Etude de l'Interaction Polymère-Ecoulement
Texte intégral
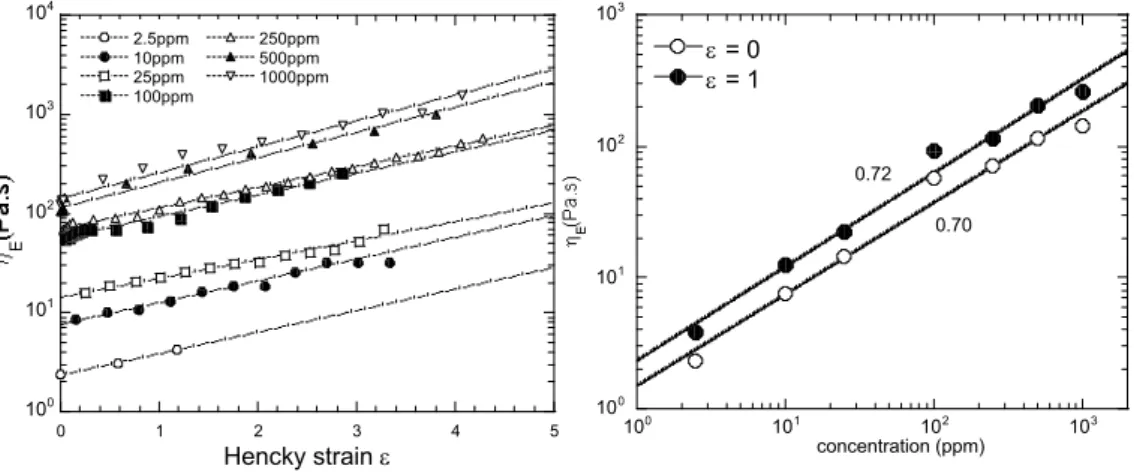
Figure





Outline
Documents relatifs
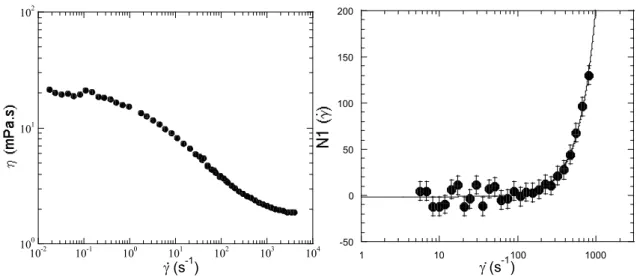
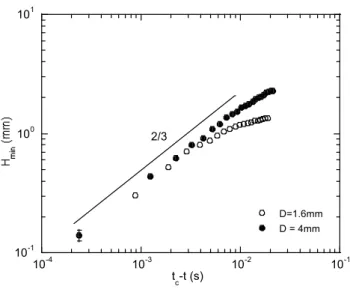
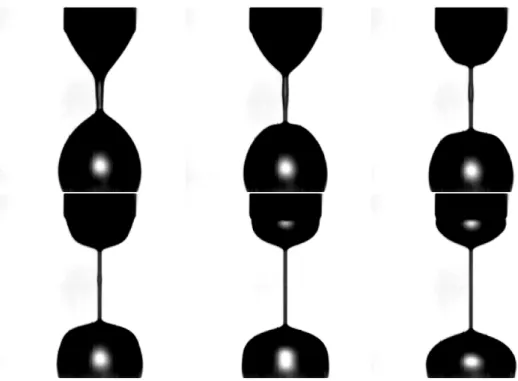
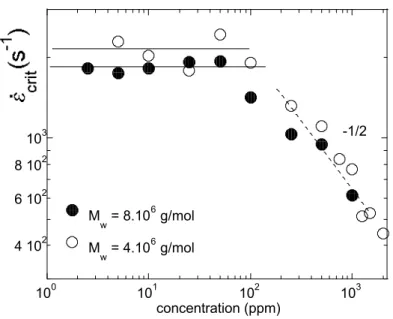
Etudions le comportement d’un polymère fluide simple (un fluide newtonien dont la viscosité ne dépend pas de la vitesse de cisaillement) soumis à une contrainte...
On ne voit pas du tout en quoi on peut détacher cette fonction du savoir de quelque chose qui en dernière analyse se décante de n’être que du – parce que ce n’est rien du tout,
Elles sont donc plus facilement assimilables par le vivant, ce qui explique pourquoi des plantes arrosées avec cette eau grandissent plus vite et plus haut que
Nous présentons dans ce travail une étude expérimentale montrant l’influence de l’ajout de polymère à une solution de tensioactifs sur la stabilité de la mousse et son
Reprenant l’idée d’une expérimentation de Kaneta et Murakami (1986), nous utilisons une machine bi- disques comportant un galet éprouvette fragile transparent, pour étudier
En outre, un pays ayant un niveau d’épargne élevé pourra bénéficier d’une croissance à long terme du PIB plus importante étant donné les ressources
Relaxation d’une chaîne de polymère enfermée dans un gel subitement
Pour tous les figures, nous remarquons une décroissance linéaire de la température en fonction de la dimension de la cellule L dans le domaine occupé par la matrice (PP).Cette
