Computational analysis of cell-cell communication in
the tumor microenvironment
MASSACHUSETS INSTITUTE
by
OFTECHNOLOGYManu Prajapati
Kumar
JUL
03
2019
B.S., University of Illinois (2014)
LIBRARIES
ARCHIVES
Submitted to the Department of Biological Engineering
in partial fulfillment of the requirements for the degree of
Doctor of Philosophy
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
June 2019
@
Massachusetts Institute of Technology 2019. All rights reserved.
Signature redacted
A u th o r ...
Department of Biological Engineering
May 9, 2019
Certified by...Signature
redacted...
Douglas A. Lauffenburger
Professor of Biological Engineering
Thesis Supervisor
Signature redacted
Accepted by ...
Forest M. White
Chairman, Graduate Program Committee
Computational analysis of cell-cell communication in the
tumor microenvironment
by
Manu Prajapati Kumar
Submitted to the Department of Biological Engineering on May 9, 2019, in partial fulfillment of the
requirements for the degree of Doctor of Philosophy
Abstract
Cell-cell communication between malignant, immune, and stromal cells influences many aspects of in vivo tumor biology, including tumorigenesis, tumor progression, and therapeutic resistance. As a result, targeting receptor-ligand interactions, for instance with immune check-point inhibitors, can provide significant benefit for pa-tients. However, our knowledge of this complex network of cell-cell interactions in a tumor microenvironment is still incomplete, and there is a need for systematic approaches to study cell-cell communication.
This thesis presents computational approaches for characterizing cell-cell com-munication networks in three different experimental studies. In the first study, we modeled metastatic triple negative breast cancer in the liver using a microphysiolog-ical system and identified inflammatory cytokines secreted by the microenvironment that result in the proliferation of dormant metastases. In the second study, we used single-cell RNA sequencing (scRNA-seq) to quantify receptor-ligand interactions in six syngeneic mouse tumor models. To identify specific receptor-ligand interactions that predict tumor growth rate and immune infiltration, we used receptor-ligand in-teractions as features in regression models. For the third study, we extended our scRNA-seq approach to include inferences of single-cell signaling pathway and tran-scription factor activity. We then identified protein-protein interaction networks that connect extra-cellular receptor-ligand interactions to intra-cellular signal transduction pathways. Using this approach, we compared inflammatory versus genetic models of colorectal cancer and identified cancer-associated-fibroblasts as drivers of a partial epithelial-to-mesenchymal transition in tumor cells via MAPK1 and MAPK14 signal-ing.
Overall, the methods developed in this thesis provide a foundational computa-tional framework for constructing "multi-scale" models of communication networks in multi-cellular tissues.
Thesis Supervisor: Douglas A. Lauffenburger Title: Professor of Biological Engineering
Acknowledgments
I am thankful to many people who made my graduate studies at MIT not only a
time of personal and professional growth, but also an enjoyable period of time I will remember fondly.
First and foremost is my advisor, Douglas Lauffenburger. From our first conversa-tion during interviews until completing my Ph.D., Doug has always made me feel like a valued student in the department and member of his lab. I have appreciated the freedom Doug has given me to explore research directions and pursue personal inter-ests, all the while knowing that Doug is ready to provide support and guidance at a moments notice. I am also immensely grateful for the connections Doug has provided me throughout my graduate studies. I have been fortunate to have the opportunities to travel to numerous conferences and visit international collaborators.
I would also like to thank members of my thesis committee: Forest White, Linda
Griffith, and Andreas Raue. My thesis committee meetings were always a positive source of encouragement and intellectual insight. I'd like to especially thank Andreas for bringing me on as an intern at Merrimack Pharmaceuticals during the summer of 2017. I enjoyed working alongside Andreas and have appreciated his continued mentorship throughout my graduate studies.
I'd also like to thank past and present members of the Lauffenburger and Griffith
labs. My decision to join the lab was in large part due to the many alumni and current students who took the time to meet with me and provide guidance on choosing a lab. Throughout the Ph.D., I have enjoyed the continuous support of past and present lab members, and my intellectual and personal growth is largely due to our daily interactions.
I'd like to thank my classmates in the BE-2014 cohort. I consider myself fortunate
to be part of such a talented group of people. I am constantly inspired by their many accomplishments, both in and out of the lab, and have enjoyed being part of such a good-natured group of people.
ge-ographically, the messages and encouragement from afar have always been a welcome source of comfort. In particular, I'd like to thank my parents and my brother, who have always supported me in all that I do.
Finally, I'd like to thank Isha for her daily support both before and throughout graduate school.
This doctoral thesis has been examined by a Committee of the
Department of Biological Engineering as follows:
Professor Forest M . W hite ...
Chairman, Thesis Committee
Professor of Biological Engineering
Professor Douglas A. Lauffenburger.
Thesis Supervisor
Professor of Biological Engineering
Professor Linda G . G riffith...
Member, Thesis Committee
Haslam and Dewey Professor of Biological Engineering
A nd reas R aue ...
Member, Thesis Committee
Director, Merrimack Pharmaceuticals
Contents
1 Introduction 17
1.1 Cell-cell communication plays a key role in health and disease . . . . 17
1.1.1 Mechanisms of cell-cell communication . . . . 18
1.1.2 Cell-cell communication activates intra-cellular signaling to
al-ter cellular behavior . . . . 21
1.1.3 Experimental systems for studying cell-cell communication . . 25
1.1.4 Techniques for measuring signal transduction in multiple cell
types... ... 26
1.2 Computational modeling of cell-cell communication . . . . 28
1.2.1 Integrating data-driven and mechanistic modeling approaches
using prior knowledge . . . . 29
1.2.2 Multiscale modeling approaches are necessary for modeling
cell-cell communication . . . . 30
1.3 Overview of thesis . . . . 32
2 A model of dormant-emergent metastatic breast cancer progression
enabling exploration of biomarker signatures 35
2.1 A bstract . . . . 36
2.2 Introduction . . . . 37
2.3 R esults . . . . 39
2.3.1 Recapitulation of dormant-emergent breast cancer metastasis
2.3.2 Differential signaling profiles of dormant and outgrowing metastatic
niches in the liver MPS . . . . 42
2.3.3 Distinct functional pathways are activated within each metastatic
niche in the liver MPS . . . . 46
2.3.4 Univariate analysis distinguished candidate biomarkers and
sig-natures of dormant and outgrowing metastatic stages 49
2.3.5 Potential diagnostic decision tree models of signaling proteins
accurately stage the metastatic niche of the liver MPS . . . . 51
. . . . 51 . . . . 57 . . . . 64 2.4 D iscussion . . . . 2.5 Supplementary Figures . . . . 2.6 Experimental Procedures . . . . 2.6.1 Cell Sources . . . . 2.6.2 Ex Vivo Hepatic MPS . . . .
2.6.3 Clinical Chemistry Assays . . . .
2.6.4 Cytochrome P450 Assay . . . .
2.6.5 Multiplex Immunoassays . . . .
2.6.6 Cancer Cell Detection and Quantification .
2.6.7 Click-iT PLUS EdU Assay . . . .
2.6.8 Ingenuity Pathway Analysis (IPA) Bioinformatics
2.6.9 2.6.10 Analysis . . . 64 . . . 64 . . . 65 . . . 65 . . . 66 . . . 66 . . . 67 ;.. 67
Experimental Design and Statistical Rationale of Analyses Statistics. . . . .
3 Computational analysis of single-cell RNA-seq identifies cell-cell com-munication associated with tumor characteristics
3.1 A bstract . . . .
3.2 Introduction . . . .
3.3 R esults . . . .
3.3.1 Single cell RNA sequencing of syngeneic mouse tumor models
3.3.2 Classification of cell types based on scRNA-seq data of syn-geneic mouse tumor models . . . .
67 68 69 70 70 72 72 75 . . . .
3.3.3 Scoring cell-cell interactions using known ligand-receptor
inter-actions . . . . 77
3.3.4 Associating cell-cell interaction scores with phenotypes of interest 80 3.3.5 Quantifying interactions in human metastatic melanoma . . . 83
3.4 D iscussion . . . . 87
3.5 Supplementary Figures . . . . 91
3.6 M ethods . . . . 97
3.6.1 Anim al models . . . . 97
3.6.2 C ell lines . . . . 97
3.6.3 Single-cell RNA sequencing of mouse syngeneic tumor models 98 3.6.4 Comparison of scRNA-seq and flow cytometry . . . . 99
3.6.5 Single cell RNA sequencing data processing . . . . 99
3.6.6 Determining gene markers for syngeneic tumor models . . . . 100
3.6.7 Fitting Gaussian mixture models to determine marker expression100 3.6.8 Training the decision tree classifier . . . . 100
3.6.9 Calculating ligand-receptor interaction scores . . . . 101
3.6.10 Human to mouse homolog conversion . . . . 102
3.6.11 Analysis of human metastatic melanoma . . . . 102
3.6.12 LASSO regression models . . . . 102
3.6.13 Computing correlations of randomized interaction scores . . . 102
4 Computational approaches to identify multi-scale cell-cell interac-tions in mouse models of colorectal cancer using scRNAseq 105 4.1 A bstract . . . . 106
4.2 Introduction . . . . 106
4.3 R esults . . . . 108
4.3.1 Single cell RNA sequencing of mouse models of colorectal cancer108 4.3.2 Classification of cell types based on scRNA-seq data of syn-geneic mouse tumor models . . . .111
4.3.3 Scoring cell-cell interactions using known ligand-receptor
inter-actions . . . . 113
4.3.4 Identifying signaling pathway activity and transcription factory activity from scRNA-seq data . . . . 118
4.3.5 Inferred intra-cellular singaling network identifies active MAPK1 and M APK 14 . . . . 121
4.4 D iscussion . . . . 124
4.5 Supplementary figures . . . . 127
4.6 M ethods . . . . 134
4.6.1 Single-cell RNA sequencing mouse models of colorectal cancer 134 4.6.2 Single cell RNA sequencing data processing . . . . 134
4.6.3 Quantification of gene expression programs . . . . 135
4.6.4 Fitting Gaussian mixture models to determine marker expression135 4.6.5 Training the cell type classifier . . . . 135
4.6.6 Calculating ligand-receptor interaction scores . . . . 136
4.6.7 Human to mouse homolog conversion . . . . 136
4.6.8 Computing PROGENy pathway activation scores . . . . 136
4.6.9 Estimating transcription factor activity using DoRothEA . . . 136
4.6.10 Identifying cell-signaling networks using CARNIVAL . . . . . 137
4.7 Data and software availability . . . . 137
5 Conclusions and Future Perspectives 139 5.1 Advancing the use of MPSs for studying cell-cell communication . . 139
5.2 Improving models of cell-cell communication using single cell RNA sequencing . . . . 142
5.2.1 Cell type identification and classification . . . . 142
5.2.2 Improving the accuracy of single cell RNA inferences . . . . . 143
List of Figures
1-1 Forms of inter-cellular signaling . . . . 20
1-2 Different kinds of intra-cellular signaling proteins along a signaling
pathway from a cell-surface receptor to the nucleus . . . . 24
2-1 Modeling dormant-emergent metastatic breast cancer in the hepatic
n ich e . . . . 4 1
2-2 Signaling profiles from growing, dormant and emergent metastatic breast
cancer cells in the hepatic niche . . . . 45
2-3 Activated biological pathways in the dormant-emergent metastatic MPS
identified by IPA . . . . 48
2-4 Candidate biomarkers and decision trees discerning metastatic breast
cancer niches in the hepatic MPS . . . . 50
2-Si Patient donor characteristics and baseline metrics of hepatic tissue . . 57 2-S2 MDA-MB-231 metastatic hepatic niche prior to and after
chemother-apeutic treatm ent . . . . 58
2-S3 Secondary treatment of emergent metastatic MDA-MB-231 cells with
the same (1 pM doxorubicin) or different chemotherapy (20 pM cisplatin) 59 2-S4 Scatterplots comparing signal abundance across the hepatic, growing
metastatic, dormant metastatic and emergent metastatic niches from
the 27- plex and Cancer Panel 2 kits . . . . 60
2-S5 Scatterplots comparing signal levels across the hepatic, growing metastatic,
dormant metastatic and emergent metastatic niches from the 40-plex
2-S6 Heatmaps depicting individual signal abundance in the hepatic,
grow-ing metastatic, dormant metastatic and emergent metastatic niches . 62
2-S7 Additional activated biological pathways in the dormant-emergent metastatic
M PS identified by IPA . . . . 63
3-1 t-SNE visualization of single cell sequencing data and cell type . . . . 74
3-2 Quantification of cell-cell interactions occurring in the tumor . . . . . 80
3-3 Interaction scores correlate with relevant characteristics of the tumor m icroenvironm ent . . . . 83
3-4 Assessing cell-cell interactions occurring in human metastatic melanoma 87 3-Si Single cell RNA sequencing of mouse syngeneic tumors and comparison to flow cytom etry . . . . 91
3-S2 Classification of cell types based on scRNAseq data of syngeneic mouse tum or m odels . . . . 93
3-S3 Characterizing known receptor ligand cell-cell interactions and their association with phenotypes of interest . . . . 95
3-S4 Analysis of interactions present in human metastatic melanoma . . . 97
4-1 Single-cell RNA sequencing of mouse colorectal tumors and identifica-tion of cell types . . . . 110
4-2 Identifying differences in receptor-ligand interactions between AOM and APC tumor models . . . . 117
4-3 Identifying differences in signaling pathway and transcription factor activity in tum or cells . . . . 120
4-4 Intracellular signaling network downstream of cell-cell communication 123 4-Si Sequencing metrics and quality control . . . . 127
4-S2 Expression of marker genes and gene signatures . . . . 128
4-S3 Training the cell type classifier . . . . 129
4-S4 pCreode trajectory analysis of wild-type epithelial cells . . . . 130
4-S5 Inferred receptor-ligand interactions for non-tumor cells . . . . 131
4-S7 DoRothEA inferences of transcription factor activity . . . . 133
Chapter 1
Introduction
1.1
Cell-cell communication plays a key role in health
and disease
A human contains trillions of cells that are traditionally divided into approximately
200 loosely defined "cell types" (e.g. immune cells, neurons, epithelial cells etc.) based on morphological, molecular, and functional properties [1, 2, 3, 4]. With few exceptions, all cells in human are genetically identical; however, each cell type ex-presses only a subset of its genes, which leads to the observed differences among each cell type [5]. Distinct regulation of gene expression by different cell types enables each cell type to have specialized functions, and this specialization requires that cells communicate with one another in order to coordinate these diverse functions. The importance of cell-cell communication is evident in numerous aspects of in vivo hu-man biology, such as the coordination among immune cells to fight infection and the differentiation and proliferation of stem cells during development. For example, cells of the immune system can recruit and activate other immune cell types by secreting cytokines and chemokines [6]. In healthy colon, tissue pericryptal fibroblasts pro-vide niche factors that promote stem cell maintenance and renewal, as well as direct differentiation along the epithelial lining [7].
dysreg-ulation of cell-cell communication is also involved in many human diseases. Human cancers in vivo do not exist as homogeneous collections of cancer cells, but rather as heterocellular systems that contain a mixture of different cell types [8, 9]. This mixture of cells, as well as the signaling molecules and extracellular matrix (ECM) surrounding the cells, collectively forms the tumor microenvironment (TME). Re-search has shown that the tumor microenvironment plays a role in many aspects of cancer biology, including carginogenesis [10], angiogenesis [11], metastatic seeding [12], and response to therapy [13]. Therefore, understanding how cell-cell commu-nication is dysregulated is important for understanding both the pathogenesis and treatment of disease.
Because aberrant cell-cell communication underlies numerous aspects of disease bi-ology, many recent therapeutics now target molecules involved in dysregulated cell-cell communication. This new therapeutic approaches can either target a specific cell-cell interaction or may even aim to deplete an entire cell type involved in dysregulated communication. For example, cancer immunotherapies target specific interactions be-tween tumor cells and immune cells that prevent the immune system from responding to the tumor [14, 15]. In Crohn's disease, anti-TNFa therapies aim to deplete an in-flammatory cytokine produced predominantly by monocytes, macrophages, and T lymphocytes that activate T cells [16]. While these therapeutic examples underscore the value of targeting cell-cell interactions, our understanding of cell-cell communica-tion in disease is still limited. Therefore, there is a need for approaches that enable the study of cell-cell communication in a generalizable manner across diseases.
1.1.1
Mechanisms of cell-cell communication
Given the importance of inter-cellular communication (i.e. cell-cell communication), cells have numerous mechanisms for sending and receiving messages. Cells send sig-nals in a variety of biochemical forms, including small-molecules, metabolites, hor-mones, peptides, fatty acid derivatives, and secreted proteins (e.g. growth factors)
[17]. These signaling molecules are typically secreted extra-cellularly; however, in
Collec-tively, these many forms of signaling molecules are commonly referred to as ligands. While there are many possible biochemical forms of signals, cells primarily sense signals from their environment via protein receptors expressed on the cell surface membrane. Receptors are often transmembrane proteins that initiate intra-cellular signaling upon binding of an extra-cellular signal [181. However, in some cases recep-tors remain bound to the intra-cellular nuclear membrane and primarily respond to small, hydrophobic signaling molecules capable of diffusing across the cell membrane. In addition to characterization based on the biochemical form of signaling molecule, cell-cell communication via secreted factors is also characterized based on the the lo-cation of target cell relative to the sending cell (Figure 1-1) [17, 19]. Endocrine signaling, in which the signaling molecules are generally hormones, occurs when the signal is transported via the circulatory system. Paracrine signaling refers to signal-ing that occurs between cells that are in close proximity and the ligand can reach its target via diffusion rather than relying on the circulatory system. Prominent exam-ples of paracrine signaling include the release of neurotransmitters from one neuron to a neighboring neuron and the secretion of growth factors during development. Autocrine signaling occurs when cells send signals that they themselves receive, a
signaling mechanism that is often used to maintain a particular cellular behavior
117].
Cells also communicate via two forms of contact-dependent mechanisms: 1) physical contact with other cells, and 2) contact with the surrounding ECM. Cell-cell contact communication occurs via membrane bound ligand and receptor interactions or via cellular junctions (adherens junctions, gap junctions, and tight junctions) [20, 21, 22]. For example, nucleated cells express MHC-I receptors that interact with T cell re-ceptors to activate an immune response. Communication dependent on interactions with the ECM occurs due to the depostion of ECM components, such as collagens, laminins, and fibronectin, which signal to surrounding cells that express adhesion re-ceptors, such as integrin receptors [23]. Both of these contact dependent signals play an important role in tissue development by inhibiting proliferation when dividing cells come into contact with either neighboring cells or surrounding ECM [24, 25].
JA) CONTACT-DEPENOENT
signelkng cell tacget cell
rembcane-toound signol molecule (81 PAAEINE localcel rmditc 4C) SYNAPTIC (D) ENDOCRINE
endocr necaN receptor
target mll synapse neuron axon o cell neurotransmitter body bloodstrearn target Cel:
Figure 1-1: Forms of inter-cellular signaling. Reproduced from [17]. (A) Contact-dependent signaling requires cells to be in direct membrane-membrane contact. (B) Paracrine signaling depends on signals that are released into the extracellular space and act locally on neighboring cells. (C) Synaptic signaling is performed by neurons that transmit signals electrically along their axons and release neurotransmitters at synapses, which are often located far away from the cell body. (D) Endocrine signal-ing depends on endocrine cells, which secrete hormones into the bloodstream that are then distributed widely throughout the body. Many of the same types of signaling molecules are used in paracrine, synaptic, and endocrine signaling; the crucial dif-ferences lie in the speed and selectivity with which the signals are delivered to their targets.
1.1.2
Cell-cell communication activates intra-cellular
signal-ing to alter cellular behavior
Complicating our ability to understand cell-cell communication is the fact that a cell is exposed to numerous ligands simultaneously. Recent studies have identified approx-imately 600 distinct protein ligands alone and over 500 corresponding receptors [26]. The combinatorial effects of these receptor-ligand interactions enables a wide-variety of cellular responses. For example, receptor-ligand interactions may elicit additive, redundant, or even opposing effects on cellular behavior [27]. Furthermore, different cell types respond differently to the same mixture of signaling molecules depending on the profile of receptors expressed on the cell surface. However, despite the number of receptors and ligands involved in communication, receptor-ligand interactions all act via a limited set of well-conserved signaling pathways [28]. Therefore, understand-ing which pathways are actively signalunderstand-ing within a cell provides insight into how the spectrum of receptor-ligand interactions occurring at a given moment are combining to affect intra-cellular behavior.
Receptors typically undergo conformational changes upon extra-cellular ligand binding that activate intra-cellular signaling pathways [28]. In general, signals are propagated through signaling pathways via protein phosphorylation cascades that eventually lead to the activation of transcription factors [291. These activated tran-scription factors then alter gene expression patterns by promoting or repressing the transcription of their target genes [30]. Ultimately, this change in gene expression drives cellular behavior such as proliferation, apoptosis, or migratrion.
Given the importance of signaling pathways in controlling cellular behavior, many pathways are named after the cellular behavior the pathway was first discovered to
elicit. For example, the transforming growth factor
#
superfamily include the ligandsTGFO-1, which was discovered based on its ability to "transform" cultured cells
into a malignant phenotype, and the bone morphogenetic protein (BMP-7), which was identified based on its ability to induce bone formation [31]. Receptors in the
transcription factors, which are tightly associated to the intracellular domain of TGF
#
receptors [32]. SMAD transcription factors then translocate to the nucleus, leading to transcriptional changes in the cell that result in changes to phenotypic behavior[33].
In the context of the immune system, the JAK-STAT pathway plays a central role in communication mediated by cytokines. The binding of ligand to cytokine receptors activates JAK kinases, which are physically associated with the cytosolic domain of cytokine receptors [34]. JAK kinases activation results in phosphorylation of STAT transcription factors, which also translocate to the nucleus and elicit altered cellular behavior [35, 36]. For example, the cytokines granulocyte colony stimulating factor
(G-CSF) and erythropoietin induce proliferation of bone marrow progenitor cells into
granulocytes and erythrocytes, respectively.
Also within the context of the immune system, cell-cell communication via se-creted chemokines drives the recruitment of immune cells (i.e. chemotaxis) to specific locations within the body. Chemokines signal via their cognate receptors, which are members of the G protein-coupled receptor (GPCR) family. GPCRs all function
by activating trimeric G proteins bound to the intra-cellular portion of the receptor [37]. These membrane bound G-proteins activate phosphoinositide 3-kinase (P13K)
and phosphoinositide-specific phospholipase C (PLC)
138].
This cascade results inproduction of secondary messenger molecules, including diacylglycerol (DAG) and
inositol-trisphosphate(IP 3) [39]. These secondary messengers induces a variety of
ef-fects, including activation of protein kinase C and realase of intracellular Ca2+, which ultimately induces chemotaxis.
Another family of cell-surface receptors, receptor tyrosine kinases (RTKs), are commonly involved in the response to secreted growth factors during development. Rather than activate a protein kinase bound to the intra-cellular domain of the recep-tor, RTKs contain a kinase domian within the intra-cellular segment of the receptor [40]. RTKs commonly activate the Ras/MAPK pathway, which consists of a cascade of protein kinases rather than the direct activation of a transcription factor [41, 42]. Members of the MAPK signaling cascade include Jun N-terminal kinases (JNKs),
p38 kinases, and ERK [43]. As before, activation of the MAPK pathway results in the activation of many transcription factors, such as ternary complex factor (TCF) and serum response factor (SRF), that mediate the phenotypic response to RTK activation.
Other well-conserved signaling pathways include the Wnt signaling pathway, which signals via Frizzled receptors and results in the activation of TCF via ,3-catenin. The Wnt signaling pathway plays a key role in the regulation of epithelial differentia-tion, and mutations that cause activation of Wnt signaling lead to the development of colrectal cancers. The NF-rB pathway is activated in immune cells downstream of inflammatory cytokines such as TNFa and interleukin 1 (IL-1). Unlike other pathways, ligand binding to receptors upstream of the NF- r'B pathway causes phos-phorylation of an inhibitor of the NF-rB transcription factor [44]. Phosphos-phorylation of the inhibitor results in its degradation, which enables NF- ,'B to translocate to the nucleus and exert its effect on cellular behavior.
General principles of signaling by cell-surface receptors 0~ 0 0 0@0 0 lpuEgeli
*W
0 on 0 0 Coll surface recepto'r Respenlug eel Modification of Modification of cellular metabolism, gene expression, function, movement development(1) Synthesis and (2) release of signaling molecules by the signaling cells;
(3) Transport of the signal to the target cells; (4) Binding of the signal by a specific receptor protein;
(5) Initial of intracellular signal transduction pathways;
(6) Specific changes in cellular functions; (7) Inactivation of the receptor;
(8) Removal of signaling molecules
Figure 1-2: Different kinds of intra-cellular signaling proteins along a sig-naling pathway from a cell-surface receptor to the nucleus Reproduced from
[28]. In this example, a series of signaling proteins and small intracellular mediators
relay the extracellular signal into the cell, causing a change in gene expression. The signal is amplified, altered (transduced), and distributed en route. Many of the steps can be modulated by other extracellular and intracellular signals, so that the final result of one signal depends on other factors affecting the cell. Ultimately, the sig-naling pathway activates (or inactivates) target proteins that alter cell behavior. In this example, the target is a gene regulatory protein.
1.1.3
Experimental systems for studying cell-cell
communica-tion
Despite the understanding that cell-cell communication plays an important role in hu-man biology, we are still limited by techniques for studying cell-cell communication. The primary complication is that hetero-cellular systems can not be accurately stud-ied by breaking up the system into its individual components (i.e. cell types). Put
another way, multi-cellular systems are non-linear, such that f(a)
+
f(b) # f(a + b).Rather, communication between cell types results in "emergent" behavior that would not be seen by studying each individual cell type in isolation [45, 46]. Therefore, stud-ies of cellular communication require either complex experimental systems containing multiple cell types or in vivo studies.
The increased appreciation of cell-cell communication has led to the development of numerous platforms for studying the heterocellular systems. While primary tis-sue samples or data gathered from clinical trials provide in vivo relevance, in vitro systems often provide a more accessible and higher throughput method for studying
heterocellular interactions
[47].
However, an important consideration whendevelop-ing in vitro systems is the relevance to in vivo biology, as in vitro finddevelop-ings do not always translate in vivo. As a result, many in vitro methods attempt to construct systems with cells in 3D matrices as opposed to more common 2D tissue culture and often utilize primary cells as opposed to immortalized cell lines.
Numerous methods exist for constructing 3D models of in vivo systems.
Nat-urally derived
/decellularized
matrices, such as collagen and Matrigel, can capturethe complexity of in vivo tissue matrices; however, native matrices are difficult to tune in a modular fashion and variability between matrices limits the ability to gain mechanistic insight [48]. In contrast to naturally derived matrices, synthetic matrices provide a tunable framework for constructing in vitro 3D microenvironments [49, 50]. In particular, synthetic hydrogels are a well-established model for tissue engineering applications. By using crosslinkers that are degradable by secreted matrix metallo-proteinases (MMPs), cells can cleave the hydrogel and reform the surrounding matrix.
Furthermore, synthetic hydrogels enable modular control of matrix stiffness and cell adhesion by incorporating adhesion peptides (e.g. synKRGD binding motif) [51, 52]. However, one limitation of both natural and synthetic matrices is the difficulty of accessing cells once they have been encapsulated within the gel.
Beyond 3D extracellular matrices, recent work has also focused on developing microphysiological systems (MPS) that aim to more accurately mimic the structure and function of specific human tissues or organ regions. MPSs typically use primary human cells in order to accurately recapitulate in vivo biology, although induced pluripotent stem cells and immortalized cell lines may be used as well [531. These systems allow for precise control of cellular, physical, chemical microenvironment via microfluidic modulation. For example, a LiverChip MPS designed for modeling the liver microenvironment uses a scaffold placed in a perfuseable bioreactor to mimic the structure of the liver sinusoid [54, 55]. The liver MPS is an all-human system that utilizes freshly isolated donor-matched hepatocytes and non-parenchymal cells
(NPCs) from partial hepatectomies.
While both 3D matrices and ex vivo MPSs provide an excellent experimental plat-form to study cell-cell communication, both approaches still have several drawbacks. While it is easy to assay secreted ligands in the extra-ceullular media or circulating effluent from these systems, it is difficult to attribute the presence of a ligand to a specific cell type. Furthermore, it is difficult to perform cellular assays using these systems, which makes measurements on intra-cellular signaling activities challenging.
1.1.4
Techniques for measuring signal transduction in multiple
cell types
Numerous methods exist for measuring signaling events from multiple cell types. Per-haps the simplest is to keep cells physically separated during the experiment and mea-sure signaling events in each cell type individually. Certain experimental setups, such as conditioned media and transwells, utilize this approach; however, these approaches prohibit the study of contact-dependent signaling [56]. Therefore, it is beneficial to
use systems in which multiple cell types are cultured together and physically isolate cell types post hoc. Fluorescence-activated cell sorting (FACS) can separate mixed populations of cells via fluorescent labels. However, FACS requires the generation of single cell suspensions, which may induce time lag and alter signaling states between the experimental end point and measurement. Furthermore, spectral overlap limits
the number of measurable quantities to < 20 [57]. To improve the number of
mea-surable parameters, mass cytometry methods use heavy-metals to label antibodies,
which are well-resolved when analyzed via mass spectrometry.
[58]
However, bothmethods are still limited by the inability to measure molecular signaling events at high throughput, and require extensive validation of antibody panels.
While FACS and CyToF both rely on antibodies for accurate detection of signaling events, mass spectrometry approaches detect signaling events in a sample at much higher throughput. To distinguish signaling events from different cell populations, mass spectrometry approaches rely on mass differences between isotopically labeled amino acids. One approach, stable isotope labeling by amino acids in cell culture
(SILAC), uses cells grown on isotopically "light" or "heavy" amino acids to label all
the proteins within a given cell type based on mass [59]. Another approach, cell type-specific labeling using amino acid precursors (CTAP), uses non-mammalian enzymes expressed in distinct cell types to convert isotopically heavy or light precursors into L-lysine [60]. This approach was recently used to identify cell type specific changes in phosphoprotein activation that occur due to interactions between pancreatic ductal adenocarinoma cells and pancreatic stellate cells [61]. While these approaches are incredibly powerful for deriving insights from experimentals using cell lines, it is not yet possible to use these mass spectrometry approaches for in vivo studies.
In recent year, single cell technologies have enabled the measurement of molecular features from individual cells [62, 63, 64]. Single-cell technologies enables the reso-lution of individual cell types based on the measured molecular features, and have been used extensively to refine our definitions of cell types and discover heterogeneity within cell types. However, beyond characterization of cell types, this technology also enables cell type specific measurements of receptor and ligand expression involved in
cell-cell communication in an in vivo context. However, the ligands assayed by single cell sequencing technologies are limited to protein products measurable via mRNA, and does not include small molecules, peptides, or hormones. Furthermore, it is not straightforward to assess intra-cellular signaling from measurements of mRNA. Therefore single cell technologies provide a valuable data source for the examining cell-cell communication, but we still lack the computational methods to fully utilize this rich data source.
1.2
Computational modeling of cell-cell
communica-tion
Computational models aim to mathematically replicate and describe the behavior of biological systems. By acheiving this goal, computaional models provide the abil-ity to test proposed biological mechanisms at a larger scale than with experimental approaches alone [65]. As a result, computational models can inform the design of additional experimental studies and provide a framework to interpret biological data
by demonstrating if a proposed theoretical model is consistent with experimental
data. Therefore, constructing computational models of cell-cell communication can help guide the discovery of underlying principles that govern cell-cell interactions.
Despite the potential utility of computationally modeling cell-cell communication, it is still unclear what the appropriate mathematical and computational frameworks are for modeling cell-cell communication networks. In particular, two main challenges must be addressed: 1) the modeling framework should account for the "multi-scale" effects of cell-cell communiction, and 2) the framework should integrate mechanistic and data-driven modeling approaches. Multi-scale modeling approaches are neces-sary to account for not only extra-cellular receptor-ligand interactions, but also the effect of receptor-ligand interactions on intra-cellular signal transduction pathways and cellular behavior. In addition, there is currently a trade-off between models that account for the full spectrum of receptor-ligand interactions and models that provide
mechanistic biological insight. In the context of computational modeling, this chal-lenge is represented by the trade-off between data-driven and mechanistic modeling approaches that can be addressed by using "prior knowledge".
1.2.1
Integrating data-driven and mechanistic modeling
ap-proaches using prior knowledge
Mechanistic modeling approaches specifically incorporate biological information, such as known biochemical reactions, when defining model structure [66, 67]. The structure of these models is often represented in the form of ordinary or partial differential equa-tions that correspond to known biological reacequa-tions. Because these models correspond to known physical processes, interpretation of these models can provide mechanistic and casual understanding of the system under examination. However, a limitation of mechanistic modeling approaches is the amount of prior knowledge required to accu-rately parameterize and structure these models. Therefore, to define a mechanistic model that sufficiently incorporates the hundreds of known receptor-ligand interac-tions at sufficient mechanistic detail for each cell type would require an intractable amount of experimental prior knowledge. Furthermore, as the number of species un-der investigation increases, mechanistic models can quickly become computationally intractable.
Data-driven models offer an alternative approach to mechanistic modeling. These models don't require knowledge of biochemical interactions between components, but rather learn statistical relationships between components. Depending on the form of the model and the data available for model fitting, these statistical relationships can take the form of correlations, conditional probability distributions, etc [68, 69]. Given a sufficient amount of data, assuming the data includes the most relevant features, the hope is that data-driven approaches can capture mathematical relationships between variables. However, the relationships learned by data-driven models, such as correla-tions, do not necessarily have biological meaning or correspond to physical properties of the system under examination. Therefore, it can be difficult to gain insight into
biological mechanisms when interpreting data-driven models.
Mechanistic modeling approaches and data-driven approaches are not mutually exclusive [70, 71]. Previous research has extensively detailed relationships between molecular species as well as annotated associations of specific molecules with biolog-ical mechanisms and functions. For example, numerous studies have attempted to catalog the target genes of known trancription factors [72]. Therefore, when analyzing a data-driven study, the levels of these target genes can used to infer transcription factor activity even in the absence of measurements of the transcription factor itself
[73]. Similar approaches have identified the transcriptional changes that occur fol-lowing activation of signaling cascades [74] using the abundance of publicly available
microarray data [75], enabling the inference of pathway activity from transcriptional
changes. Together, these methodologies enable data-driven approaches to incorporate prior biological knowledge, and increase the ability of data-driven models to provide mechanistic insight.
1.2.2
Multiscale modeling approaches are necessary for
mod-eling cell-cell communication
Constructing multi-scale models that account for all facets of cell-cell communication is difficult due to the discrepancy in the form of computational models used to describe each scale of the biological system (i.e. extra-cellular vs. intra-cellular). The field of multi-scale modeling attempts to address this problem by construct computational frameworks that integrate models operating at different "scales" of biology [76, 77, 78]. The most common framework for modeling cellular behavior is through agent-based models. Agent agent-based models simulate the behavior of individual agents (i.e. cells) by defining a set of rules that govern agent behavior, such as rules that describe
cell-cell and cell-environment interactions [79]. For example, a recent study
exam-ined how interactions between tumor cells and tumor associated macrophages affect tumor outgrowth [80]. This model simulated diffusion of soluble factors using partial differential equations and the secretion of soluble factors and behaviors of cells in
response to these factors are based on rules derived from experimental data. While agent based models account for extra-cellular interactions, they are not a suitable framework for modeling intra-cellular signal transduction via signaling pathways and transcription factors. Ordinary differential equation (ODE) models are often used to model intra-cellular signaling cascades, although other approaches such as artificial neural networks and logic models are also used. To integrate these disparate model-ing frameworks, multi-scale models often use an agent based model to describe the presence of extra-cellular stimuli, and the state of the extra-cellular environment is used as input to an ODE model that simulates the intra-cellular response. Based on the results of the intra-cellular simulation, the agent takes phenotypic action (i.e. proliferates, migrates) and the state of the extra-cellular environment is updated [811. However, while this multi-scale approach is powerful modeling approach, a major dif-ficulty in constructing these models is that the scope of the model quickly grows to
an intractable level
[70].
Beyond the parameters necessary to specify each scale of themodel, multi-scale models also require parameters that link each scale of the model. Therefore, constructing multi-scale models requires not only experimental evidence to formulate each individual scale of the model, but also evidence to define how each scale is linked to each other.
As an alternative to agent based models, another approach for modeling cell-cell communication is to use a graph theoretical approach to model extra-cellular commu-nication. In this approach, each cell type can be treated as a node in a network (i.e. computational graph), with weighted, directed edges between each node correspond-ing to the sendcorrespond-ing and receivcorrespond-ing of signals via ligand-receptor interactions. Compli-cating this conceptualization is the fact that each cell type processes and responds to input signals differently [82]. Therefore each node the the cell-cell communication network requires an internal "sub-network" that describes how the cell type responds to inputs. Further adding to the difficult of this approach is the heterogeneity within a given cell type. This heterogeneity means that cells of a given cell type do not act uniformly, but behave differently depending on their "cell state". Therefore, a suitable computational framework will need to predict how each node (i.e. cell type)
responds to input signals.
The development of single-cell sequencing technologies has provided a novel source of data for generating models of cell-cell communication networks in tissue microenvi-ronments. However, because of the complexity of fully modeling cell-cell communica-tion, current approaches typically focus on a limited set of receptor-ligand interactions and a limited set of cell types. For example, recent studies have focused on cytokine interactions between a limited set of immune cells [83, 84]. Other studies have focused on broader sets of interactions, but only focused on CAF-melanoma interactions [85] or only quantified the number of interactions without examining the biological effect of identified interactions [86]. Other studies have attempted to comprehensively de-scribe receptor-ligand interactions occuring between all cell types in a tissue; however, these approaches are still descriptive rather than quantitative in nature [87]. These approaches all represent important steps towards the goal of modeling cell-cell com-munication networks; however, there is still a gap between current approaches and a generalizable framework for quantitatively modeling receptor-ligand interactions, the downstream signal transduction, and the resultant cellular outcomes.
1.3
Overview of thesis
This thesis has focused on developing computational approaches to model cell-cell communication. Despite the importance of cell-cell communication in biological sys-tems, approaches for quantitatively modeling this communication are still lacking. Studies of cell-cell communication are often limited in scope, either focusing on par-ticular cell types of interest or limiting the molecular species under consideration. The aim of this work has been to provide generalizable methodologies for studying cellular communication mediated by receptor-ligand interaction, the effect of com-munication on intra-cellular signal transduction, and how comcom-munication ultimately affects phenotypic cellular behavior.
Chapter two describes a study using an in vitro MPS of the liver microenviron-ment to study cell-cell communication in metastatic breast cancer. Breast cancer
mortality predominantly results from dormant micrometastases that emerge as fatal outgrowths years after initial diagnosis. In order to gain insights concerning factors associated with emergence of liver metastases, we recreated spontaneous dormancy in an all-human ex vivo hepatic microphysiological system (MPS). Multiplexed pro-teomic analysis of the MPS effluent enabled identification of both key factors and processes that correlated with the various tumor cell states and candidate biomark-ers for actively proliferating (either primary or secondary emergence) vbiomark-ersus dormant metastatic cells in liver tissue. Given the minimal tumor burden, these markers likely represent changes in the tumor microenvironment rather than in the tumor cells.
A computational decision tree algorithm applied to these signatures indicated the
potential of this MPS for clinical discernment of each metastatic stage from blood protein analysis.
Chapter three describes an approach for identifying ligand-receptor interactions in tumor microenvironments using single-cell RNA sequencing data. This chapter focuses on quantitatively modeling inter-cellular signaling and constructing predictive models describing how cell-cell communication relates to phenotypic outcomes, such as tumor growth and immune infiltration. The study was performed using six different mouse syngeneic models in order to identify receptor-ligand interactions that occur consistently across different tumor types. By identifying conserved mechanisms of cell-cell communication, the goal of this study was to identify receptor-ligand interactions that may be potential therapeutic targets.
Chapter four describes an extension of the cell-cell communication approach de-veloped in chapter three to include analyses of intra-cellular signaling pathway and transcription factor activity. This chapter explores approaches for integrating models of cell-cell communication at the cellular scale with intra-cellular models of signaling at the molecular scale. For this study, we again used single-cell RNA sequencing mea-surements of colorectal tumors, which are composed of multiple cell types, including malignant, immune, and stromal cells. We developed an approach to computation-ally characterize cell-cell communication mediated by ligand-receptor interactions at a single-cell level. In addition, we used functional genomic approaches to infer
sig-naling pathway and transcription factor activity using single-cell RNA sequencing data. Finally, with the combined information regarding inter-cellular receptor-ligand communication and intra-cellular pathway and transcription factor activity, we used a network inference tool in order to mechanistically determine protein-protein inter-action networks that explain how inter-cellular communication affects intra-cellular signaling.
Overall, this thesis presents computational approaches for modeling receptor-ligand mediated cell-cell communication. While this work has focused on the study of tumor microenvironments, the methods described in this thesis are applicable to any multi-cellular tissue of interest. Furthermore, this work presents approaches for inte-grating modeling of extra-cellular cell-cell communication with models of intra-cellular signaling. Integrating these two spatial scales of modeling advances our current ap-proaches for constructing multi-scale models of biological systems, a longstanding goal of systems biology research.
Chapter 2
A model of dormant-emergent
metastatic breast cancer progression
enabling exploration of biomarker
signatures
The contents of this chapter were published as:
Amanda M. Clark, Manu P. Kumar, Sarah E. Wheeler, Carissa L. Young, Raman Venkataramanan, Donna B. Stolz, Linda G. Griffith, Douglas A. Lauffenburger, and Alan Wells. A model of dormant-emergent metastatic breast cancer progression en-abling exploration of biomarker signatures. Mol. Cell. Proteomics, 17(4):619-630,
2018
See the online publication for all references to supplementary material not included in this thesis.
Contributions
A.M.C., M.P.K., S.E.W., D.A.L., and A.W. designed research; A.M.C., M.P.K., S.E.W., C.Y., and D.S. performed research; A.M.C., M.P.K., C.L.Y., D.S., D.A.L.,
and A.W. analyzed data; A.M.C., D.A.L., and A.W. wrote the paper; R.V., D.S., and
L.G.G. contributed new reagents/ analytic tools; M.P.K., R.V., L.G.G., and S.E.W.
reviewed manuscript; D.A.L. and A.W. provided financial support;
2.1
Abstract
Breast cancer mortality predominantly results from dormant micrometastases that emerge as fatal outgrowths years after initial diagnosis. To gain insights concern-ing factors associated with emergence of liver metastases, we recreated spontaneous dormancy in an all-human ex vivo hepatic microphysiological system (MPS). Two populations formed after seeding this MPS with small numbers (<0.05% by cell count) of the aggressive MDA-MB-231 breast cancer cell line: actively proliferat-ing ('growproliferat-ing'; EdU+) and spontaneously quiescent ('dormant'; EdU-). Followproliferat-ing chemotherapeutic treatment, the proliferating cells were eliminated and only quies-cent cells remained. This residual dormant population could then be induced to a proliferative state ('emergent'; EdU+) using inflammatory stimuli. Proteomic analy-sis of the MPS effluent identified key factors and processes that correlated with the various tumor cell states, and candidate biomarkers for actively proliferating (either primary or secondary emergence) versus dormant metastatic cells in liver tissue. Dor-mancy was found to be associated with signaling reflective of cellular quiescence even more strongly than the original tumor-free liver tissue, whereas proliferative nodules presented inflammatory signatures. Given the minimal tumor burden, these markers likely represent changes in the tumor microenvironment rather than in the tumor cells. A computational decision tree algorithm applied to these signatures indicated the potential of this MPS for clinical discernment of each metastatic stage from blood protein analysis.
2.2
Introduction
Once breast cancer advances to clinically evident metastatic disease, death invariably ensues. Upon diagnosis, the vast majority of breast cancer patients present with no evidence of disseminated disease. However, tumor cells escape into the circula-tion early during primary tumor development [89] and in some instances establish as small, clinically silent dormant micro-metastases in secondary ectopic sites, which emerge years later as lethal, clinically overt metastatic growths [90]. As a result, following removal of the primary mass, prophylactic chemotherapy is often admin-istered to eradicate any undetected disseminated tumor cells circulating throughout the body. While this approach has reduced recurrence and mortality by a third, there is significant morbidity and even mortality in the universal application of adjuvant chemotherapy. Furthermore, the established dormant micro-metastases are typically
resistant to such treatments, which mainly act upon actively cycling cells
191,
92].Triple-negative breast cancer (TNBC) is a salient example wherein 25% of patients die from recurrence within 5-years of diagnosis despite prophylactic treatment [93]. With respect to ectopic sites, evidence of breast to liver metastases is particularly
foreboding with a median survival of 4 - 23 months after detection [94, 95, 96].
This treatment paradox has driven the search for defined non-invasive biomarkers or molecular signatures of secondary dissemination and outgrowth. It is imperative
to discern the status of these micro-metastases - whether such cells are beginning to
emerge as lethal macro-metastases or simply remaining as dormant, clinically silent cells/nodules. This is challenging as the vanishingly small number of cells at the earliest stages are unlikely to produce sufficient signals for detection within the body. It is precisely this dilution of signals that has obstructed the development of can-cer screening protocols for early detection using tumor cell-derived biomarkers. We propose that it is most fruitful to detect surrogate biomarkers that reflect the home-ostasis of the tumor microenvironment being one of either suppressive dormancy or active outgrowth. As the surrounding tissue will be orders of magnitude greater than the actual tumor cell count early in emergence, the dilution of candidate biomarkers
in whole body fluids should be proportionally less. To date, only a handful of reliable
biomarkers have been approved
[97]
and these markers are usually correlative andnot mechanistically related to disease in ways that would inform therapeutic options. It is difficult to predict recurrence, yet pinpointing novel biomarkers as tools for the early detection and monitoring of metastatic recurrence would be clinically beneficial.
The surrounding tumor microenvironment, particularly the inflammatory/ immune system, plays a key role in regulating metastatic resistance and recurrence [981. How-ever, our understanding of the underlying mechanisms is limited, especially with
respect to the drivers of emergence. Efforts have been hindered by the absence
of pre-clinical human models that simultaneously capture the complexities of the chemoresistance exhibited by dormant metastatic cells/nodules and their subsequent emergence in a physiologically relevant ectopic niche. Such models would enable discovery of candidate biomarkers mechanistically related to disease state, and eval-uation of therapeutic efficacy in real-time. The latter is of particular importance as metastatic disease is presently incurable. Further, the capability to evaluate the effi-cacy of new therapeutics in an all-human pre-clinical context is needed to drive more rapid progress in precision medicine.
Modeling these micro-metastases requires "mesoscale' tissues with organ-to-tumor ratios reflective of the human situation of early and often cryptic metastases. Our
3D ex vivo all-human microphysiological system (MPS) is thus attractive for such
investigations. Using the liver as the ectopic metastatic site, we are able to establish micro-metastases in a relatively large mass of resident hepatic cells (both parenchymal and non-parenchymal [NPC]) wherein the tumor cells initially comprise <0.05% of the tissue [99, 100]. Herein, we report that not only can we recreate dormant-emergent metastatic progression of breast cancer cells, but that this MPS has the potential to identify candidate biomarkers for dormant and actively outgrowing tumor cells by detecting signals derived from the ectopic tissue.
2.3
Results
2.3.1
Recapitulation of dormant-emergent breast cancer
metas-tasis progression in a liver MPS
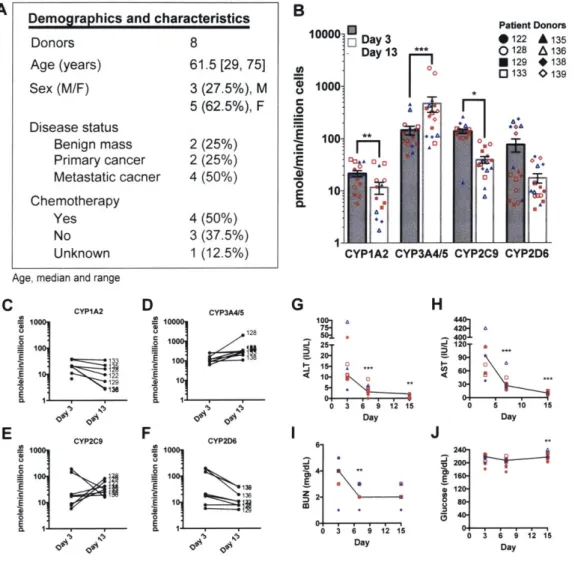
Key to the use of primary cell-derived organs is the functional consistency of cells over time and between donors. The demographics and characteristics of the patient donors utilized within this study are summarized in supplemental Fig. 2-SlA and supplemental Table S1. Hepatic niche function and health was unaffected by patient donor background and was maintained throughout the experiments (supplemental Fig. 2-S1B-J).
Experiments using the ex vivo hepatic MPS were conducted as outlined in Fig.
2-lA. MDA-MB-231 (labeled with RFP) cells were seeded on day 3, treated on day 7 with proliferation-targeting chemotherapy for 72 hours, left untreated for the
sub-sequent 72 hours, and then exposed to an inflammatory stimulus on day 13 for 48 hours. Consistent with our previous work [99, 100], on day 7, four days after col-onizing the hepatic niche, MDA-MB-231 cells were predominantly actively growing
(RFP+/EdU+) with a small population of quiescent dormant cells (RFP+/EdU-)
(Fig. 2-1B,C left panel, supplemental Fig. 2-S2A, supplemental Table S2). Treat-ment of the MPS with chemotherapy resulted in eradication of the vast majority of actively proliferating MDA-MB-231 cells (Fig. 2-iB, supplemental Fig. 2-S2B, supplemental Table S2), leaving behind non-proliferating breast cancer cells by day
13 (Fig. 2-iC, middle panel). This indicated that within the ex vivo hepatic niche,
there was a pre-existing dormant subpopulation of the normally highly aggressive MDA-MB-231 cells as these cells had not progressed through S phase (EdU-) prior to chemotherapy.
Distinct from and as an advance on our previous studies [99, 1001, the main question herein was to determine if these persisting cells were indeed viable, reversibly-growth arrested cells and not merely (pre-)apoptotic or senescent cells that remained trapped within the 3D hepatic tissue. To investigate this, the persisting cells were
![Figure 1-1: Forms of inter-cellular signaling. Reproduced from [17]. (A) Contact- Contact-dependent signaling requires cells to be in direct membrane-membrane contact](https://thumb-eu.123doks.com/thumbv2/123doknet/14431348.515237/20.917.142.767.116.668/cellular-signaling-reproduced-contact-contact-dependent-signaling-requires.webp)
![Figure 1-2: Different kinds of intra-cellular signaling proteins along a sig- sig-naling pathway from a cell-surface receptor to the nucleus Reproduced from [28]](https://thumb-eu.123doks.com/thumbv2/123doknet/14431348.515237/24.917.138.776.162.564/figure-different-cellular-signaling-proteins-pathway-receptor-reproduced.webp)