc. Elles contiennent également des billes particulières
appe-lées GEM (Gel bead in EMulsion). Ces GEM sont recouvertes de séquences adaptatrices uniques
2. Ces deux méthodes ont leur avantages et leur inconvénients. La première méthode utilise très peu de volume réactionnel et est automatisée, ce qui limite la variabilité technique, contrairement à la seconde méthode. Cependant, le système C1 ne permet pas de mettre plusieurs conditions dans une plaque pour rendre compte de la variabilité inter-plaque. Seul un multi-marquage fluorescent préalable des cellules peut permettre de mélanger plusieurs conditions dans une plaque en passant par la technique C1. Une identification du marquage, et donc de la condition expérimentale de la cellule, se fait par l’image de la micro-puce entre l’isolation et la lyse.
est unique pour chaque cellule, ce qui facilite le regroupement des fragments d’ADNc par cellule
lors de la lecture des fragments. Les séquences UMI sont associées à chaque fragment d’ADNc
pour limiter le biais d’amplification. Enfin, la séquence polyT permet la fixation des ARNm par
complémentarité avec leurs queues polyA. Une fois la lyse des cellules commencée, les ARNm vont
se fixer à la surface des GEM par leur queue polyA. Les séquences barcode, UMI et ARNm sont
ensuite converties en ADNc. Les ADNc sont identifiés par leur barcode et regroupés par cellule. Ils
sont ensuite séquencés et alignés sur le génome de référence. Une matrice d’expression est ainsi
obtenue avec le nombre de fragments d’ADNc pour chaque gène et pour chaque cellule [228].
Tout comme la RTqPCR en cellule unique, cette technique permet, en plus de la mesure de
la SEG, la caractérisation de types cellulaires sans a priori et l’inférence de réseaux de régulation
génique (travaux en cours dans l’équipe).
Aujourd’hui, l’évolution impressionnante et la diversité de ces récentes techniques rendent
l’analyse de la variabilité de l’expression génique accessible à tous les chercheur.e.s.
1.1.3.3
Les limites et biais de mesure de la variabilité
Ces mesures d’expression génique peuvent comprendre des biais dus à la mesure propre ou à
l’hétérogénéité morphologique des cellules qui conduisent potentiellement à des quantités d’ARN
ou de protéines différentes, indépendantes de la SEG. Pour le premier cas, la mesure de la SEG
notamment par RTqPCR et séquençage à haut débit en cellule unique trouve sa limite dans
les gènes faiblement exprimés. En effet, les ARN en faible quantité sont difficilement capturés
par la reverse-transcriptase lors de la RT. Les zéros peuvent alors être de faux négatifs. Il a été
estimé que seulement 10 à 20% des ARN étaient détectés [99]. D’autre part, pour normaliser les
données en RTqPCR et en séquençage en cellule unique, des ARN synthétiques, appelés Spikes,
sont ajoutés de manière homogène dans chaque puit de cellule. L’utilisation de ces spikes pour
la normalisation des données sous-entend que ces ARN synthétiques sont amplifiés et séquencés
de la même manière que pour les ARN endogènes. Or, des études récentes montrent que ce n’est
pas toujours le cas [84]. Enfin, l’hétérogénéité de taille des cellules ou de leur position dans le
RTqPCR en cellule unique. D’un côté le système C1 qui permet une isolation, lyse,
RT et pré-amplification. De l’autre côté, le FACS qui permet l’isolation des cellules
(1 = Détecteurs des différents faisceaux de lumière pour recueillir des données sur la
morphologie des cellules ou leur fluorescence si les cellules sont préalablement
mar-quées ; 2 = Rupture du flux par vibration pour créer des gouttelettes qui contiennent
des cellules individuelles ; 3 = Les gouttelettes sont chargées selon les conditions
sé-lectionnées ; 4 = Champs électrostatique permettant le déplacement perpendiculaire
des gouttelettes ; 5 = Les gouttelettes non chargées (non sélectionnées) vont aux
déchets). Les étapes de lyse, RT et pré-amplification se font ensuite manuellement.
Ces deux étapes amènent à une plaque 96 puits avec dans chaque puit, les ADNc
d’une cellule. Ils sont ensuite quantifiés par le système Biomark qui donne accès à
une matrice de valeur d’expression de 96 gènes d’intérêt et pour les 96 cellules.
147, 23, 103, 143, 122]. De nombreuses études tentent de trouver des solutions pour mesurer ces
paramètres et intégrer leurs influences dans la mesure de la SEG [177, 23, 179, 207]. Cependant,
très peu de méthodes universelles (tous types cellulaires confondus) et non toxiques pour les
cellules existent dans la littérature. Je détaillerai nos recherches à ce sujet dans le chapitre 3.
1.1.3.4
L’analyse de la variabilité
Toutes ces techniques nous donnent accès à des distributions de la quantité d’ARN ou de
protéines, d’un gène ou de plusieurs gènes donnés, pour chaque cellule mesurée. Pour pouvoir
quantifier la SEG à partir de ces distributions, des assomptions ont été faites.
Les distributions Gamma
L’hypothèse dominante actuellement utilisée est que les
distribu-tions de valeurs d’expression des gènes suivent une loi de distribution Gamma [1, 76]. Les raisons
de cette approximation sont les suivantes : la transcription se fait de manière aléatoire durant
les bursts. La taille des bursts (durée ou intensité) est variable. Les distributions d’ARNm et
po-tentiellement celles des protéines prennent une forme de distribution Gamma, sous l’hypothèse
que les gènes sont plus souvent/longtemps dans un état inactif et que la dégradation des ARN
pendant un burst est faible [1].
Les outils de quantification
A partir de ces distributions d’expression, nombreux sont les
outils utilisés pour quantifier la SEG. Le plus connu est le coefficient de variation (
CV =
σμ)
[146]. Il représente la variance relative au nombre de produits du gène (protéines ou ARN) avec
σ l’écart-type de la distribution de la quantité de produits et μ sa valeur moyenne. En deuxième
position vient le Fano Factor (
F =
σμ2). C’est la variance de la distribution sur la moyenne des
valeurs d’expression. Dans la littérature, cet outil est souvent décrit comme pouvant quantifier la
différence entre une distribution donnée et une distribution qui suit la loi de Poisson [150, 222].
Il faut cependant faire attention à l’utilisation de ces deux outils. Ils n’évoluent pas de la même
manière selon la nature de la variation du bruit (fréquence ou taille des bursts). Par exemple,
d’après Victor Wong en 2018, si la variation du niveau de SEG provient d’un changement dans la
mais le coefficient de variation (ici noté comme une mesure du "bruit") ne va pas changer [222].
Un troisième outil de mesure de la SEG est la variance normalisée (
NV =
σ2μ2
). Elle représente la
variance de la distribution normalisée sur la moyenne au carré [211]. Plus récemment, un nouvel
outil est apparu dans la littérature, indépendant de la moyenne des valeurs d’expression : l’entropie
de Shannon. Initialement, l’entropie servait en physique pour mesurer le "désordre" d’un système.
Dans ce cadre, plus le désordre augmente, plus l’entropie augmente. En 1948, Claude Shannon
a repris cet outil pour l’appliquer dans le cadre de la théorie de l’information [185]. Dès lors,
cet outil est devenu une mesure de la quantité d’informations délivrée par un système. Plus le
message fourni dans chaque information (par exemple une suite de lettres alphabétiques) donnée
est le même, c’est à dire homogène, plus l’entropie est minimale. A l’inverse, plus le message
fourni est incertain (désordonné et aléatoire), plus l’entropie est grande (Figure 1.3). Il est utilisé
aujourd’hui pour la mesure de la SEG [168, 195]. De la même manière, plus l’expression d’un
gène donné entre cellules est homogène, plus sa valeur d’entropie est faible, et à l’inverse, plus ce
niveau d’expression est hétérogène et aléatoire, plus sa valeur d’entropie augmente. Par ailleurs,
un dérivé de l’entropie de Shannon, plus adapté à la biologie et notamment pour la comparaison
des genomes est le Jensen-Shannon-divergence (JSD) [188, 85]. Il permet de mesurer la différence
entre les distributions d’expression de gènes [47].
L’ensemble de ces outils de mesures de la SEG permettent aujourd’hui de quantifier la
varia-bilité de l’expression génique dans beaucoup de processus biologiques, améliorant notre
compré-hension des multiples rôles de la SEG.
1.1.4
Les rôles connus dans les processus biologiques
Il n’est aujourd’hui plus possible de nier l’implication de la SEG dans de nombreux processus
biologiques. Depuis l’acceptation de la théorie moderne du "non déterminisme" visant à montrer
que du désordre peut ressortir de l’ordre, la littérature à ce sujet a réellement explosé [59].
Les rôles biologiques de la stochasticité ont tout d’abord été démontrés chez les organismes
cellulaires procaryotes [109, 3] et plus récemment chez les eucaryotes [118, 93]. Ce que nous
AAAAAAAA
AAAA
BB
DD
A
B
DD
B
A
B
D
Forte entropie Entropie moyenne Entropie faibleA
B
D
A A
A
B
B
B
B
D
D
D
D
B
A
Figure 1.3 – Illustration de l’utilisation de l’entropie par Claude Shannon dans le
cadre de la théorie de l’information.
connaissons à l’heure actuelle, c’est que la SEG est impliquée à tous les niveaux. Elle se révèle
nécessaire lors des premiers stades de développement chez l’homme mais aussi dans d’autres
organismes [176]. Elle a aussi un rôle important lors des sélections adaptatives de différents types
de cellules allant de la bactérie aux cellules cancéreuses [3, 111, 159]. Plus récemment, son
implication dans le destin cellulaire [219, 174] ou encore la différenciation [168, 134, 181, 195]
a été mis en évidence mais son rôle précis reste encore très peu connu. Dans ce paragraphe, à
travers plusieurs exemples, l’étendue des rôles connus et des implications de la SEG dans plusieurs
processus biologiques vont être décrits.
Commençons par les premières étapes de la vie d’un organisme. Durant le développement
embryonnaire humain, les premières divisions des cellules souches possèdent un caractère
sto-chastique et mènent à la formation de tissus embryonnaires transitoires composés de cellules de
phénotypes variés. Pour permettre la formation des différents tissus qui composent notre
orga-nisme, une étape de réduction du bruit et de stabilisation des phénotypes (conservés pour la suite
du développement) est nécessaire [176]. Dans d’autres espèces comme C. elegans, il a été montré
que la SEG augmentait dans des nématodes portant des mutations sur les gènes impliqués dans le
développement intestinal. Cette hétérogénéité a pour but d’augmenter la variabilité du phénotype
concerné par la mutation dans la population ce qui rend la pénétrance de la mutation incomplète
[164]. Chez la drosophile, il a été montré l’existence d’un équilibre entre la stabilité de son
dé-veloppement (équilibre entre croissance cellulaire et prolifération) et des variations stochastiques.
ment, mène à une grande variabilité dans la taille des cellules des ailes de la drosophile, ayant
pour conséquence une augmentation de l’asymétrie phénotypique et de la variabilité individuelle
[49]. Chez les bactéries, comme chez d’autres espèces, la SEG peut servir de force motrice à leur
adaptation dans des milieux extrêmes. Cette stratégie, appelée bet-hedging, consiste à modifier
aléatoirement les phénotypes de chaque cellule au sein d’une même population pour assurer la
survie d’au moins quelques cellules, en cas de changement brutal de l’environnement, et donc de
la population à long terme [159]. C’est le même principe pour les bactéries qui se retrouvent dans
un milieu contenant un antibiotique [3]. De la même manière, l’augmentation du niveau de SEG
dans les cellules cancéreuses rend certaines cellules résistantes aux traitements. Ainsi, des cellules
résistantes vont survivre et se développer, tandis que les autres vont être éliminées par le
traite-ment [111, 121]. Dans ces exemples, la SEG permet d’enrichir le pool de phénotypes disponibles
à l’échelle d’une population ou d’un tissu à génome comparable. Cette variation, souvent due à
une augmentation du niveau de bruit dans l’expression des gènes, est une stratégie nécessaire à
l’adaptation et la survie des cellules face à leur environnement.
Le rôle de la variabilité de l’expression des gènes est également impliqué dans le destin cellulaire.
En 2012, il a été montré que la SEG augmentait précocement lors de la reprogrammation de
cellules différenciées en cellules souches pluripotentes induites [24]. Une telle implication dans le
destin cellulaire commence à voir le jour [29, 203]. Par exemple, en 1991, il a été montré que
dans la rétine de l’oeil de drosophile, la quantité de protéines Notch produite est très variable
d’une cellule à une autre. Selon le niveau de protéines Notch sécrété par une cellule, sa cellule
voisine pourrait alors soit devenir une cellule épidermique soit au contraire un neurone [75].
La SEG intervient également dans le destin des cellules immunitaires. Par exemple, la division
asymétrique des cellules immunitaires naïves lors d’une infection ne pouvait expliquer seule la
disparité phénotypique qui existe entre ces cellules une fois activées et différenciées. Il existe en
effet une dimension stochastique au cours de l’expansion clonale de ces cellules capable d’expliquer
leur hétérogénéité observée in vivo [62, 44, 63].
testé différentes molécules, initialement utilisées dans le domaine de la santé, sur leurs cellules
immunitaires humaines dans le but de sélectionner des substances capables de modifier le niveau
de variation du bruit [35]. Ces molécules ont été testées sur des lymphocytes humains infectés par
le virus de l’immunodéficience humaine (VIH). La conséquence majeure de ces traitements a été
d’augmenter la probabilité de réactivation du virus, en le "forçant" à sortir de sa phase latente.
Ici, il ne s’agissait pas de simplement constater l’implication de la SEG dans le destin du VIH,
mais de la modifier expérimentalement [165, 35]. Cette étude nous a ainsi ouvert les portes du
potentiel contrôle de certains processus biologiques par la variation expérimentale de la SEG.
Dans les processus de différenciation, nous savons depuis peu qu’il existe une variation du
niveau de SEG [134, 181, 195, 168]. Plus récemment, il a été aussi montré chez les cellules
souches embryonnaires de souris qu’une augmentation de l’hétérogénéité inter-cellulaire conduisait
à une réduction des capacités de pluripotence des cellules et à une augmentation de la probabilité
d’engagement dans un lignage [136]. Cependant, ce lien entre la SEG et d’autres processus de
différenciation reste peu décrit dans la littérature. Des théories sur la dynamique du processus de
différenciation et l’implication de la SEG commencent à être publiées [80]. Une des théories les
plus connues est la suivante : chaque cellule a une position dans un espace déterminée par le niveau
d’expression de chacun de ses gènes. Cet espace est donc un espace à n dimensions où n est égal
au nombre de gènes composant le système. Si le phénotype de ces cellules n’est pas transitoire,
elles se retrouvent alors dans un état stable, dit "attracteur" [79]. Donc des cellules d’une même
population, placées dans un environnement homogène, sont disposées non loin l’une de l’autre
dû à un niveau de SEG basal. Pour s’adapter à un nouvel environnement, une modification
du niveau d’expression de leurs gènes est nécessaire afin qu’elles puissent se déplacer dans cet
espace et explorer différentes directions
3. Cette phase de transition exploratoire nécessite donc une
augmentation de la SEG [96, 112, 227]. Les cellules expriment alors leurs gènes de manière plus
aléatoire les unes par rapport aux autres pour suivre chacune une trajectoire différente jusqu’au
3. Ces directions sont cependant contraintes par le réseau de régulation de gènes sous-jacent. Cette contrainte se traduit par le fait que les trajectoires restent limitées dans l’espace. Dans le cas d’un choix de lignage, cette contrainte est nécessaire pour qu’une cellule emprunte une trajectoire parmi toutes celles qui lui sont permises dans ce lignage et pas celles d’un autre lignage.