Bioconjugated oligonucleotides: recent developments and thera-peutic applications
Texte intégral
Figure
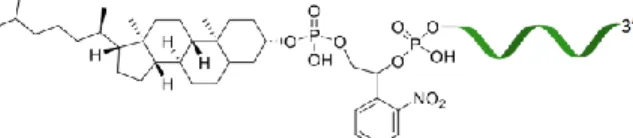
Documents relatifs
In this thesis, I present new evidence on upper mantle seismic anisotropy around La Réunion hotspot – that is proposed to be fed by a deep-rooted mantle plume – and its
Etre indigène numérique, utilisateur de facebook et futur enseignant de FLE ou comment la scénarisation pédagogiquechez les apprentis-enseignants inhibe l’exploitation de
On sait d´efinir une involution de l’ensemble des classes de repr´esentations admissibles irr´eductibles, qui g´en´eralise l’involution introduite par Ze- levinsky dans le cas
first line marking the onset of convection within the mobile layer, the second marking the onset time of unzipping, and the last line indicating the time taken by stable
of K 2 PdCl 4 was obtained with uracil as the opposite base, but only when the metal-ion-mediated base pair was placed at a terminal position.[26] Presumably both of these Pd
The peptide translocation experiments showed that, under our experimental conditions, RW9 was able to cross the membrane of LUVs containing liquid disordered domains but not in LUVs
In order to investigate the stability of RICK compared to CADY-K, we analyzed the siRNA release of formulated nanoparticles (CPP:siRNA-Cy3b, R = 20) by fluores- cence
Single-stranded donor oligonucleotides (ssODNs) with short homology arms, ss1s (43-mer) and ss2s and ss3s (58-mer), and the 58-bp DS3 duplex (formed by hybridization of ss3s with
