by Yongwon Jung M.Sc., Chemistry
Korea Advanced Institute of Technology and Science, 2000
SUBMITTED TO THE DEPARTMENT OF CHEMISTRY IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY IN BIOLOGICAL CHEMISTRY AT THE
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
September 2005
© Massachusetts Institute of Technology, 2005 All rights reserved
Signature of Author: I fI U I J Department of Chemistry August 22, 2005 a '% Certified by -1-x -= I -' ' Stephen J. Lippard Arthur Amos Noyes Professor of Chemistry Thesis Supervisor Accepted by: IMASSACHUSETTS INSTITU OF TECHNOLOGY OCT 1 2005 LIBRARIES ) Robert W. Field Chairman, Departmental Committee on Graduate Studies
ARCHIVES
1
_ __ __ I rm
This doctoral thesis has been examined by a Committee of the Department of Chemistry as follows:
JoAnne Stubbe Novartis Professor of Chemistry
Committee Chairman
' -
(
Q'ephen J. Lippard
Arthur Amos Noyes Professor of Chemistry Thesis Supervisor
Catherine L. Drennan Associat herine L. Drennan Associate Professor of Chemistry
al-Cellular Responses against DNA Damaged by Platinum Anticancer
Drugs
By Yongwon Jung
Submitted to the Department of Chemistry on August 16, 2005 In Partial Fulfillment of the Requirements for the
Degree of Doctor of Philosophy in Chemistry
ABSTRACT
The anticancer activity of platinum-based drugs such as cisplatin, carboplatin, and oxaliplatin is mediated by their ability to attack DNA such that generated adducts trigger numerous cellular responses. A better understanding of these processes is critical for developing more effective therapeutic approaches, which can increase the anti-cancer activity of the drugs while minimizing side effects and extending successful treatment to a wider range of human cancers. Chapter 1 provides the current comprehension of early cellular responses to platinum-DNA adducts. The event primarily occurs through platinum-DNA adduct recognition by a number of cellular proteins.
Among proteins that recognize platinum-DNA lesions, one class constitutes
proteins that selectively recognize severely distorted DNA generated by the platinum adduct formation. The TATA-binding protein (TBP) and high mobility group protein
HMGB1, both highly abundant and vital proteins, are reported to bind
cisplatin-damaged DNA and to be involved in mediating the cytotoxic activity of platinum-based agents. Chapters 2 and 3 discuss the structural and kinetic properties of both proteins binding to platinated DNA. The TBP protein recognizes the TATA box element of transcriptional promoters and recruits other initiation factors. TBP binds with high affinity (Kd = 0.3 nM) to DNA containing site-specific cisplatin 1,2-intrastrand d(GpG)
cross-links. The ko and koff values for the formation of these TBP complexes are 1-3 x 105 M-ls-1 and -1-5 x 104 sec-l, respectively, similar to the corresponding values for the
formation of a TBP-TATA box complex. When TBP was added to an in vitro nucleotide excision repair (NER) assay, it specifically shielded cisplatin-modified 1,2-(GpG) intrastrand cross-links from repair. HMGB1, a highly conserved non-histone
DNA-binding protein, interacts with specific DNA structural motifs such as those
encountered at cisplatin damage, four-way junctions, and supercoils. The full-length HMGB1 protein binds to DNA containing a 1,2-intrastrand d(GpG) cross-link mainly through domain A with a dissociation constant Kd of 120 nM. Interaction of the C-terminal tail with the rest of the HMGB1 protein was examined by EDC cross-linking experiments. The acidic tail mainly interacts with domain B and linker regions rather than domain A in HMGB1.
Another group of proteins that encounter platinum-damaged DNA are involved with DNA function and therefore are in frequent contact with DNA. DNA, RNA
polymerases and histone proteins inevitably run into platinum-DNA adducts.
Transcription inhibition by DNA adducts of cisplatin is considered to be one of the major routes by which this anticancer drug kills cancer cells. Stalled RNA polymerases at platinum-DNA lesions evoke various cellular responses such as nucleotide excision repair, polymerase degradation, and apoptosis. The consequences of RNA polymerase blockage by platinum lesions are discussed in the next two chapters. T7 RNA polymerase and site-specifically platinated DNA templates immobilized on a solid support were used. Polymerase action is inhibited at multiple sites in the vicinity of the platinum lesion. The stalled polymerase can be dissociated from the DNA by subsequent polymerases initiated from the same template. The immediate consequences of human RNA polymerase (Pol) II arrest at the site of DNA damaged by cisplatin were
studied in whole cells and cell extracts, with a particular focus on the stability of stalled Pol II and its subsequent ubiquitylation. Pol II was completely blocked by a cisplatin intrastrand cross-link and the stalled polymerase was quite stable in nuclear extracts as well as in cisplatin-treated HeLa cells. The stalled Pol II proteins were transcriptionally
active and capable of resuming transcription beyond the DNA adduct following its chemical removal from the template. A series of experiments revealed that lysines other than Lys-48 of ubiquitin are involved in Pol II ubiquitylation following DNA damage. Only a fraction of ubiquitylated Pol II dissociates from damage sites and that it is
rapidly destroyed by proteosomes.
In the final chapter, hapten-conjugated platinum compounds were studied in an effort to follow DNA damaged by platinum agents in living cells. Platinum complexes containing a desthiobiotin moiety (DTB-Pt) with different linkers were synthesized and characterized in vitro and in cells. DNA damaged by DTB-Pt was strongly interacted with streptavidin-coated beads. Moreover, less than 1 fmol of DTB-Pt DNA adduct can be detected by using a simple dot blot analysis.
Thesis Supervisor: Stephen J. Lippard Title: Arthur Amos Noyes Professor of Chemistry
Acknowledgments
For the past five years I have dreamed about writing acknowledgments of my thesis and I still cannot believe I am doing it right now! There are so many people I want to thank for their contribution to this thesis. First, I need to thank my advisor, Steve Lippard. I feel very fortunate to have the opportunity to work in his lab filled with wonderful people. Steve has always trusted me and provided me the infinite freedom and an extra laboratory funding to explore bioinorganic chemistry. He is an amazing scientist who puts a great amount of effort to educate his students, which I will always respect. I would also like to thank Professor JoAnne Stubbe for helpful discussions as my committee chair, and Professor Cathy Drennan for her guidance to be a crystallographer.
I joined the Lippard lab with Katie Barnes, Emily Carson, and Sungho Yoon, forming the best class ever in this lab history! Emily has been always a leader of this group. I think she does not know how much I admire her for everything she did. Katie has claimed as my "older American sister" since I have four Korean sisters already. I really appreciate that she has taken care of me in countless ways from the beginning. And there is my big brother Sungho. He is the best basketball player I have ever played with and is also the best inorganic chemist as well! We have been called as "The twin" in the basketball court.
It has been a pleasure to get to know every former and current member of the Cisplatin subgroup. Seth Cohen and Yuji Mikata helped me to start working in the lab. Min Wei always encouraged me when I was (seldom) depressed. I wish my very best for Alissa Dangel, Olga Burenkova, Ariel Haskel and Sumi Mukhopadhyay. Christiana
Zhang was the happiest person in our subgroup and Dong Wang was the best
photographer. I am confident that Evan Guggenheim, Big Evan, will take good care of our subgroup with help from Katie Lovejoy, Datong Song, and Dong Xu after Katie and I leave. Dong, you can do it! I have also had the opportunity to mentor two great MIT undergraduates Sarah Simmons and Cindy Yuan. I wish them good luck in following their dream.
I was fortunate to work with many incredible people over years in the Lippard lab. From Dongwhan Lee, Scott heldebrand, Josh Farrell, Matt Clark, Adna Ambundo, Lisa Chatwood to Liz Nolan, Andy Tennyson, Jeremy Kodanko, Rayane Moreira, and
Simone Friedle, it is amazing how a single lab can have these many awesome people. Especially my great baymates deserve a huge amount of thanks. Erik Dill, Viviana Izzo, and Mike McCormick are now new bosses in MY biobay. I hope Jessica Blazyk will find new home some time soon in other than Boston. Liz Cadieux is the nicest person I have ever met and she will be the best Mom as well. And finally to Matt Sazinsky, thanks for having meals and going on many trips with me, and just being my friend. I was also lucky to have two Korean lab mates, Mihee Lim and Yoojin Kim. I will not forget many nights we had Korean food together.
I cannot talk about my life at MIT without all the sports I played. First, I want to thank to my IM basketball crew: Chris Chang, Leslie Murray, and Laurance Beauvais. We did make playoffs! Almost all lab members played ice hockey with Jane Kuzelka as our captain. We needed to play D- league. D league is too competitive. Playing softball with Brian Wong, Todd Harrop, and many others was great. And after five years of playing, I think I became a real member of the Lippard lab volleyball team at last.
There are also many non-Lippard lab friends to whom I am grateful for assistance and support, without which I would never be able to survive this far. One of my Korean classmates, Seungjib Choi, has been an invaluable teacher and friend for me. We must have had literally thousands cups of coffee together. I also need to thank to many of my basketball friends for five years although I still don't know some of their names. Finally, but not the least, Yoon-aa Choi has made my last year at MIT so much fun and filled it with happiness. She always supported me through the tough times by giving me confidence, even when she also tried to figure out a demanding graduate life at MIT. Most of all she helped me to index this thesis for 2 hours!
Well I already said this thesis is dedicated to my family. But I will try a little more here to express my appreciation to them. My Mom and Dad have always believed in me doing whatever I wanted with endless support. I know how much you care about me and pray for me. And to my four sisters, thanks for your consistent trust and just about everything you did for your so called "only brother". I am blessed with this great family. I promise I will a better person so you can be proud of me. I will be back soon!
Table of Contents Abstract ... 3
D edication ...
...
6
Acknowledgments
...
Table of Contents ... List of Tables ... 15 List of Schemes ... 16 List of Figures ... .. 17 Glossary of Terms ... 19 Chapter 1. Direct Cellular Responses to Platinum-Induced DNA Damage ... ... 21Background and Focus ... 22
Biological Target of Platinum-based Anticancer Drugs ... 23
Cellular uptake and efflux of platinum complex ... 24
DNA: primary target for platinum drug ... 25
The nature of platinum-DNA adducts ... 26
Platinum Adducts Interaction with DNA Function Proteins ... 27
The effects of platinum adduct interaction with DNA polymerase ... 27
The effects of platinum adduct interaction with RNA polymerase ... 30
The effects of platinum adduct on chromatin: nucleosome structure and
modification
...
32
Repair of Platinum-damaged DNA ... 34
Nucleotide excision repair ... 34
Mismatch repair ... 37
DNA recombination ... 39
Proteins Binding To Platinum-damaged DNA ... 39
Repair proteins ... 40
NER proteins ... 40
Mismatch repair proteins ... 42
DNA-PK
...
42
HMG domain proteins ... 44
S S R P 1 ... ... 47
Other Cellular Proteins ... 49
TBP
...
...
... . ... 49
p53
...
49
PA
RP-1
.
. ...
.. ...
51
Y B -1 ... ... 5 2 Concluding Remarks ... 52References
...
... 54
Chapter 2. Kinetic Studies of the TATA-Binding Protein Interaction with Cisplatin-Modified D N A ... 80Introduction . . . ... ... 81
Experimental Procedures ... 83
Preparation of Yeast TBP ... ... ... ... 83
Preparation of Oligonucleotides Probes ... ...83
Electrophoretic Mobility Shift Assay (EMSA) ... 84
Kinetic EMSA Analysis ... ... 84
Competition Assay ... 85
Excision Repair Assay ... 8...6...86
Footprinting Assay ... 86
R esults ...
...
87
Kinetics of TBP Binding to the TATA Box and Cisplatin-Damaged DNA ... 87
Flanking Sequence Preference of TBP Binding to Cisplatin-Damaged DNA ...88
Cisplatin-Damaged DNA Sequesters TBP from the TATA Box ... 89
TBP Blocks NER of the Cisplatin 1,2-d(GpG) Cross-Link ... 89
TBP Binding Mode to Cisplatin-Damaged DNA ... 90
D iscussion ... ... ... ... ...90
TBP Binding to Cisplatin-Damaged DNA ... 90
Flanking Sequence Dependence of TBP Binding to Cisplatin-Damaged DNA...92
Biological Implications of TBP Binding to Cisplatin-Damaged DNA ... 94
Acknowledgements
...
96
Chapter 3.
The Nature of Full-Length HMGB1 Binding to Cisplatin-Modified DNA ... 116
Introduction
...
117
Experimental Procedures ... 119
Construction of Expression Vectors ... 119
Expression and Purification of HMGB1 Proteins ... 119
Preparation of Oligonucleotides Probes ... 120
Electrophoretic Mobility Shift Assay (EMSA) ... 120
Footprinting Assay ... 121
EDC Cross-Linking ... 121
Results
... ...
122
Footprinting Analysis of HMG Box Protein Binding to Cisplatin-Modified
DNA
... .. ...
122
Mutation of Intercalating Residues in HMGB1 ... 122
Effect of the C-terminal Acidic Tail ... 123
Discussion
...
125
Full-Length HMGB1 Binding to Cisplatin-Modified DNA; Two Tandem HMG
Boxes
...
125
The C-terminal Tail in HMGB1 ... 127
Implications for the Mechanism of HMGB1 Function ... 129
Roles of HMGB1 in Cisplatin Action ... 131
Conclusion
...
132
References
...
133
Chapter 4. Multiple States of Stalled T7 RNA Polymerase at DNA Lesions Generated by Platinum Anticancer Agents ... 144
Introduction
...
145
Experimental Procedures ... 146
Materials
...
146
Construction of Site-Specifically Platinated DNA Templates ... 147
Promoter-Independent In Vitro Transcription ... 148
Multi-Round In Vitro Transcription ... 149
Platinum Removal by Cyanide Ion Treatment ... 149
Results
...
150
Promoter-Dependent In Vitro Transcription on Platinated Templates ... 150
Transcription Bypass Through Platinum Binding Sites ... 151
Promoter-Independent In Vitro Transcription ... 151
UTP-Specific Incorporation by T7 RNA Polymerase at the Site of a Cisplatin 1,2-Intrastrand d(GpG) Cross-Link ... ... ...153
Multi-Round In Vitro Transcription ... 154
Restarting Transcription Following Platinum Removal by Cyanide Ion
Treatment
...
154
Discussion
...
155
Promoter-Dependent and -Independent Transcription Inhibition at Platinum
Cross-Links
...
155
A Closer Look at Polymerase Blockage by Platinum Adducts ... 157
The Effect of Multiple Polymerases at the Site of a Platinum-DNA
Cross-Link
...
160
Resumption of Transcription of T7 RNA Polymerase Stalled at a Platinum Binding Site ... 161
Conclusion
...
162
Acknowledgments
...
163
References
...
165
Chapter 5. RNA Polymerase II Blockage by Platinum DNA Damage: Polyubiquitylation of Stalled Polym erase ... 179
Introduction
...
180
Experimental Procedures ... 182
Materials
...
182
Construction of DNA Templates ... 183
Preparation of HeLa Nuclear Extract ... 184
In Vitro Ubiquitylation Assays in a HeLa Nuclear Extract ... 186
Cellular Protein Fractionation . . ... 187
HA-tagged Ubiquitin Expression in HeLa Cells ... 188
Immunoprecipitation
.
...
189
Results ..
.... ...
... ... .... ...
190
Stability of RNA Polymerase II Stalled at a Platinum-DNA Lesion ... 190
Dynamic State of Stalled RNA Polymerase II: Backtracking and Transcription
Resum
ption . ...
.... ...
. ...
193
Polyubiquitylation of Stalled RNA Polymerase II: In Vitro Ubiquitylation ...194
Polyubiquitylation of Stalled RNA Polymerase II in HeLa Cells . ... 196
Discussion
...
... ..
...
99
Stability of RNA Polymerase II Stalled at a Platinum DNA Lesion ... 199
Dynamic State of Stalled RNA Polymerase II: Backtracking and Transcription Resumption ... 200
Polyubiquitylation of Stalled RNA Polymerase II . ... ...201
Polyubiquitylation of Stalled RNA Polymerase II: Effect on Pol II ... 203
Conclusion
... ...
204
Acknowledgement
...
205
References ... .. ... ... 206
Chapter 6. Following Cisplatin: Hapten-Conjugated Platinum Complex ... 220
Introduction
... ...
221
Experimental Procedures ... 222
Desthiobiotinamido-hexylamine-Boc (DTB6Nboc) (1) (Scheme 6.1) . ... 222
Desthiobiotinamido-hexylamine (DTB6N) (2) . ... 222 Diboc-aminoethyl-Gly (3) ... 2... 223 DTB9diamine (4) ... 223 D TB9Pt (5) ... 224 Cbz-6-amino-hexanol (6) (Scheme 6.2) ... 224 Cbz-6-amino-hexanal (7) ... 225 Cbz-6-amino-hexyl-diamine-diboc (8) . . ...225 DTB6diamine (9) ... 225
DTB6Pt (10) ... 226
1, 4-Diazidobutane (11) (Scheme 6.3) ... 226
1, 4-Azidoaminobutane (12) ... 227
Pt4N3 (13) ... 227
DNA Blot and Desthiobiotin Detection ... 227
Cytotoxicity Assay ... 228
Results and Discussion ... 228
Synthesis of Desthiobiotin-Conjugated Platinum Agents ... 228
Characterization of Desthiobiotin-Conjugated Platinum Agents ... ...229
Synthesis of Azide-Conjugated Platinum Agent ... 231
Conclusion
...
232
Acknowledgements
... ...
232
References
...
233
Biographical Note ... 239
List of Tables
Table 1.1. Key human proteins that bind to cisplatin-modified DNA ... 72 Table 2.1. Duplex DNA probes and abbreviations ... 102
Table 2.2. Calculated and observed (ESI-MS) molecular weights for platinated
oligonucleotides
...
103
Table 2.3. Kinetic and thermodynamic parameters for TBP binding to each probe ...104 Table 3.1. Affinities of HMGB1 Proteins Toward Cisplatin-Modified DNA ... 135 Table 4.1. The complete sequences of DNA and RNA fragments employed in this
study
...
169
List of Schemes
Scheme 6.1. Synthesis of DTB9Pt ... 235 Scheme 6.2. Synthesis of DTB6Pt ... ... ... ... 236 Scheme 6.3. Synthesis of Pt4N3... 237
List of Figures
Figure 1.1. Cisplatin and related platinum-based anticancer drugs ... 73
Figure 1.2. Platinum DNA adducts: formation and structures ... 74
Figure 1.3. Schematic representation of transcription inhibition by platinum lesions and consequent outcomes ... 75
Figure 1.4. The effect of platinum damage on chromatin structure and function ... 76
Figure 1.5. The mechanism of nucleotide excision repair of platinum lesions ... 77
Figure 1.6. The roles of proteins binding to platinated DNA in cisplatin anticancer
action ...
78
Figure 1.7. Direct cellular responses to platinum adducts: overall picture of current
understanding
...
79
Figure 2.1. EMSA experiment to determine kon ... 105
Figure 2.2. EMSA experiment to determine koff . ... 106
Figure 2.3. EMSA of TATAMLP and cisplatin-damaged probes with TBP ... 107
Figure 2.4. Kinetic EMSA data for cisplatin-damaged probes binding to TBP...108
Figure 2.5. Competition EMSA assay between TGGC and TATAMLP ... 109
Figure 2.6. Competition EMSA assay between AGGA and TATAMLP ... 110
Figure 2.7. Effect of TBP on excision repair assay of cisplatin-DNA intrastrand cross-links in HeLa cell extract ... 111
Figure 2.8. Hydroxyl radical footprint of TBP ... 113
Figure 2.9. Schematic diagram of TBP-DNA complex interactions as revealed by
footprinting
...
115
Figure 3.1. Schematic representation of HMGB1 and sequence of duplex DNA probes and abbreviations ... 136
Figure 3.2. Footprint analysis of the interaction between HMGB1 proteins and
35AGGA
...
137
Figure 3.3. EMSA analysis of HMGB1 and AB165 binding to 25TGGA ... 138
Figure 3.4. EMSA analysis of HMGB1 and AB165 mutants binding to 25TGGA...139
Figure 3.5. Footprint analysis of the interaction between HMGB1 mutant proteins and
35AGGA
...
140
Figure 3.6. CNBr cleavage analysis of EDC cross-linked HMGB1 ... ...141
Figure 3.7. EDC cross-linking of HMGB1 bound to cisplatin-modified DNA...142
Figure 4.1. Construction of DNA templates ... 170
Figure 4.2. Gel-electrophoresis analysis of promoter-dependent
in vitro
transcription
...
171
Figure 4.3. Gel-electrophoresis analysis of transcription bypass ... 172
Figure 4.4. Gel-electrophoresis analysis of promoter-independent transcription ... 173
Figure 4.5. Analysis of T7 RNA polymerase blockage by various platinum
adducts
...
174
Figure 4.6. Analysis of nucleotide incorporation by T7 RNA polymerase ... 175
Figure 4.7. Gel-electrophoresis analysis of transcription by multiple polymerases ...176
Figure 4.8. Gel-electrophoresis analysis of platinum removal from DNA ... ...177
Figure 4.9. Structures of the template strand of a T7 RNA polymerase elongation
complex
...
178
Figure 5.1. Construction of the DNA template ... ... 209
Figure 5.2. The complete sequences of DNA fragments employed to construct 99-95
DNA
...
210
Figure 5.3. Schematic representation of expression vector construction for HA-tagged
ubiquitin
...
211
Figure 5.4. Gel electrophoresis analysis of in vitro transcription in HeLa nuclear
extracts
...
212
Figure 5.5. Western blot analysis of RNA polymerase II ... 213
Figure 5.6. Gel electrophoresis analysis of in vitro transcription in HeLa nuclear
extract
...
214
Figure 5.7. Western blot analysis of in vitro ubiquitylation of RNA polymerase II...215
Figure 5.8. Western blot analysis of ubiquitylation of RNA polymerase II in HeLa
cells
...
216
Figure 5.9. The effect of MG132 on ubiquitylation of RNA polymerase II in HeLa
cells
...
217
Figure 5.10. Subcellular localization of ubiquitylated RNA polymerase II in HeLa
cells
... ...
218
Figure 5.11. Model depicting the consequences of RNA polymerase II blockage by cisplatin adducts ... 219
Abbreviations 1,2-d(GpG) 1,2-d(ApG) AAG AAS AdML bp BSA carboplatin cisplatin CNBr CPD CS CTR DACH DCC DNA-PK DNP DSB DTB IDTT IEDC EDTA EMSA en ERCC1 ESI-MS FACT FBS GGR HA HM(G IC5 0 MMR MTT NER oxaliplatin PARP PBS PCNA Pol II PMSF PVDF FRPA SSRP1 TBP TCR TLS cis-[Pt(NH 3)2{d(GpG)-N7(1 )-N7(2)}] cis-[Pt(NH3) 2 { d (ApG)-N7(1)-N7(2) } ]
3-methyladenine DNA glycosylase atomic absorption spectroscopy adenovirus major late
base pair
bovine serum albumin
cis-diammine(1,1-cyclobutanedicarboxylato)platinum(II) cis-diamminedichloroplatinum(II)
cyanogen bromide
cyclobutane pyrimidine dimmer cockayne syndrome
copper transporter diaminocyclohexane dicyclohexylcarbodiimide DNA-dependent protein kinase dinitrophenyl
double strand break desthiobiotin
dithiothreitol
1-ethyl-3(3-dimethylaminopropyl)carbodiimide ethylenediaminetetraacetic acid
electrophoretic mobility shift assay ethylene diamine
excision repair cross complementation group 1 electrospray ionization mass spectrometry facilitates chromatin transcription
fetal bovine serum global genome repair hemagglutinin
high mobility group
concentration required to kill 50% of treated cells mismatch repair
[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide] nucleotide excision repair
(1R,2R-diaminocyclohexane)oxalatoplatinum(II) poly(ADP-ribose)polymerase
phosphate buffered saline
proliferating cell nuclear antigen RNA polymerase II
phenylmethylsulfonylfluoride polyvinylidene fluoride
Replication protein A
structure-specific recognition proteins 1 TATA binding protein
transcription coupled repair translesion synthesis
transplatin tsHMG UV-DRB XP YB-1
trans-diamminedichloroplatinum(II)
testis specific HMGUV-damage recognition protein xeroderma pigmentosum
Chapter 1
BACKGROUND AND FOCUS
Since the serendipitous discovery of the anti-cancer activity of cisplatin, the drug has been used extensively in cancer chemotherapy (1). Platinum-based drugs such as cisplatin, carboplatin, and oxaliplatin, are widely used against various solid tumors including ovarian, cervical, bladder, and nonsmall cell lung cancer (2). Cisplatin is particularly effective in the treatment of testicular cancer with a cure rate of over 90% and nearly 100% when tumors are discovered early (3). The clinical use of cisplatin,
however, is restricted by dose-limiting side effects including nephrotoxicity,
emetogenesis and neurotoxicity (4). Moreover, many tumor cells are inherently resistant or often acquire the resistance to platinum-based drugs, which further limits the use of these drugs (5). For over three decades, continuous efforts have been made to alleviate these limitations with a primary focus on the development of new platinum drugs. Over 3000 platinum compounds have been synthesized and tested for their biological activity. Of these, however, less than 30 compounds have entered clinical trials (6).
Development of new anticancer platinum drugs has encountered difficulties to
overcome the drawbacks of cisplatin in actual clinical tests. At present, only four platinum drugs are registered as marketed drugs and only one compound (oxaliplatin) has been approved by the FDA since cisplatin (Figure 1.1)(5,7,8).
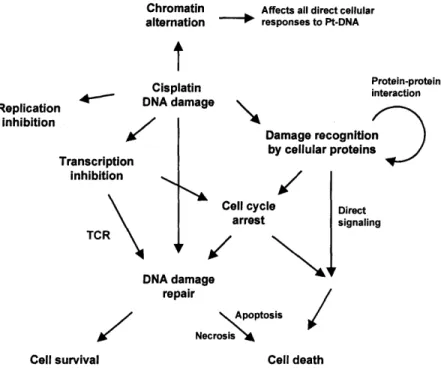
A better understanding of cellular responses to platinum drugs is being sought not only to develop novel platinum-based anticancer agents but also to find more effective cancer therapies with existing drugs. It is now generally accepted that DNA is the main biological target of cisplatin. The complicated mechanism of the anticancer action of cisplatin includes cellular uptake and transport of the drug to the nucleus, DNA adduct formation, and adduct interaction with damage-response proteins (9).
platinum-DNA damage and cellular proteins lead to cell-cycle arrest and repair of DNA damage, the result of the process deciding the fate of treated cells (10). Knowledge of the precise mechanism of cisplatin action is still lacking. In particular, there is a considerable gap in our understanding of how platinum-DNA damage initiates various cellular signaling pathways.
The present review focuses on the current comprehension of early cellular responses to DNA adducts. The event primarily occurs through platinum-DNA adduct recognition by a number of cellular proteins, which triggers many signaling pathways (11). Proteins that encounter platinum-DNA lesions can be divided into two classes. One class comprises proteins that selectively recognize severely distorted DNA generated by the adduct formation with platinum agents. The other group of proteins are involved with DNA function and therefore are in frequent contact with DNA. Hence, these proteins, such as DNA and RNA polymerases and histone
proteins, inevitably encounter platinum-DNA adducts. Here we provide recent
information available for the interactions of platinum-DNA adducts with cellular
proteins and the effects of the adducts on proteins that are involved in various DNA-related processes. The topics discussed will offer a useful guidance to link
platinum-DNA damage with subsequent cellular pathways, and will ultimately provide a
valuable basis for the development of better therapeutic strategies with platinum-based agents (12,13). Other aspects of cellular processes mediating cisplatin cytotoxicity or developing resistance against the drug are reviewed elsewhere (14-16).
BIOLOGICAL TARGET OF PLATINUM-BASED ANTICANCER DRUGS
DNA has been a main biological target of platinum-based anticancer drugs. These drugs generate many kinds of DNA adducts. Moreover, various platinum agents
display different adduct profiles, due to their unique structural and kinetic properties for DNA binding. Understanding the nature of these platinum adducts is important to successfully discuss how these adducts are recognized and processed by cellular proteins.
Cellular uptake and efflux of platinum complex. Upon administration to the
bloodstream, cisplatin maintains a relatively stable neutral state, due to a high
concentration of chloride ion (100 mM), until the drug enters the cell. Passive diffusion has long been considered as a main mechanism for cisplatin uptake. The argument is supported by the fact that the platinum concentration is the rate limiting factor for drug accumulation inside cells, and the uptake is not saturable (17-19). In addition, the uptake of cisplatin is not inhibited by its structural analogues (20). Alternatively, however, some evidence indicates a role of active transporters for cisplatin uptake and efflux (20). For example, multiple studies demonstrated that reactive aldehydes inhibit cisplatin accumulation in cells, possibly by modifying membrane proteins (21,22). Very
recently, a series of studies have indicated the direct linkage between copper
transporters and the uptake and efflux of platinum compounds (23). The first evidence was the association of copper-transporting P-type adenosine triphosphatase (ATP7B), a key player in copper homeostasis, with cisplatin resistance (24). The direct connection of copper transporter to the uptake of cisplatin, however, was first discovered in a transposon mutagenesis experiment in yeast (25). Yeast cells lacking copper uptake protein Ctrl show increased resistance to cisplatin and decreased accumulation of the
drug. The same results are also observed in mouse embryo fibroblast cells.
Furthermore, a later study confirmed that Ctrl mediates the uptake of other platinum drugs including cisplatin analogues (26). More studies with ATP7B as well as ATP7A,
another copper transporting protein, suggested that the proteins modulate cisplatin
levels in cells, presumably by provoking drug efflux (27-30). It is evident now that proteins managing copper homeostasis participate in regulating sensitivity of platinum-based drugs, likely through controlling the platinum level in the cell (31-34). More studies are needed to fully understand how the cellular level of platinum drug is managed by passive diffusion and copper homeostasis proteins, or possibly by other unidentified transporters.
DNA: primary target for platinum drug. Once cisplatin enters the cell, the low concentration of chloride ion (4 mM) facilitates hydrolysis of the compound to produce [Pt(NH3)Cl(OH2)]+, which is an active form of the drug (14). This cationic
mono-aquated platinum compound reacts with various cellular components including proteins, RNA, DNA, membrane phospholipids, microfilaments, and thiol-containing molecules. Controlling cisplatin hydrolysis and transporting activated cisplatin to biological targets are among the key elements to be appreciated to comprehend the mechanism of action of the drug and these subjects have been extensively discussed elsewhere (35). It is now generally accepted that DNA is the primary target of cisplatin among many poteintial cellular targets. Cisplatin-treated bacteria show phenotypes that are characteristic of those treated by DNA-damaging agents (9). More conclusive proof came from the experiments with DNA repair-deficient cells (36,37). Cells lacking in DNA repair are more sensitive to cisplatin. In addition, levels of platinum atoms bound to proteins and RNA are too low to exhibit significant inhibitory effects on the targets (38). There is some evidence, however, that non-DNA targets are involved in drug action to some extent (5).
The nature of platinum-DNA adducts. The reaction of cisplatin with cellular components is proposed to be controlled kinetically rather than thermodynamically. This hypothesis explains the fact that cisplatin binds to DNA in the nucleus instead of reacting with S-donor ligands such as glutathione and methionine, which form more stable platinum complexes (35). Mono-aquated cisplatin [Pt(NH3)CI(OH2)]+ (tl/2 of
formation reaction: -2 h) readily modifies DNA through binding to the N7 atom of a guanine or adenine base to form a monofunctional adduct (tl/2 -0.1 h) (9,39). The second
chloride ligand is hydrolyzed with a half life of -2h, and eventually a bifunctional adduct (intra- or interstrand cross-link) is formed. Adducts analysis of purified DNA treated with cisplatin or DNA isolated from cisplatin-treated patients demonstrates that damaged DNA contains approximately 65% 1,2-d(GpG), 25% 1,2-d(GpA), and 5-10% 1,3-d(GpNpG) intrastrand cross-links as major forms (11). A small percentage of
interstrand cross-links and monofunctional adducts is also found. Transplatin, a
clinically inactive isomer of cisplatin (Figure 1.1), is unable to form a 1,2 intrastrand cross-link, owing to its stereochemistry. Platinum drugs such as carboplatin and oxaliplatin contain different leaving ligands from the chloride ions of cisplatin and therefore exhibit different kinetics for DNA binding and generate disparate adduct profiles from cisplatin (8).
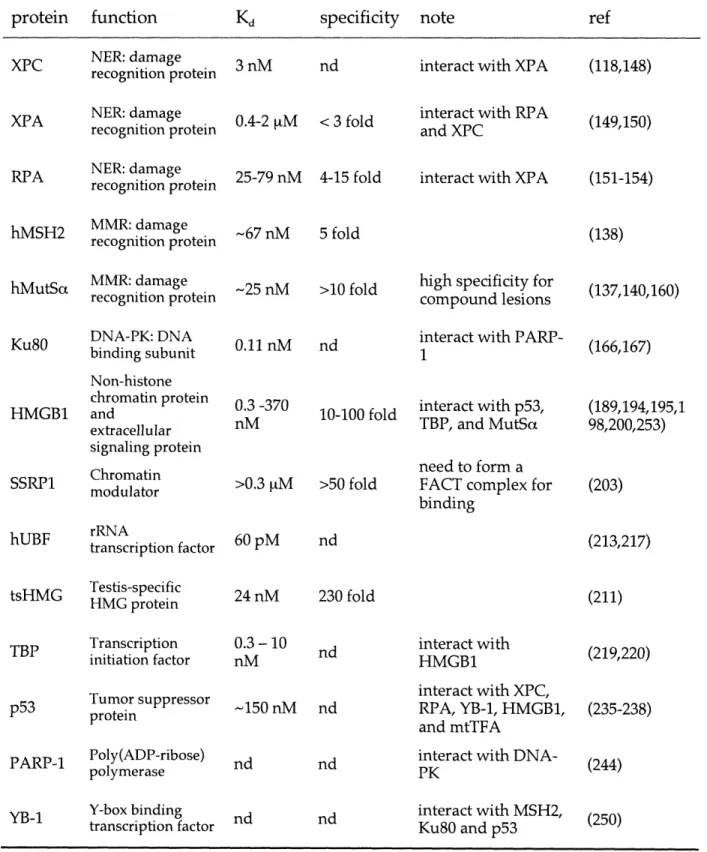
The formation of cisplatin adducts significantly alters the structure of target DNA. Early biochemical studies demonstrated unwinding and bending of DNA as well as duplex destabilization induced by cisplatin lesions (40,41). Detailed structures of platinum adducts have been extensively studied (9,42). Structures of duplex DNA containing 1,2- and 1,3-intrastrand cross-links are illustrated in Figure 1.2 (43-45). Major platinum adducts (intrastrand cross-links) unwind the duplex DNA in the vicinity of the platinum site, bending it towards the major groove, and generate a widened and
shallow minor groove. On the other hand, the interstrand DNA cross-link formed by cisplatin affords a helix bending towards the minor groove with the platinum moiety located in the minor groove. Although these platinum adducts display some degree of structural similarity, it is clear that each adduct distorts the duplex DNA in a distinct way. In addition, the structures of DNA adducts formed by platinum drugs with carrier ligands different from the amines of cisplatin show some variations from the discussed structures of cisplatin-DNA adducts (8). Different platinum adducts are believed to be distinctly recognized and processed by cellular proteins, suggesting also different roles
in mediating cisplatin cytotoxicity.
PLATINUM ADDUCTS INTERACTION WITH DNA FUNCTION PROTEINS
Once the platinum drug forms a DNA adduct, it interferes with essential DNA functions. The inhibition of replication and transcription is a key cytotoxic ability of the drug. Events that occur following the interaction of a platinum adduct with DNA polymerases and RNA polymerases, however, contribute to more than just cytotoxicity of cisplatin. In addition, recent studies discovered the influence of cisplatin adducts on the properties of chromatin.
The effects of platinum adduct interaction with DNA polymerase. The inhibition of DNA synthesis by cisplatin was discovered early and believed to contribute to the cytotoxicity of cisplatin. The activities of partially purified human DNA polymerases a and 3 are inhibited by cisplatin treatment of the DNA template (46). Cisplatin-induced inhibition of DNA replication is also observed in vivo in green monkey CV-1 cells
different platinum adducts to block various DNA polymerases followed (48,49). Most
bifunctional adducts, intra and interstrand cross-links, effectively inhibit DNA
polymerases, while monofunctional adducts cannot block the polymerases. T4 and T7 DNA polymerases, DNA polymerase I and III are blocked by platinum adducts, bypassing the lesion only -10% of the time (48). Despite the evident inhibition of DNA synthesis by cisplatin based on these reports, murine leukemia L1210 cells treated with cisplatin progress through the S phase of the cell cycle and are blocked only in the G2 phase (50). DNA replication continues even in the cells that do not divide. Furthermore, a study with Chinese hamster ovary cell lines both proficient and deficient for DNA excision repair demonstrated that the inhibition of DNA synthesis is dependent only on the concentration of cisplatin and not on the sensitivity of the cell line to the drug (51). Only the content of cells arrested in the G2 phase correlates with cell line sensitivity to cisplatin. It is likely that direct inhibition of DNA replication by cisplatin-DNA damage cannot fully explain the unique properties of this anticancer agent (52).
Mammalian cells possess the ability to synthesize DNA while ignoring various DNA lesions. The process, called translesion synthesis (TLS), demands specialized DNA polymerases, which are less stringent than the major replicative DNA polymerases and can accommodate damaged bases (53). In eukaryotes, the Y-family DNA polymerases
(, , K, and Rev 1) and DNA polymerase , a member of the B family, perform DNA replication across DNA lesions (54). Translesion synthesis through cisplatin-DNA adducts has been an interesting aspect of DNA synthesis in cisplatin-treated cells due to its closer correlation to drug sensitivity (55). Cisplatin-resistant cells exhibit more of TLS than drug-sensitive cells (56-59). The process also has a critical role in the mutagenic property of cisplatin because of the nature of TLS, which carries out both error-prone and error-free DNA synthesis (54). The mutagenicity of cisplatin is closely related to the
evolution of resistance of cell lines against the drug. In fact, the reduced ability to replicate cisplatin-damaged DNA decreases the rate at which the cells obtain resistance to cisplatin. For example, suppression of human DNA polymerase involved in TLS, such as polymerase Rev 1 (60) or C (61), increases the sensitivity of cells to cisplatin and reduces the rate of appearance of cisplatin resistance at the population level.
DNA polymerases that are shown to bypass replication across cisplatin adducts
in vitro, include DNA polymerase 13, t, and 1, while polymerases a, , and are unable to perform TLS past platinum adducts (62-65). Each DNA polymerase displays a distinct specificity in its lesion-bypass property, such as the bypass ability, fidelity, and extension ability. For example, DNA polymerase rl bypasses platinum adducts most efficiently in error-free TLS, which was proved both in vivo and in vitro (59,66). Polymerase t is the most error-prone enzyme, mediating mainly frame-shift mutations (67). Currently, the identities of DNA polymerases that are responsible for TLS past platinum adducts in vivo are not clear. Moreover, two DNA polymerases often work together to complete TLS (54). Although polymerase ~ is unable to bypass certain DNA lesions including those from platinum agents, the enzyme has the ability to extend TLS once nucleotides are inserted opposite DNA adducts by other polymerases (54,61).
Immediate cellular responses to a stalled replication fork at the site of platinum-DNA lesion are still unclear. Recent studies indicate that proliferating cell nuclear antigen (PCNA) plays a key role for the TLS process by recruiting TLS DNA polymerases to the site of stalled replication forks (53). It was proposed that, following replication fork blockage, Radl8 binds to exposed single-stranded DNA at the fork and
together with Rad6 mediates mono-ubiquitylation of PCNA. Mono-ubiquitylated
with the stalled replicative DNA polymerase. The efficiency and fidelity of TLS depends on the nature of the adduct and the recruited polymerase. Oxaliplatin has different properties towards replication bypass in its platinated DNA from cisplatin both in vivo and in vitro, presumably due to its bulky diaminocyclohexane (DACH) carrier ligand (Figure 1.1) (42). This behavior of oxaliplatin is thought to contribute to its distinct anticancer activity compared to cisplatin.
The effects of platinum adduct interaction with RNA polymerase. Early in vitro studies reported the ability of cisplatin adducts to inhibit transcription elongation by various RNA polymerases including wheat germ RNA polymerase II (Pol II) and E. coli, T7, and SP6 RNA polymerases (68,69). Similar to the inhibition of DNA synthesis, RNA polymerases are strongly blocked by bifunctional adducts and not by mono-functional adducts. Direct transcription inhibition by cisplatin and transplatin is observed in human and hamster cell lines that are transfected with a plasmid containing a reporter gene and pre-modified by platinum compounds (70,71). A higher level of transplatin adducts is required to inhibit transcription to the same degree as cisplatin adducts. Accumulated data indicate a close relation between transcription inhibition by cisplatin and the drug's anticancer activity.
RNA polymerases are believed to encounter platinum lesions at a relatively early stage in the DNA damage-response process. Approximately 100 copies of RNA polymerase I are constantly transcribing the rRNA gene in the cell (72). Although the inhibition of RNA polymerase I by platinum adducts has not been directly studied, it is speculated that the damage can block this polymerase (73). RNA polymerase II transcribes most eukaryotic genes and is one of the most abundant proteins with -300,000 copies in a single cell (74). A photobleaching experiment revealed that 25% of
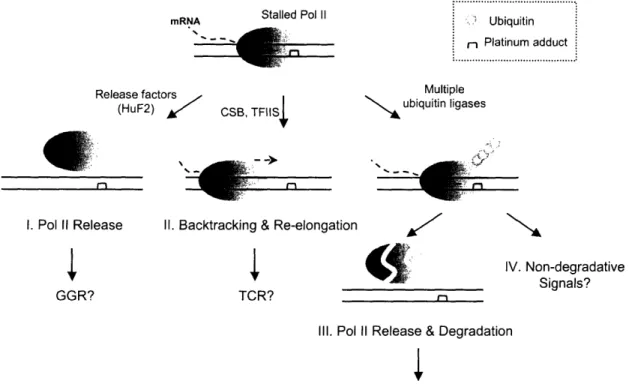
this enzyme is persistently associated with cellular DNA to generate mRNA (75,76). RNA polymerase II has been a major focus of the field studying cellular responses to DNA damage including those by platinum drugs because of its dual roles in the process. Arrested polymerase at the site of platinum lesion not only functions as a damage recognition factor, triggering transcription-coupled repair (TCR) (77), but also mediates programmed cell death (78).
Our knowledge of cisplatin adduct-induced blockage of RNA polymerase II has been greatly advanced over the past several years. The DNA probes containing a
site-specific platinum lesion are employed in in vitro transcription assays with human cell ,extracts or partially purified human transcription factors (79,80). Platinum 1,2-(GpG) and 1,3-(GpTpG) intrastrand cross-links strongly block the elongation complex. A study 'with T7 RNA polymerase revealed that polymerase action is inhibited at multiple sites in the vicinity of the platinum lesion, the nature of which can be altered by the concentration of NTPs and types of platinum adducts (81). The elongation complex is able to proceed into the site of platinum damage, where the polymerase inserts an incorrect nucleotide UTP, rather than a correct nucleotide CTP, opposite a cisplatin 1,2-(GpG) cross-link. The fate of stalled RNA polymerase II at a platinum lesion is also closely examined, which can provide a useful insight into the mechanism of TCR. Solid-phase in vitro transcription experiments have been employed in multiple studies (80,82). Stalled polymerases are fairly stable but can be released from DNA in an ATP-dcependent manner by cellular release factors including human release factor 2 (HuF2) (73,80,83). A differently designed in vitro experiment indicated that a considerable level of stalled polymerase II proteins can remain strongly associated with damaged DNA in cell extracts (82). This result was supported by a cell fractionation experiment using cisplatin-treated HeLa cells, which demonstrated an increased level of
chromatin-associated polymerase II proteins following DNA damage. These polymerases are able to backtrack from the damage sites, cleave the transcripts, and re-elongate. Various
cellular proteins, including CSB and TFIIS, are thought to mediate this process (Figure 1.3) (80,84). Nucleotide excision repair (NER) can occur in vitro at the DNA damage site with polymerase remaining on the DNA (83).
In mammalian cells, cisplatin treatment facilitates RNA polymerase II
degradation following ubiquitylation of the protein (82,85,86). In vitro transcription
experiments with a cisplatin-damaged plasmid also demonstrate ubiquitylation of
polymerase II in a transcription-dependent manner (87). Although ubiquitylation-mediated polymerase degradation is required for DNA damage repair in yeast (88), the role of this process in human cells is unclear. Recent experiments both in cell extracts and living cells suggest that polyubiquitylation of polymerase II following cisplatin treatment can occur through Lys-6, Lys-48, Lys-63, and possibly other lysines of ubiquitin (82,89). Ubiquitylation may trigger non-degradative signals or affect the
properties of stalled polymerase in addition to its degradative roles (90). RNA
polymerase II degradation is prevented by the proteosomal inhibitor MG132 with a subsequent increase in the relative amount of ubiquitylated polymerase. Fractionation of polymerase II from cells co-treated with MG132 and cisplatin indicates that this additional ubiquitylated polymerase is mostly unbound or only loosely associated with chromatin (82). Only a fraction of ubiquitylated polymerase II dissociates from damage sites and is destroyed rapidly by proteosomes (Figure 1.3).
The effects of platinum adduct on chromatin: nucleosome structure and modification.
In a eukaryotic nucleus, DNA is not bare and rather incorporated into nucleosomal fiber, with each nucleosome comprising of a core histone octamer, to form chromatin.
Alteration of chromatin properties significantly affects various DNA metabolic
processes, such as replication, transcription, and repair. It must be appreciated that platinum drugs modify cellular DNA in chromatin in vivo, and therefore platinum
adducts on chromatin will be processed differently from those in free DNA. For example, nucleotide excision repair (NER) of nucleosomal DNA containing a site-specific platinum lesion is significantly less efficient than that of free DNA containing the same platinum lesion in cell extracts (91,92). Furthermore, the repair efficiency of damaged nucleosomes alters depending on the post-translational modification of histone proteins.
The effects of chromatin structure on the reactivity of platinum drugs to DNA have been studied by using reconstituted chromatin as well as various human cell lines (93). The general conclusion is that the linker DNA of chromatin is the preferential
target for platinum drugs (94-96), although the effect is diminished at high drug
concentrations (96). Overall increase in cisplatin-adduct formation is observed in human cancer cell lines when the cells were treated with arginine butyrate, which inhibits histone deacetylases, affords hyperacetylation of histone proteins, and therefore allows chromatin unfolding (97). Platinum drugs clearly bind more favorably to an open form of chromatin. Structural changes of chromatin by transcription activation (98) or protein binding (99) also modulate cisplatin binding to DNA in human cells.
The influence of cisplatin modification on chromatin has also been investigated both in vivo and in vitro. In early studies, chicken erythrocyte nuclei and nucleosomal core particles are treated with cisplatin and the obtained chromatin or nucleosomes are digested by microccocal nuclease (100) and DNase I (101). Digestion profiles indicate that cisplatin binding does not significantly alter the DNA structure of nucleosomal core particle but rather affects the higher order structure of chromatin. This finding is
supported by later observation that chromatin remodeling and transcription factor
binding are severely impaired by cisplatin modification (71), possibly due to altered
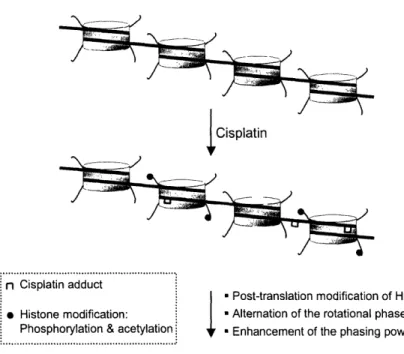
chromatin structure. Cisplatin treatment in HeLa cells induced post-translational
modification of histones H3 (phosphorylation) and H4 (hyperacetylation) (102), modifications known to modulate chromatin structure. It is unclear at this point if these modifications were direct cellular responses to cisplatin binding to chromatin or indirect results from down-stream cellular pathways following cisplatin treatment. Recently, hydroxyl-radical footprinting was employed for structural analysis of a
nucleosome containing a site-specific cisplatin intrastrand cross-link. Cisplatin
modification not only changes the rotational phase of DNA wrapping around the histone octamer but also increases the phasing power (103). Enhanced phasing of the nucleosome by cisplatin lesions may explain drug's effect on the higher order structure of chromatin (Figure 1.4).
REPAIR OF PLATINUM-DAMAGED DNA
Following platinum-induced DNA damage, cellular repair systems immediately act on damage and continuously function until the fate of drug-treated cells is decided. Knowledge of the repair mechanism of platinum-damaged DNA provides essential clues to understand cellular responses to platinum-based anticancer drugs.
Nucleotide excision repair. NER is a primary process for repair of platinum-damaged DNA. Bacterial and mammalian cells deficient in NER are more sensitive to platinum drugs (9,104). For example, xeroderma pigmentosum (XP) cell lines, lacking one of the components of the NER process, have increased sensitivity to cisplatin treatment and cell extracts obtained from these cell lines exhibit no repair activity to cisplatin-induced
DNA damage (105,106). Cisplatin-resistant tumor cell lines show higher levels of the genes producing NER proteins such as XPC, XPA, and ERCC1 (107), with a concomitant
higher repair activity of their cell extracts (108), compared to their wild types.
Moreover, enhanced expression of XPC and ERCC1 mRNA is observed in ovarian cancer tissues obtained from patients clinically resistant to platinum compound (109). It is suggested that the exceptional sensitivity of testicular tumors to cisplatin is credited to lower levels of several repair proteins, such as XPA, ERCC1, and XPF, in these cells
(110,111). Recently, the enhanced sensitivity to cisplatin of human cancer cells is reported when ERCC1 is suppressed by small interfering RNA (siRNA) (112,113).
The molecular mechanism of NER to remove platinum adducts from DNA has been extensively studied (Figure 1.5) (114). During the early stage of NER, platinum lesions are recognized by different mechanisms for two sub-pathways of NER,
transcription-coupled repair (TCR) and global genomic repair (GGR). Stalled RNA polymerase II acts as a damage recognition system to initiate TCR as discussed above (77). Cockayne syndrome (CS) proteins, CSA and CSB, are believed to participate in this process although the exact roles of the proteins are unknown (115). For GGR, damage recognition is initiated by XPC-HR23B (116,117). After the initial recognition of DNA damage, TCR and GGR are thought to follow the same events since subsequent NER proteins are required for both GGR and TCR except XPC-HR23B. TFIIH, XPA, and RPA are the next set of proteins to join the damaged DNA. Although the exact binding order of these proteins is controversial, proteins may be cooperatively recruited to the damage site (117,118). In a subsequent step, XPB and XPD helicases, components of TFIIH, unwind the DNA in a process that requires ATP. XPC-HR23B is released when
endonuclease XPG binds to this unfolded DNA. Another structure-specific
occurs to remove platinated oligonucleotides, which are 24-32 nucleotides in length. Excised oligonucleotides, containing a platinum lesion, and dual incision factors are released from DNA. RPA, however, remains associated with the incised DNA and possibly recruits DNA resynthesis factors such as PCNA and replication factor C (RFC) to fill the gap (Figure 1.5) (117).
Recently, multiple studies investigated dynamic behaviors of several NER factors such as XPF-ERCC1 (119), TFIIH (120), and RPA and PCNA (121) in living cells. The data consistently indicate that each component of NER diffuses freely and participates in repair processes randomly instead of existing as a repair holo-complex. Most noticeably, dynamic targeting of RPA and PCNA to sites of cisplatin DNA damage is examined in Rat-1 and U20S cells expressing GFP fused to these proteins (121). Cisplatin treatment readily induces the relocalization of PCNA and RPA into discrete foci, while platinum DNA lesions are relatively dispersed throughout the nucleus. PCNA and RPA levels recruited to repair foci are proportional to the platinum adducts level. Proteins at repair foci are highly immobile and turned over only on the order of minutes.
The repair of different DNA adducts generated by cisplatin has been investigated in cell-free extracts as well as reconstituted NER systems. In vitro studies with a repair excision or repair synthesis assay revealed that cisplatin 1,3-(GpNpG) intrastrand cross-links are more efficiently repaired by NER than 1,2 intrastrand cross-cross-links (122,123). The cisplatin interstrand cross-link, however, is not repaired in the same fashion. Other platinum compounds have also been tested for NER of their DNA lesions, which are different from corresponding cisplatin lesions. Intrastrand DNA adducts generated by cisplatin, oxaliplatin, and JM216 (Figure 1.1) are similarly repaired in vitro by NER (124), suggesting that the carrier ligand does not affect the repair efficiency. Although
monofunctional adducts of cisplatin and [Pt(dien)Cl]+are not substrates of NER, several clinically active trans-compounds such as trans-[PtCl2(NH3)(thioazole)] (125) and
trans-[PtCl
2(iminoether)
2] (126) form monofunctional adducts, which are successfully
removed by the NER system. Monofunctional adducts of these compounds produce a local conformational distortion at the site of DNA damage similar to cisplatin intrastrand cross-links. The efficient repair of DNA adducts generated by a trinulcear platinum complex has also reported (127).
Mismatch repair. Numerous studies indicate that the mismatch repair (MMR) process closely correlates with cisplatin resistance (128). Cisplatin-resistant cell lines, which possess the property inherently or acquire it after drug treatment, are often defective in MMR (129,130). Cancer or mouse model cell lines deficient in MMR are several times more resistant to cisplatin than corresponding MMR proficient cells (58). On the other hand, no clear correlation of MMR deficiency with cisplatin resistance was reported in multiple studies (16). The MMR process is likely only one of the pathways linked to cisplatin action, and the influence of MMR on platinum cytotoxicity will vary for every experimental condition. The MMR system eliminates base-base mismatches, and deletion and insertion mutations (131). In eukaryotic cells, hMutScc (MSH2-MSH6
heterodimer) initiates MMR by binding to single mismatches and small
insertion/deletion loops, and hMutS3 (MSH3-MSH6 heterodimer) starts MMR through recognition of insertion/ deletion loops of different sizes. Following damage recognition by hMutSa, hMutLc (MLH1-PMS2 heterodimer) and PCNA are recruited to the site of DNA mismatch to proceed the repair. Several exonucleases and helicases, the
replication machinery, and DNA ligase I are subsequently recruited to degrade the error-containing strand and fill the gap.
MMR proteins reportedly contribute to the cytotoxicity of cisplatin by two distinct pathways (132). First, as a repair process, MMR proteins actively repair the
newly synthesized DNA opposite platinum adducts, which is generated from
translesion synthesis (TLS) past the platinum lesion as mentioned above. The process can cause a futile repair of cisplatin damage, which ultimately leads to cell death (58). Defects in MMR proteins not only increased cisplatin resistance but also enhanced
replicative bypasses of cisplatin adducts (58). MMR proteins are proposed to associate with the replication machinery and to recognize mismatches efficiently on platinum-damaged DNA (131). Binding of MMR proteins to cisplatin-platinum-damaged DNA also appears to initiate cellular signals, which lead to programmed cell death (apoptosis) (133). These cellular pathways triggered by MMR proteins are independent from the repair process since certain mutations in hMutS homologs cause mismatch repair deficiencies but do not interfere with the signaling functions of MMR proteins (134,135).
Direct interactions of MMR proteins, especially MutS proteins, with cisplatin DNA adducts were studied in vitro. Bacterial MMR protein MutS (136) and its
eukaryotic homologues hMutSc (137) and hMSH2 (a component of hMutSa
heterodimer) (138) specifically bind to the major cisplatin adduct, a 1,2 intrastrand cross-link. Interestingly, hMSH2 and MutS preferentially recognize the cisplatin-modified DNA over oxaliplatin-cisplatin-modified DNA. Defects in MMR do not affect resistance of oxaliplatin (58), suggesting that the interaction of MMR protein with DNA adducts is important to mediate MMR functions in response to DNA damage. In a recent study, binding properties of MutS (139) and hMutSa (140) to duplex DNA containing cisplatin
compound lesions, which possess various mismatches opposite a cisplatin 1,2-(GpG) intrastrand cross-link, were investigated. Cisplatin compound lesions, formed by misincorporation of a base opposite the site of platinum adducts, are better substrates for MutS binding, the affinities of which are changed by the nature of the mismatches.
DNA recombination. The roles of recombinational repair for protecting cells from cisplatin treatment have been observed in E. coli (15,141). Many recombination-deficient
strains show enhanced sensitivity to cisplatin compared to wild type cells.
Recombinational repair is independent from NER since cells containing double mutations in both NER and recombination proteins are more sensitive to cisplatin than cells with either mutation (141). Spontaneous and cisplatin-induced recombination are also observed in E. coli (142). Impaired recombination DNA repair in yeast (143) and prostate cancer cells (144) enhances sensitivity to cisplatin. In mammalian cells, the disruption of homologous recombination repair (HR) increases cisplatin sensitivity, whereas the knockout of the nonhomologous endjoining (NHEJ) does not affect cell's sensitivity to the drug (145). Presently, how recombinational repair proteins specifically
encounter platinum adducts is unclear. It was recently proved that collapsed replication
forks by strong obstacles on DNA including possibly platinum adducts, recruit
recombination proteins to restore the process (146).
PROTEINS BINDING TO PLATINUM-DAMAGED DNA
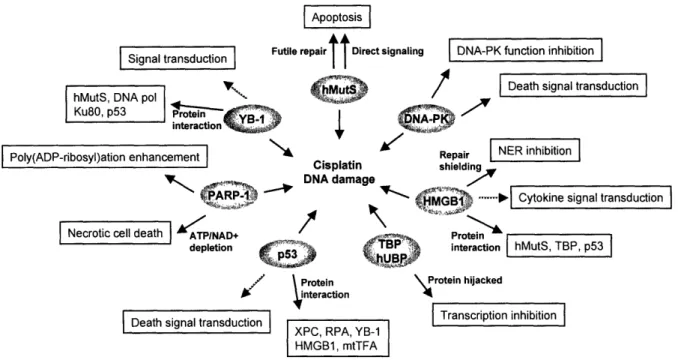
Platinum modification distorts the structure of duplex DNA in a distinct way. A variety of cellular proteins specifically recognize this unique form of DNA structure. These proteins include those involved in repair processes, proteins containing HMG domains, and many others. The interaction of the proteins to platinum-damaged DNA
plays a key role in early cellular responses to platinum drugs. Continuous efforts have been made to identify such proteins and characterize their interaction with cisplatin adducts.
Repair proteins.
Damage recognition proteins in various cellular DNA repair processes are reported to bind to cisplatin-damaged DNA. Their general roles in cisplatin action are evident as discussed above, with loss of their functions generally leading to the enhanced
sensitivity of cells to the drug. Some repair proteins, however, possess distinct
properties to initiate various signaling pathways.
NER proteins. Proteins that initiate the NER process are clear candidates to interact with
cisplatin DNA adducts. Numerous studies indicate that XPC-hHR23B, XPA, RPA, and TFIIH recognize platinum adducts cooperatively at an early stage of NER (117,147). Among these, XPC-hHR23B, XPA, and RPA all are reported to bind specifically to duplex DNA containing a cisplatin intrastrand cross-link (11). Moreover, these proteins interact with each other, which affects additionally their binding to cisplatin adducts.
XPC-hHR23B, a human homolog of yeast Rad4 and Rad23 proteins, shows the highest binding affinity (Kd = -3 nM) to cisplatin 1,3-intrastrand adducts (148). XPC physically interacts with XPA, but the interaction does not contribute to the stability of the XPC-platinated DNA complex. The XPC-XPA interaction appears to be inhibited by the presence of platinated DNA (118). The XPA protein consists of 273 amino acids (-31 kDa) and contains a zinc finger motif. Although XPA is clearly involved in the NER
damage recognition process, it has the lowest binding affinity (-2 tM under
physiological conditions) to cisplatin-damaged DNA (149,150). XPA, however, interacts with RPA and the XPA-RPA complex displays a greater binding affinity to duplex
cisplatin-damaged DNA than either XPA or RPA alone (151). XPA modulates RPA-DNA interaction by enhancing the stability of the ternary complex and inhibiting strand
separation of the target DNA.
RPA is a heterotrimeric protein consisting of 70, 34, and 14 kDa subunits, and an essential component of DNA repair, replication, and homologous recombination. The protein was identified as one of the cisplatin-damaged DNA recognition proteins from the fractionation experiment of human cell extracts by using cisplatin-DNA affinity chromatography (152). RPA specifically recognizes duplex cisplatin-damaged DNA (Kd = 25-79 nM) with about 4-15 fold preference over undamaged DNA, but its binding to single-stranded DNA is also very strong with Kd values in the sub-nanomolar range (151,153). It is proposed that, upon binding to cisplatin-modified DNA, RPA denatures the duplex DNA in the vicinity of the lesion and binds to single-stranded DNA opposite the lesion (154). RPA binds to DNA containing a cisplatin 1,3-intrastrand cross-link 1.5-2 fold better than to DNA containing a 1,1.5-2-intrastrand cross-link, possibly due to low thermal stability of 1,3-adduct compared to that of 1,2-adduct. As mentioned above, XPA enhances RPA binding to platinated DNA (Kd = ~0.5 nM), but does not affect RPA binding to single-stranded DNA (151). The p34 subunit of RPA becomes
phosphorylated in response to DNA damage in vivo as well as in vitro (155). RPA hyperphosphorylation inhibits its duplex DNA binding but this form of the protein still possesses its binding specificity to platinated DNA (153).
XPE-deficient cells display the mildest disorder among XP variants, and these cells still hold 40 to 60% of the repair capacity of normal cells (11). An early study demonstrated that protein extracts of XPE-deficient cells lack a nuclear factor that binds specifically cispaltin-damaged DNA (156). This nuclear factor, called XPE binding factor or UV-damage recognition protein (UV-DRB), is a complex with two subunits of 127
and 48 kDa, and recognizes a broad range of DNA damage (157), with its role in damage repair unknown. The protein is induced by cisplatin treatment (158), and cisplatin-resistant cells express increased levels of XPE binding factor (159).
Mismatch repair proteins. Damage recognition proteins in MMR, hMutSa (MSH2-MSH6)
and bacterial MutS, are reported to bind to cisplatin-modified DNA. The hMutSa heterodimer consists of the MutS homologue hMSH2 and hMSH6 (GTBP/pl60). Purified hMSH2 protein specifically recognizes DNA globally modified by cisplatin (Kd = 67 nM) or [Pt(en)2C12], but not to DNA modified by transplatin and [Pt(dien)Cl]* (138). The protein also binds selectively to 100-bp duplex DNA containing a site-specific cisplatin 1,2-d(GpG) intrastrand cross-link. Human MutSa binds to cisplatin 1,2-d(GpG) (Kd = -25 nM) but not transplatin 1,3-d(GpTpG) adducts (160). hMutSa shows a higher binding affinity to mismatched DNA duplexes than to cisplatin adducts (137). As
discussed above, cisplatin compound lesions, such as DNA with a CT sequence opposite a cisplatin 1,2-d(GpG) cross-link site (Pt-GG/CT), are the best binding substrate for hMutSa (140). The interaction of bacterial MutS to platinum adducts was also investigated (136,139). The protein preferentially recognizes globally cisplatin-modified DNA (Kd = -57 nM) over oxaliplatin-cisplatin-modified DNA (Kd = -120 nM) (136). A recent study demonstrated that MutS binds to 24-bp duplex DNA containing a cisplatin 1,2-d(GpG) adduct (Kd = 36 nM) with only 1.5 fold specificity over undamaged DNA, and shows no specific binding to other cisplatin lesions such as 1,2-d(ApG), 1,3-d(GpCpG), and interstrand cross-links (139). Similar to hMutSa, MutS strongly binds to cisplatin compound lesions, displaying almost 86-fold better binding affinity to Pt-GG / CT site than to Pt-Pt-GG / CC site.
![Figure 1.2. Platinum DNA adducts: formation and structures. (A) Cisplatin is activated by hydrolysis in the cell and the activated form of cisplatin, [Pt(NH 3 )CI(OH 2 )] + , binds covalently to the N7 position of purines](https://thumb-eu.123doks.com/thumbv2/123doknet/14214131.482479/74.918.131.769.134.645/platinum-formation-structures-cisplatin-activated-hydrolysis-activated-covalently.webp)