On the kinetics of the nitrate reduction in concentrated nitric acid
Texte intégral
Figure
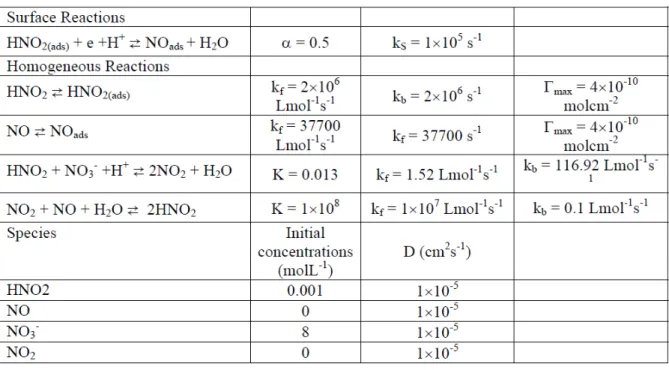
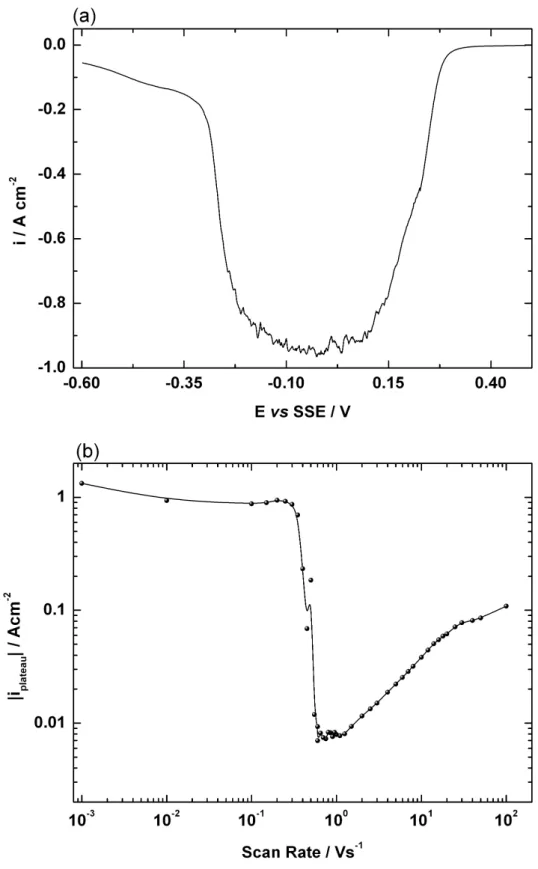
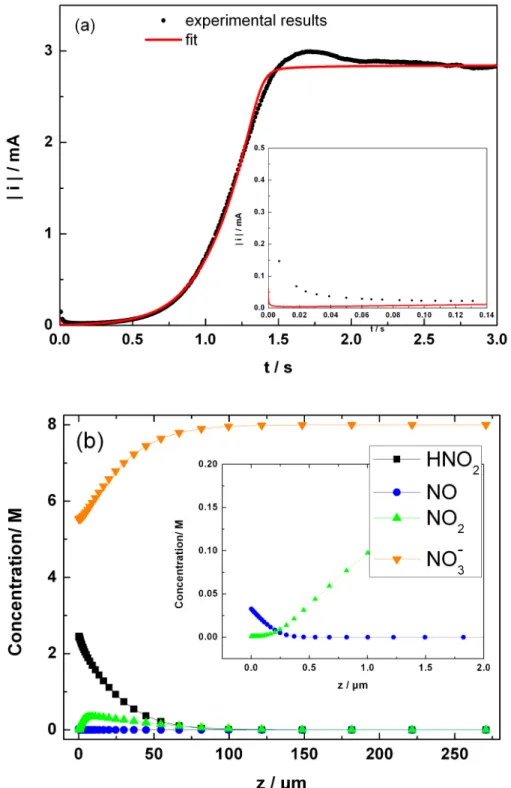
Documents relatifs
L’accès à ce site Web et l’utilisation de son contenu sont assujettis aux conditions présentées dans le site LISEZ CES CONDITIONS ATTENTIVEMENT AVANT D’UTILISER CE SITE WEB..
In this paper we prove a global-in-time existence and uniqueness (up to possible fattening) result for the crystalline mean curvature flow valid in all dimensions, for
The main purpose of this letter is to demonstrate that the cloud phase assumption made in previous cirrus clouds re- trievals using infrared split window signatures is not
Parmi la diversité des fluides hydrothermaux pouvant être impliqués dans la formation de gisements d’or, il semble que les fluides magmatiques soient les plus efficaces
-9 ـِؾإج مآه ثامىلٗم ةعاضلا ةصىجلا تلماكلا : ضبلا ًم ءاكوب مآه ثامىلٗم لزاص تؿؾاالإا ذمؿٌ تب٢اغمب ثاُلمٗلا ةعىهب ةغمخؿم غٞىٍو تمىلٗالإا يف
SHD 1M1316 Mémoire sur la reconnaissance de la côte d’Oran par Tatareau capitaine SHD 1M1316 Mémoire sur la reconnaissance de la côte d’Oran par Tatareau capitaine au
are prone to illegitimate rearrangements and chromosomal translocations observed in human T-cell acute lymphoblastic leukemia (T-ALL) often involve the site of the TCR
