HAL Id: hal-03051654
https://hal.archives-ouvertes.fr/hal-03051654
Submitted on 10 Dec 2020
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
Siyang Xia, Luciano Cosme, Joel Lutomiah, Rosemary Sang, Marc Ngangue,
Nil Rahola, Diego Ayala, Jeffrey Powell
To cite this version:
Siyang Xia, Luciano Cosme, Joel Lutomiah, Rosemary Sang, Marc Ngangue, et al.. Genetic structure
of the mosquito Aedes aegypti in local forest and domestic habitats in Gabon and Kenya. Parasites
& Vectors, 2020, 13, �10.1186/s13071-020-04278-w�. �hal-03051654�
RESEARCH
Genetic structure of the mosquito Aedes
aegypti in local forest and domestic habitats
in Gabon and Kenya
Siyang Xia
1*, Luciano V. Cosme
1, Joel Lutomiah
2, Rosemary Sang
2, Marc F. Ngangue
3,4, Nil Rahola
3,5,
Diego Ayala
3,5and Jeffrey R. Powell
1Abstract
Background: The mosquito Aedes aegypti is a devastating disease vector transmitting several important human arboviral diseases. In its native range in Africa, the mosquito can be found in both the ancestral forest habitat and anthropogenic habitats such as villages. How do the different habitats impact the population genetic structure of the local mosquito populations?
Methods: To address this question, we simultaneously sampled Ae. aegypti from the forest and local villages in La Lopé, Gabon and Rabai, Kenya. The mosquitoes were genotyped at 12 microsatellite loci and a panel of ~25,000 single nucleotide polymorphisms (SNPs), which allowed us to estimate their genetic ancestries and the population genetic structure related to habitats and sampling sites.
Results: In the context of the global population genetic structure of Ae. aegypti, clustering analysis showed that mosquitoes from the same locality (La Lopé or Rabai) have similar genetic ancestry, regardless of their habitats. Further analysis at the local scale also found no strong genetic differentiation between the forest and village mos-quitoes in both La Lopé and Rabai. Interestingly, these results from our 2017 samples from Rabai, Kenya contrast to the documentation of genetic differentiation between village and forest mosquito collections from 1975–1976 and 2009. Between-habitat measures of genetic difference (Fst) vary across the genome, with a peak of high divergence observed at the third chromosome only in the La Lopé populations.
Conclusion: Collectively, these results demonstrated that there is little genetic isolation between forest and village habitats, which suggests possible extensive gene flow between them. From an epidemiological perspective, the for-est habitat could act as a refuge for mosquitoes against vector control programmes in the domfor-estic settings. Moreo-ver, sylvatic populations could play a role in zoonotic pathogen transferred to humans. Therefore, future studies on disease transmission and vector control planning in the study area should take natural populations into consideration. Keywords: Aedes aegypti, Forest and domestic habitat, Domestication, Africa, Population genetic structure
© The Author(s) 2020. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://crea-tivecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver ( http://creativecommons.org/publicdo-main/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
Open Access
*Correspondence: siyang.xia@yale.edu
1 Department of Ecology and Evolutionary Biology, Yale University, New
Haven, Connecticut, USA
Background
Anthropogenic habitats such as villages and urban areas have been exploited by many species, including dis-ease vectors [1, 2]. Among them, one of the most suc-cessful and notorious examples is the mosquito, Aedes
aegypti, the main disease vector of several arboviral
diseases, including yellow fever, dengue, chikungunya and Zika [3, 4]. Therefore, the mosquito has become a major public health concern in tropical and subtropical regions worldwide [5]. This mosquito is native to sub-African forests and invaded domestic habitats before spreading to other continents in the last 400–500 years, likely associated with human movements such as the slave trade [6–12]. Populations of Ae. aegypti outside of Africa exhibit strong adaptations to the domestic envi-ronments, for example, a strong preference for biting humans and breeding in human-made containers [13]. These domestic adaptations likely help the mosquitoes to reside permanently around humans, which contrib-utes to their high efficacy as human disease vectors.
Genetic data accumulated during the last few dec-ades consistently suggested strong genetic differentia-tion between the ancestral populadifferentia-tions of Ae. aegypti in Africa and the derived populations outside of Africa [7–
10, 14]. These two genetic groups roughly match the con-ventional description of the two subspecies, Ae. aegypti
formosus (Aaf) in Africa with darker body color and Ae. aegypti aegypti (Aaa) outside Africa with lighter body
color [15, 16]. Although subspecies identification is not always clear-cut, here we will use these terms to repre-sent the two major genetic clusters of Ae. aegypti. Aaf can be frequently found in the forest in Africa, while Aaa is specialized in domestic habitats in other continents [9]. However, this match between genetic clusters, geo-graphical locations, and habitats is not perfect: morpho-logically and genetically defined Aaf has been found in many villages and cities (i.e. domestic habitats) in Africa [10, 17–19]. These findings raise an interesting question: what is the impact of habitat shift on the genome of Ae.
aegypti?
Habitat differences, in theory, could lead to divergent adaptive selection and reduced gene flow allowing inde-pendent genetic drift, both contributing to genetic dif-ferentiation. While the latter generates genomic-wide differentiation, the former could lead to accelerated rates of divergence in certain parts of the genome that are either directly under selection or tightly linked to the genomic regions under selection [20]. These regions may include genes that mediate the interaction between the animals and their environments, such as genes related to host preference, oviposition, or responses to anthropo-genic stressors (e.g. insecticide) in Ae. aegypti [21–25]. On the other hand, if the mosquitoes are predisposed to
invade different habitats and there is frequent gene flow between habitats, genetic differentiation between habi-tats will be weak, which could explain the genetic simi-larities between the forest and domestic Aaf populations in Senegal and Cameroon [17–19]. If this is the case, the exploitation of the different habitats is not due to or not necessarily dependent on genetic specialization.
To address this question and to further our under-standing of the domestication history of Ae. aegypti, we examined the genetic structure of Ae. aegypti in La Lopé, Gabon (Central Africa) and Rabai, Kenya (East Africa). Both locations have Ae. aegypti living in nearly sympatric forest and village habitats, and the two loca-tions are sufficiently distant from each other that they represent two independent habitat shift events. The mos-quito populations in La Lopé were previously sampled in 2014 and genetically classified as Aaf [10]. The story in Rabai, Kenya is more complicated. Previous studies have found that mosquitoes living inside residential houses in Rabai are likely descendants of Aaa outside of Africa that were introduced to the east coast of Kenya [9, 11, 12]. They lived only indoor and remained genetically distinct from the Aaf populations in the nearby forest [26–28]. A third type of Ae. aegypti population in Rabai has been proposed as “peridomestic” living in human-disturbed areas such as gardens between the huts and forests, and is similar to what has been observed in village settings at La Lopé. However, the mosquito populations in Rabai, Kenya had not been monitored genetically since 2011 [12]. Re-examining the genetic structure of the forest and village mosquitoes in Rabai could provide updates on this unique case in the evolutionary history of Ae. aegypti. More importantly, if this co-existence of the forest, peri-domestic, and domestic types prevails, it would present a contrast to La Lopé, Gabon (no evidence of external introduction) to study the genomic evolution associated with habitat difference.
Specifically, the primary goals of this study are to exam-ine: (i) whether forest and village Ae. aegypti in each of the two localities shared similar genetic ancestry; (ii) how genetically differentiated mosquitoes are in different hab-itats; (iii) whether certain regions on the genome show parallel habitat-associated genetic differentiation in the two locations (La Lopé and Rabai). We used 12 micros-atellite markers and a high-throughput SNP microarray containing ~25,000 SNP loci, both of which have proven to provide high resolution in detecting the population structure of Ae. aegypti globally as well as pinpointing the source of local invasion [9, 10, 29–32]. This study adds to our knowledge of the evolution of mosquitoes living in local forest and domestic habitats inside Africa and pro-vides insights on “domestication” history in Ae. aegypti.
Methods
Mosquito collections
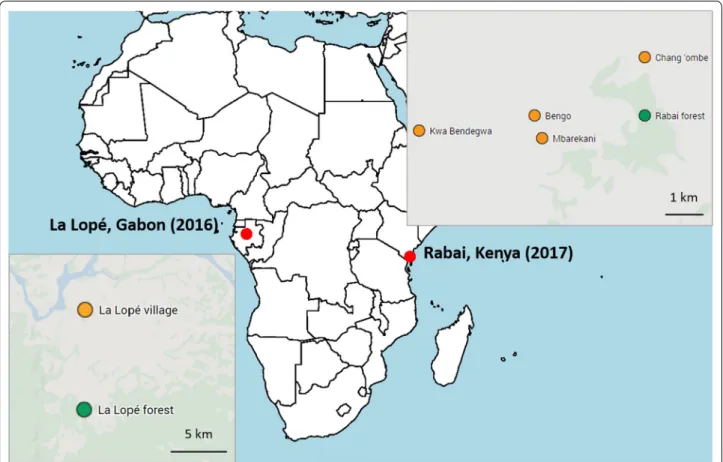
Mosquitoes were collected from natural breeding sites as larvae in La Lopé, Gabon in November and Decem-ber 2016 and in Rabai, Kenya in April and May 2017. The sampling sites in La Lopé were at the edge of a con-tinuous tropical rainforest in the La Lopé National Park, where there is a small village (i.e. La Lopé village) about 5–13 km from the forest (Fig. 1). We collected forest Ae.
aegypti larvae from five rock pools near small streams
and two bamboo traps. The seven containers were less than 7 km away from each other. Because of the low abundance of Ae. aegypti larvae found in the forest, we supplemented the larvae collection with human land-ing catches in the forest. We also collected Ae. aegypti in La Lopé village from 11 artificial containers like tires and construction bricks scattered no more than 2 km away from each other. In Rabai, Kenya, Ae. aegypti were found in tree holes in the forest and artificial containers in four villages (Kwa Bendegwa, Bengo, Mbarekani, and Chang’ombe) about 3–7 km away from the forest (Fig. 1). The 16 tree holes that produced Ae. aegypti were within a 1.5 km by 0.6 km forest patch. Within each village, the mosquito breeding sites (3–9 containers per village) were no more than 1 km apart. Because of the relatively low number of breeding sites produced Ae. aegypti, we col-lected all available samples and later used genomic meth-ods to identify and remove siblings (described in the following sections).
In both La Lopé and Rabai, the mosquitoes were reared to adults in the field for species identification. We kept
Ae. aegypti specimens individually in 80% ethanol at − 20
°C until shipped back to our laboratory at Yale Univer-sity. Each sample was labeled as one of the three habitat types according to the location of the collection sites: forest, peridomestic (outdoor in the village), and domes-tic (indoor in the village). In Rabai, we also recorded the geographical coordinate of each individual, which is not available in La Lopé.
DNA extraction and genotyping
We extracted whole genomic DNA from mosquito sam-ples using the Qiagen Blood and Tissue kit (Qiagen, Hilden, Germany) following the manufacturer instruc-tions. All individuals were amplified and genotyped at 12 microsatellite loci, according to Brown et al. [7]. A sub-set of samples was submitted to the Functional Genom-ics Core at the University of North Carolina, Chapel Hill, where they were genotyped using an SNP microar-ray from Affymetrix designed specifically for Ae. aegypti that contains 50,000 loci [29]. Data were analyzed in the Axiom Analysis Suite v.3.1. (Affymetrix, Santa Clara, CA, USA) to generate the genotype calling. We performed
genotype calling separately for each analysis that includes different sets of samples as best practice. The SNP data were then filtered in PLINK v 1.9 [33, 34] to remove: (i) loci that have more than 10% missingness; (ii) samples that have more than 10% missing loci; (iii) loci that fail Hardy-Weinberg tests with a threshold of 0.00001 in any population [32]; (iv) loci within a genomic window of 75 kb (moving across the genome) that have a variance inflation factor larger than two; (v) loci that have a minor allele frequency smaller than 1% which could result from genotyping error; and (vi) samples whose expected het-erozygosity values deviate more than ± 3 standard devia-tions from the mean of all samples, which may indicate low quality DNA or high inbreeding [35].
Female Ae. aegypti could lay multiple eggs in one con-tainer. Therefore, individuals sampled from the same container could be siblings. To identify them, after the filtering, we calculated individual pairwise kinship coef-ficient k as described in Loiselle et al. [36] using the SNP data in SPAGeDi [37]. Siblings were determined when k > 0.1875, as in Schmidt et al. [38]. Only one individual per sibling group was retained in the dataset. We then filtered the SNP data again using the same criteria described above in PLINK to generate the final dataset for analysis. We removed the same individuals in our microsatellite dataset as in the SNP data. We also ran SPAGeDi using the microsatellite dataset, which contains more individu-als. The analysis resulted in many negative kinship coef-ficients when individuals were paired with themselves, which suggested that the results of the analysis may not be reliable. Therefore, we did not remove any additional individuals from the microsatellite dataset.
The final number of samples in the microsatellite and SNP dataset are summarized by sampling sites and habi-tats in Table 1. Sample sizes were constrained by the mosquito density in the field as well as the cost for gen-otyping. In Rabai, the relatively small sample sizes after partitioning all samples by both sampling sites and habi-tats restricted their use for many analyses. Therefore, in the following analyses, Rabai samples were grouped by either sampling sites or habitats. For example, when we analyzed the Rabai sample by habitats, we combined the peridomestic samples from all four villages, and domes-tic samples from all villages, respectively (the second row in Table 1). Alternatively, when we analyze data by locations, we combined the peridomestic and domestic samples within each village (last column in Table 1). The pooling of samples by habitats was inspired by previous studies in the Rabai area showing the mosquitoes indoor and outdoor are genetically distinct [11, 39]. It is possi-ble that geographical separation between villages (Fig. 1) could lead to some hidden genetic differences between villages, which may confound this pooling by habitat. To
test this possibility, we thus performed alternative pool-ing by locations. The results indicate that there is little apparent population structure by location (see below). Because analysis based on a large number of genomic loci is generally less affected by relatively small sample size [40], in this study, we focused more on the SNP-based analysis and used microsatellite data to provide further validation of our findings.
Finally, for simplicity, in the rest of the manuscript, we use “populations” to merely refer to groups of mosquitoes collected from the same habitat or sampling site without any prior implication of their genetic identities.
Inferring genetic ancestry
To first examine the genetic ancestry of the forest and village Ae. aegypti in both localities, we clustered our microsatellite and SNP data from La Lopé and Rabai with a representative global genetic dataset of Ae. aegypti as references. The reference global microsatellite dataset contains 32 populations, reported in Gloria-Soria et al. [9]. The SNP dataset contains 28 populations worldwide
used in Kotsakiozi et al. [10], with the addition of the “Rabai-in, Kenya” population, which is the same as the “Rabai-in, KE” in the microsatellite panel (Table 1 in [9]). This population was collected inside huts (i.e. domes-tic) in Rabai in 2009 and 2011 and contained individuals classified as Aaa genetically. We also updated the name of the population “Kisumu, Kenya” which was originally mislabeled as “Kahawa Sukari” in [10], in order to be consistent with the microsatellite data. Detailed informa-tion of all populainforma-tions is summarized in Addiinforma-tional file 1: Table S1, and their geographical locations are shown in Additional file 1: Figure S1.
For the microsatellite data, we performed a Bayes-ian clustering analysis using STRU CTU RE v 2.3, which generated the probability of each individual attributing to each genetic cluster [41]. We randomly subsampled 30 individuals from each population when there are at least 30 samples available (23 out of 37 populations). For the rest of the populations, all but one have at least 16 individuals. We tested a different number of clusters (K) ranging from 1 to 10 and conducted 20 replicates for each
K with 500,000 MCMC iterations and 100,000 burn-in.
Fig. 1 Sampling localities in La Lopé, Gabon and Rabai, Kenya. The green area in the two inset maps roughly represents the forest area. The orange
and green dots indicate sampling sites in the villages and the forest, respectively. The continental map was generated in R using rworldmap, and the two inset maps are from Google Maps (https ://www.googl e.com/maps)
The optimal number of clusters was determined by the delta K method [42] in STRU CTU RE HARVESTER v 0.6.94 [43]. The 20 replicates of each K were summa-rized and visualized as bar plots using CLUMPP [44] in
pophelper v 2.2.8.1 [45] in R v 3.5.0 [46]. In addition to the STRU CTU RE analysis, we performed PCA using the same microsatellite data in the R package adegenet v 2.1.1 [47, 48].
The SNP data of all populations were filtered the same way as described in the last section. We performed the “snmf” function (sparse non-negative matrix factoriza-tion) in the R package LEA to estimate the individual ancestry coefficients [49], which produced similar results as STRU CTU RE. Five to twelve individuals per popula-tion were included except the Rabai domestic 2017 popu-lation, which contained 18 samples. The full SNP dataset after filtering contains 23,767 loci. We performed 20 replicates for each K from 1 to 34. The best K was deter-mined by having the lowest cross-entropy. Finally, the SNP data was also analyzed by PCA in adegenet.
Population structure of La Lopé and Rabai mosquitoes
We next focused on only mosquitoes from the La Lopé 2016 samples or Rabai 2017 samples, which are the main target populations of this study. We first examined how differentiated the mosquitoes living in different habitats or different sampling sites are from each other. The SNP datasets for La Lopé and Rabai consist of 17,694 and 23,068 loci after filtering and removing siblings. Pairwise genetic distance (Fst) and genetic differentiation were
evaluated in GENEPOP v 4.7 [50, 51] using either the microsatellite or the SNP dataset. Expected heterozygo-sity (He) and observed heterozygoheterozygo-sity (Ho) were calcu-lated in adegenet (microsatellite) and SPAGeDi (SNP). Using the microsatellite data only, we also calculated
allelic richness and private allelic richness using HPRARE v1.1 [52]. For Rabai samples, we performed all analyses by pooling habitats and pooling sampling sites, respec-tively (Table 1). Even after pooling peridomestic and domestic individuals, the sample sizes of three Rabai vil-lages are too small for using microsatellite data to esti-mate many metrics reliably. Therefore, when we grouped Rabai samples by locations, we only reported the descrip-tive metrics calculated from the SNP data (Additional file 1: Tables S2, S3).
In addition to these descriptive metrics, we estimated the genetic ancestry of the mosquitoes using STRU CTU RE (microsatellite) and LEA (SNP), as described above. We also applied principal components analysis (PCA) in
adegenet (microsatellite) and LEA (SNP) to summarize
the genetic variability in our samples and visualize the genetic distances between individuals.
To further increase the power to detect any genetic structure in our fine spatial scale (3–13 km) within La Lopé or Rabai, we supplemented our population-level analysis with individual-based approaches. Specifically, we compared the kinship coefficient k calculated by SPAGeDi (see above) across different categories of indi-vidual pairs: within forest (both indiindi-viduals were col-lected in the forest), forest-village (one individual came from the forest and the other from the village), within village (both individuals were from the same village), and between villages (the two individuals were from different villages). The last category only applies to the Rabai sam-ples, and both peridomestic and domestic habitats were considered as the village. We did not further distinguish mosquito pairs from the same breeding sites or different breeding sites, because there are very few pairs from the same breeding sites. In addition to the categories by loca-tions, for analysis of the Rabai samples, we also applied an alternative method to categorize the individual pairs by habitats, which resulted in six categories (forest-forest, forest-peridomestic, forest-domestic, peridomestic-peri-domestic, peridomestic-peridomestic-peri-domestic, domestic-domestic). Differences between categories were tested by analysis of variance (ANOVA) with post-hoc multiple comparisons by t-test and Holm correction for the P-values. We used bootstrap with 1000 iterations to estimate the P-value of each test to account for the non-independence of the pairwise data.
The relatedness measures could be affected by geo-graphical distances as individuals collected further apart are more likely to be less related. Information about the exact sampling location of each mosquito was available in Rabai but not La Lopé. Therefore, for the Rabai dataset, we examined this potential distance effect using a Man-tel test in adegenet between the individual pairwise kin-ship coefficients (estimated with SNPs) and individual
Table 1 Sampling sites, habitats, and the number of samples in
the microsatellite and SNP dataset
a Number of samples in the microsatellite data (number of samples in the SNP
data)
b The forest and the four villages in Rabai, Kenya
c Number of samples when grouping Rabai samples by habitats d Number of samples when grouping Rabai samples by sampling sites
Sampling sites Forest Peridomestic
(village) Domestic (village) Total La Lopé 47 (11)a 36 (8) 83 (19)
Rabai total 60 (11)c 22 (12)c 40 (18)c 122 (41)
Kaya Bomu forestb 60 (11) 60 (11)d
Chang’ombeb 13 (6) 15 (3) 28 (9)d
Mbarekanib 5 (3) 7 (4) 12 (7)d
Bengob 2 (1) 11 (7) 13 (8)d
pairwise geographical distance, with 999 permutations. To remove this possible distance effect in the Rabai data, we applied a linear regression model. The model used pairwise kinship coefficients as the dependent variable and the pairwise geographical distance as the predic-tor. We then extracted the residuals from the model and used it to compare between categories described above. By using the residuals, we controlled for possible distance effects on kinship. We did not apply this linear model on the La Lopé dataset as, unfortunately, the geographical information was not available. Instead, we used the raw values of kinship estimates for the La Lopé analysis. If habitat does predict genetic clustering, we would expect reduced gene flow and hence lower genetic relatedness between the forest- and village-living mosquitoes.
Lastly, we constructed a phylogeny that consisted of both La Lopé and Rabai samples with Aedes
mascaren-sis as the outgroup, using 22,287 SNPs in IQTree v 1.6.12
[53]. IQTree conducts automatic model selection (option “MFP”) [54], ascertainment bias adjustment (option “ASC”) [55], and model violation check (option “bnni”). We performed ultrafast bootstraps (n = 1000) to estimate node certainty [56]. Nodes with support values smaller than 80 were collapsed using R package ape v 3.5.3 [57,
58].
Detecting heterogeneous differentiation across the genome
Habitat-associated genetic differentiation, if it exists, could vary across the genome. To examine this possibil-ity, we took advantage of the genome-wide distribution of the SNP loci and calculated Fst between forest and
vil-lage Ae. aegypti on each small segment of the genome (i.e. a genomic “window”). We then slid this “window” through the whole genome, i.e. a genome scan. For sim-plicity, peridomestic and domestic samples were grouped as village samples. We implemented this sliding-window analysis in VCFtools v 0.1.14 [59] with a window size of 1,000,000 bp and a sliding step of 10,000 bp (i.e. mov-ing the window for 10,000 bp between two consecutive tests). The mean number of SNPs per window is 12.3 in the La Lopé dataset and 16.0 in the Rabai dataset. Win-dows with fewer than three SNPs were removed, which represented 17.3% and 15.9% of all windows in La Lopé and Rabai populations, respectively.
Results
Inferring genetic ancestry
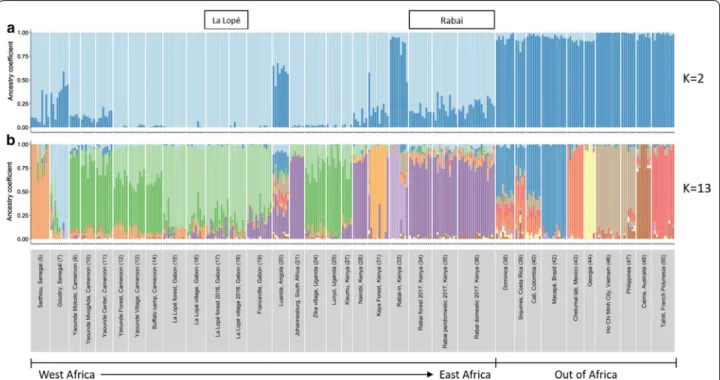
LEA and STRU CTU RE analysis confirmed that the 2016– 2017 collections of Ae. aegypti in La Lopé and Rabai were genetically similar to most other African populations. The best number of clusters was 13 for the LEA analy-sis using SNP data and two for the STRU CTU RE analyanaly-sis
using microsatellite data. However, in both analyses, the first split of all samples (i.e. allowing only two clusters or
K = 2) was between most African populations, including
our La Lopé and Rabai samples, and populations outside of Africa (Fig. 2a; Additional file 1: Figure S2a), with only a few exceptions (discussed in detail in Gloria-Soria et al. [9] and Kotsakiozi et al. [10]). This split roughly repre-sents the strong genetic differentiation between Aaf and
Aaa, as suggested in previous studies [7, 9, 11].
The snmf analysis in LEA with K = 13 (the best num-ber of clusters, Fig. 2b) demonstrated a more detailed population structure within Africa. La Lopé popula-tions and Rabai populapopula-tions were significantly geneti-cally distinct, consistent with the hypothesis that they represent independent habitat shift events. The forest and village mosquitoes in La Lopé from 2016 showed very similar genetic ancestries. They also resembled the samples collected from the same location in 2014 (Fig. 2b). As for Rabai samples, all mosquitoes from the 2017 collection clustered together, along with the populations from Nairobi, Kenya, and Johannesburg, South Africa (Fig. 2b). Clustering analysis showed little difference between individuals from different habitats (i.e. forest, peridomestic and domestic). Only minimal traces of the genetic ancestry differing from the main
Aaf cluster existed in the 2017 Rabai samples, which is
the ancestry similar to the Rabai indoor samples from 2009 and 2011 and some Asian-Pacific populations (i.e. an Aaa signal). These findings based on the SNP data were consistent with STRU CTU RE analysis with microsatellite data (Additional file 1: Figure S2, K = 5 to show more genetic clusters within Africa, despite K = 2 being the best model). In addition, PCA with microsat-ellite data and SNP data both showed that the 2016 La Lopé samples and the 2017 Rabai samples overlap with the majority of African populations (Additional file 1: Figure S3), which is again consistent with snmf and STRU CTU RE analysis results.
Population genetic structure of La Lopé and Rabai populations
Focusing separately on the La Lopé 2016 and Rabai 2017 populations, the expected and observed genetic het-erozygosity (He and Ho) of mosquitoes from different habitats or sampling sites were comparable (Additional file 1: Table S2). The average allelic richness of the 12 microsatellite loci was also similar between habitats in La Lopé and Rabai, respectively. Private allelic richness also did not differ largely between habitats in Rabai, yet in La Lopé, the forest population had more private alleles than the village population. The pairwise genetic differences (Fst) ranged from 0.0066 to 0.0336 when using
data (Additional file 1: Table S3). LEA and STRU CTU RE analysis consistently suggested K = 1 as the best model for both La Lopé and Rabai populations. When examin-ing results with a larger K (Fig. 3; Additional file 1: Fig-ures S4, S5, K = 2 as an example), both SNP-based and microsatellite-based analysis did not find evidence of genetic differences between individuals from different habitats or sampling sites, which was also supported by the PCA (Fig. 3; Additional file 1: Figures S4, S5).
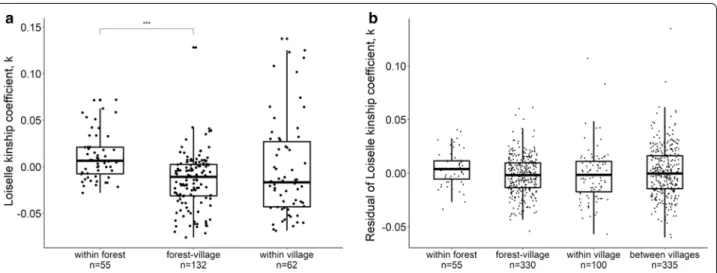
In La Lopé, pairwise kinship coefficients were higher in the “within forest” category than the “forest-village” category (Fig. 4a, ANOVA for all three categories of individual pairs: F = 10.31, bootstrap P < 0.001, post
hoc comparison-test: bootstrap P < 0.001).
Compari-sons between these two categories and the “within vil-lage” did not show a statistically significant difference (Fig. 4a). Due to the absence of geographical infor-mation associated with each mosquito individual, we could not examine the potential effect of geographi-cal distance in La Lopé. Such data were available for the Rabai samples. We found a negative correlation between geographical distance and kinship coefficients (r = −0.172, Mantel test P = 0.001, Additional file 1: Figure S6a). After removing the effect of distance, ANOVA did not show a significant difference between
categories by locations (Fig. 4b, F = 1.992, bootstrap
P = 0.097) or categories by habitats (Additional file 1: Figure S6b, F = 1.337, bootstrap P = 0.211). None of the pairwise comparisons between categories were sig-nificantly different.
The phylogeny constructed from the SNP data shows that mosquitoes from La Lopé and Rabai formed two monophyletic clades, and the split between them was highly supported (Additional file 1: Figure S7). Within the La Lopé clade, village individuals were nested within the forest individuals and form three groups. There was very little well-supported structure in the Rabai clade and no clear pattern of clustering by either habitats or sampling sites (Additional file 1: Figure S7).
Detecting heterogeneous differentiation across the genome
The sliding-window analysis revealed a peak of Fst
between the La Lopé forest and La Lopé village popula-tions on chromosome three, centered roughly at nucle-otide position 85,800,000 and spanning from position 84,990,000 to 86,960,000 (Fig. 5a). The region was anno-tated with 16 genes on VectorBase [60] (Additional file 1: Table S4). This region does not stand out in the same analysis for Rabai forest versus village samples (Fig. 5b).
Fig. 2 Genetic ancestry of the La Lopé and Rabai populations reported here (boxed names above figure) with a reference panel of the global
populations of Ae. aegypti, estimated using 23,767 SNP loci in LEA with K = 2 (a) and K = 13 (b). Each bar represents one individual. Different colors represent different genetic clusters, and the proportion of a color indicates the probability of an individual assigned to that genetic cluster. Population names and regions are listed on the x-axis. The numbers in the parentheses correspond to the population index in Additional file 1: Table S1 (column “ID”)
Discussion
The initial results of this study first show that mosquitoes in La Lopé, Gabon and Rabai, Kenya have genetic ances-try more similar to most other African Ae. aegypti pop-ulations than poppop-ulations outside of Africa. Within this general African ancestry group (Aaf), clustering analy-sis and the phylogenetic tree suggest a clear distinction between these two locations (Fig. 2; Additional file 1: Fig-ures S2, S7). This finding suggests that the La Lopé and Rabai populations likely evolved independently when col-onizing different habitats (forest vs villages). This is con-sistent with the conclusion of Brown et al. [7] suggesting multiple independent domestication events in Africa.
Considering each location separately, there is no strong genetic differentiation between mosquitoes in the forest and village habitats in La Lopé or Rabai. Although tests based on microsatellite loci resulted in statistically sig-nificant differences in a few pairs of populations (Addi-tional file 1: Table S3), the Fst values of these pairs are
small when compared with the genetic variation of Ae.
aegypti across Africa [9, 10]. Tests using the SNP data also found no significant difference between populations
in different habitats (Additional file 1: Table S3). Fst
esti-mates could be affected by our pooling of samples across the four villages in Rabai that are geographically sepa-rated (Fig. 1, Table 1). However, Fst between villages are
generally small and not statistically significant (Addi-tional file 1: Table S3), and heterozygosity measures are comparable across different habitat groups (Additional file 1: Table S2). This evidence, combined with the lack of population structure in the clustering analysis, PCA analysis, and phylogenetic analysis (Additional file 1: Figures S4–S7), suggested that pooling samples did not strongly confound our analysis or conclusions. Another potential suggestion of possible genetic differentiation is the higher relatedness between forest individuals in La Lopé (Fig. 4a). However, this may be confounded by the effects of geographic distance. Unfortunately, we do not have the mosquitoes’ geographical coordinates in La Lopé to test this hypothesis. The individual pairs within the La Lopé village have similar levels of relatedness as the across-habitat pairs, which does not support the hypothesis that a barrier of gene flow exists between the forest and village mosquitoes. Collectively, we conclude
Fig. 3 Genetic structure of the La Lopé (a and b) and Rabai (c and d) mosquitoes grouped by habitats, generated using 17,694 SNP loci and 23,068
SNP loci, respectively. a, c Results of the LEA analysis with K = 2. The habitats are labeled on top of each bar plot. b, d PCA biplots by LEA showing the first two principal components (PCs). The numbers in parentheses on the axes indicate the percentage of total variation explained by the PCs. The eclipses were drawn with an 80% confidence level
that there is little solid evidence supporting habitat-asso-ciated genetic divergence in either La Lopé or Rabai.
This genetic similarity between habitats may suggest frequent gene flow. This hypothesis is supported by the finding that between-habitat individual pairs have simi-lar levels of relatedness compared to within-habitat pairs, except for one comparison (Fig. 4, see discussion above for this exception). An alternative but not mutually exclu-sive hypothesis is that the habitat shift (i.e. invasion into a new habitat) happened relatively recently, so there has not been enough time to accumulate detectable genetic differentiation. It is also possible that habitat shifts and/ or expansion happen periodically. For example, forest mosquitoes may invade villages seasonally during the dry season seeking standing waters stored in households
to lay eggs [11]. It is likely that village mosquitoes in La Lopé and Rabai originated from the nearby forest habi-tats, as the forest is the ancestral habitat for the species [11, 12]. Yet, the data from the present study cannot rule out the possibility that village mosquitoes move to the forest. Regardless of the direction of migration, the lack of habitat-associated local genetic structure suggests that moving into different environments does not necessarily require, or result in, immediate genetic evolution.
Aedes aegypti were collected in both localities before
our 2016 and 2017 collections. The 2014 and 2016 sam-ples in La Lopé clustered together in LEA and STRU CTU RE analysis (Fig. 2; Additional file 1: Figure S2), which suggests temporal stability. However, in Rabai, the previ-ously documented genetically distinct indoor population
Fig. 4 Individual pairwise kinship coefficients of mosquito samples from La Lopé (a) and Rabai (b). Each point represents one mosquito pair. a For
La Lopé samples, mosquito pairs are grouped into three categories with the number of pairs labeled on the x-axis. Statistically significant differences between groups are determined by ANOVA with multiple comparisons by t-tests, and indicated by the brackets (significance levels: *P < 0.05, **P < 0.01, ***P < 0.001). b Residuals of the kinship coefficients in Rabai after removing distance effects grouped into four categories, with the number of pairs labeled on the x-axis. The boxplots show the median (the horizontal bar), interquartile range (IQR, the box), and 1.5 × IQR above and below the IQR (the vertical bar)
Fig. 5 Genetic difference (Fst) across the genome between the forest and village mosquitoes from La Lopé (a) and Rabai (b). Each point represents
(collected in 2009 and 2012) was not detected in 2017 (Fig. 2; Additional file 1: Figure S2). All Rabai 2017 sam-ples showed strong genetic similarity to the “Rabai-out” population in Brown et al. [7] and Gloria-Soria et al. [9] (Additional file 1: Figure S2), which contains forest and peridomestic samples from 2009 and 2012. This result suggests that mosquitoes in the forest and peridomestic habitats remain mostly stable while the indoor domes-tic mosquitoes were likely assimilated or replaced by the former. A significant recent change in the Rabai vil-lages is that villagers now have access to a centralized covered water source preventing mosquito breeding. This has eliminated the need to store water in open clay pots inside each hut, which were the source of indoor samples in the earlier studies [13, 26, 27, 61]. This likely caused the disappearance of Aaa in Rabai. However, it is important to note that the 2017 samples were taken over a single two-week period; it is conceivable that Aaa in Rabai varies seasonally and/or still exists in small pockets we did not sample. In addition, we cannot rule out the possibility that the temporal changes in Rabai were due to invasion from external sources. An ongoing project is focusing on better understanding the causes of these temporal variations of Rabai populations and identifying possible sources of external invasions. The findings will be reported in a future manuscript.
The genomic scan revealed variation in Fst between the
forest and village mosquitoes along the genome, includ-ing a strikinclud-ing high peak on the third chromosome in the La Lopé comparison. Most of the annotated genes in the peak region do not have an apparent connection with habitat adaptation (Additional file 1: Table S4). One gene of possible interest is a cytochrome P450 gene (AAEL003890 on VectorBase, or CYP6AG8 [60, 62]), which could be related to detoxication and insecticide resistance [63–67]. Further studies specifically focusing on this gene are needed to further confirm whether this gene was under diverging selection pressure related to habitats. Other possibilities that cause this peak of diver-gence include unaccounted external gene flow into the La Lopé populations, as well as possible chromosome inver-sions that accumulate genetic divergence [68–70]. This divergent genomic region was not found in the analysis of the Rabai populations (Fig. 5b) or the La Lopé samples from 2014 (data not shown). Therefore, it is likely specific to the La Lopé populations in 2016 instead of represent-ing a general mechanism of adaptation to the forest or human-made habitats.
Analysis in this study using a panel of ~17,000–25,000 SNP loci and 12 microsatellite loci reached the same general conclusions, which again proves the robustness of using these genetic markers in studies of the popula-tion genetic structure of Ae. aegypti [32, 71]. Multiallelic
loci like microsatellite and biallelic loci like SNP could behave differently due to their different nature (e.g. muta-tion rates) [68]. The congruence between them further supports the lack of detectable genetic differentiation between mosquitoes in the forest and anthropogenic habitats. When comparing the two types of markers, the SNP data has the advantage of allowing individual-based analysis, such as estimating kinship coefficients, which could contribute to addressing fine-scale questions. For example, in this study, we observed decreasing kinship between mosquito individuals further away from each other (Additional file 1: Figure S6a). In another recent study, Jasper et al. [72] developed a new methodology to use SNP based relatedness measures and spatial data to estimate dispersal in Ae. aegypti. Furthermore, the genome-wide SNP data allow explorations of variations across the genome and thus opens the opportunity to identify the genetic basis associated with any environ-mental changes. An intriguing demonstration is a recent study by Endersby-Harshman et al. [25], which found a genomic region strongly related to insecticide resistance. The sliding window Fst analysis used here also provides
some hints in this regard. Similar analyses could further benefit from using whole-genomic sequencing data pro-viding a higher resolution.
Conclusions
The genetic similarities between Ae. aegypti collected in forests and villages in La Lopé and Rabai suggest that colonizing different habitats does not necessarily accompany or require substantial genomic differentia-tion. This is consistent with previous observations of locally domesticated Aaf in multiple locations in Africa [17–19]. The finding is relevant for disease monitoring and control. Although this study did not explicitly test migration, the lack of genetic differentiation and the proximity of forest and village Ae. aegypti suggested a possibility of gene flow between habitats. If this is the case, mosquito surveillance and control could benefit from considering the sylvatic mosquito populations. For instance, control programmes like insecticide application usually focus on mosquito populations closely associated with humans, but these alone would not be effectual if the sylvatic habitats are reservoirs of potentially domestic-living Ae. aegypti. Also, if mosqui-toes readily move between habitats, they could poten-tially introduce new pathogens or strains of viruses from the forest reservoirs to the human communities, which could lead to unexpected epidemiological conse-quences. In addition, this absence of habitat-associated genetic barriers in Africa raises interesting questions about the evolutionary history of Ae. aegypti. For exam-ple, when and how did the domestic adaptations come
about in the process of Ae. aegypti becoming a human specialist if switching habitats does not necessarily link to genomic differentiation? Why do the Aaa popula-tions outside of Africa rarely move back into sylvatic habitats with only a few exceptions, such as in the Car-ibbean and Argentina [73, 74]? Will the village- and urban-living Aaf in Africa evolve to be more domesti-cated in parallel with their American and Asian coun-terparts? Addressing these questions could deepen our understanding of the global invasion of Ae. aegypti and their successful utilization of the domestic habitats. It could also provide insights into improving mosquito control, especially in African countries where habitat shifts occur repeatedly.
Supplementary information
Supplementary information accompanies this paper at https ://doi.
org/10.1186/s1307 1-020-04278 -w.
Additional file 1: Table S1. Information on the global panel of Ae. aegypti
populations included in this study. Table S2. Allelic richness, private allelic richness, expected heterozygosity (He) and observed heterozygosity (Ho) of La Lopé and Rabai samples grouped by habitats or sampling sites.
Table S3. Pairwise Fst of La Lopé and Rabai samples grouped by habitats
or sampling sites. Table S4. Annotated genes in the region of the Fst peak in La Lopé populations. Figure S1. Geographical locations of the global panel of Ae. aegypti populations included in this study. Figure S2. Genetic ancestry of the La Lopé and Rabai populations with a reference panel of global populations of Ae. aegypti. Figure S3. PCA of the La Lopé and Rabai populations with the global panel of populations. Figure S4. Genetic structure of the Rabai mosquito samples grouped by sampling sites.
Fig-ure S5. Results of STRU CTU RE analysis and PCA using microsatellite data. Figure S6. Individual pairwise kinship coefficients of mosquito samples
from Rabai. Figure S7. Phylogeny of the La Lopé and Rabai mosquitoes.
Abbreviations
Aaa: Aedes aegypti aegypti; Aaf: Aedes aegypti formosus; ANOVA: Analysis of vari-ance; MCMC: Markov chain Monte Carlo; PCA: Principal components analysis; Fst: Fixation index; He: Expected heterozygosity; Ho: Observed heterozygosity;
SNPs: Single nucleotide polymorphisms.
Acknowledgements
We thank Institut de Recherche pour le Développement (IRD) and the research Unit ESV-GAB at the Centre International de Rrecherches médicales de Franceville (CIRMF) in Gabon, and Kenya Medical Research Institute (KEMRI) in Kenya for their supports during the field sampling in La Lopé and Rabai. We also want to thank Tom Chiodo for performing the lab work to gener-ate the microsgener-atellite data, and Andrea Gloria-Soria for providing comments and suggestions on analysis and the first draft. We appreciate all the helpful discussions and suggestions from Gisella Caccone, Panagiota Kotsakiozi, John Soghigian, Norah Saarman, Evlyn Pless, Maud Quinzin, Stephen Gaughran, and Evelyn Jensen. We are grateful for the advice and discussion from Lindy McBride, Stephen Stearns, John Carlson, and Alvaro Sanchez. Lastly, we thank the Functional Genomics Core at the University of North Carolina, Chapel Hill for generating the SNP data.
Authors’ contributions
SX and JRP designed and conceptualized the study. SX, DA, NR, MFN, RS, and JL coordinated the fieldwork and collected mosquito samples. SX carried out the molecular lab work to generate the data. SX and LVC performed the analysis. SX drafted the manuscript. JRP coordinated the entire study, provided funding, and contributed to interpreting results. All authors provided
critical feedback on the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by NIH RO1 AI101112 to JRP.
Availability of data and materials
The microsatellite and SNP data of the new samples from Gabon and Kenya were stored on VectorBase.org with project ID VBP0000625. The microsatellite and SNP data of the reference panel can be found on VectorBase.org with project ID VBP0000138 and VBP0000295.
Ethics approval and consent to participate
The field mosquito collection in La Lopé was approved by the CENAREST under the national research authorization AR0013/16/MESRS/CENAREST/CG/ CST/CSAR. The forest specimens collected in the La Lopé National Parks were authorized under the national park entry authorization AE16008/PR/ANPN/ SE/CS/AEPN. Lastly, human-landing catches were approved by the National Research Ethics Committee of Gabon under authorization 0031/2014/SG/ CNE. We obtained oral and written consent documents from all volunteers participating in the human-landing catches. The study in Rabai was approved by the Kenya Medical Research Institute Scientific and Ethical Review Unit (KEMRI/SERU/3433).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Author details
1 Department of Ecology and Evolutionary Biology, Yale University, New
Haven, Connecticut, USA. 2 Arbovirus/Viral Hemorrhagic Fever Laboratory,
Center for Virus Research, Kenya Medical Research Institute (KEMRI), Nairobi, Kenya. 3 CIRMF, Franceville, Gabon. 4 ANPN, Libreville, Gabon. 5 MIVEGEC, IRD,
CNRS, Univ. Montpellier, Montpellier, France. Received: 17 March 2020 Accepted: 3 August 2020
References
1. Gottdenker NL, Streicker DG, Faust CL, Carroll C. Anthropogenic land use change and infectious diseases: a review of the evidence. EcoHealth. 2014;11:619–32.
2. Ramasamy R, Surendran SN. Mosquito vectors developing in atypical anthropogenic habitats: global overview of recent observations, mecha-nisms and impact on disease transmission. J Vector Borne Dis. 2016;53:91. 3. Li MI, Wong PS, Ng LC, Tan CH. Oral susceptibility of Singapore Aedes
(Stegomyia) aegypti (Linnaeus) to Zika virus. PLoS Negl Trop Dis. 2012;6:e1792.
4. WHO. A global brief on vector-borne diseases. Geneva: World Health Organization; 2014. https ://www.who.int/campa igns/world -healt h-day/2014/globa l-brief /en/
5. Kraemer MU, Sinka ME, Duda KA, Mylne AQ, Shearer FM, Barker CM, et al. The global distribution of the arbovirus vectors Aedes aegypti and
Ae. albopictus. Elife. 2015;4:e08347.
6. Bennett KL, Shija F, Linton YM, Misinzo G, Kaddumukasa M, Djouaka R, et al. Historical environmental change in Africa drives divergence and admixture of Aedes aegypti mosquitoes: a precursor to successful worldwide colonization? Mol Ecol. 2016;25:4337–54.
7. Brown JE, McBride CS, Johnson P, Ritchie S, Paupy C, Bossin H, et al. Worldwide patterns of genetic differentiation imply multiple ‛domesticationsʼ of Aedes aegypti, a major vector of human diseases. Proc Biol Sci. 2011;278:2446–54.
8. Crawford JE, Alves JM, Palmer WJ, Day JP, Sylla M, Ramasamy R, et al. Population genomics reveals that an anthropophilic popula-tion of Aedes aegypti mosquitoes in West Africa recently gave rise to
American and Asian populations of this major disease vector. BMC Biol. 2017;15:16.
9. Gloria-Soria A, Ayala D, Bheecarry A, Calderon-Arguedas O, Chadee DD, Chiappero M, et al. Global genetic diversity of Aedes aegypti. Mol Ecol. 2016;25:5377–95.
10. Kotsakiozi P, Evans BR, Gloria-Soria A, Kamgang B, Mayanja M, Lutwama J, et al. Population structure of a vector of human diseases: Aedes aegypti in its ancestral range. Africa. Ecol Evol. 2018;8:7835–48.
11. Powell JR, Tabachnick WJ. History of domestication and spread of Aedes
aegypti-A Review. Mem Inst Oswaldo Cruz. 2013;108:11–7.
12. Brown JE, Evans BR, Zheng W, Obas V, Barrera-Martinez L, Egizi A, et al. Human impacts have shaped historical and recent evolution in Aedes aegypti, the dengue and yellow fever mosquito. Evolution. 2014;68:514–25.
13. McBride CS, Baier F, Omondi AB, Spitzer SA, Lutomiah J, Sang R, et al. Evo-lution of mosquito preference for humans linked to an odorant receptor. Nature. 2014;515:222–7.
14. Failloux A-B, Vazeille M, Rodhain F. Geographic genetic variation in populations of the dengue virus vector Aedes aegypti. J Mol Evol. 2002;55:653–63.
15. Mattingly P. Taxonomy of Aedes aegypti and related species. Bull World Health Organ. 1967;36:552.
16. Mattingly P. Genetical aspects of the Aëdes aegypti Problem: I. Taxonomy and bionomics. Ann Trop Med Parasitol. 1957;51:392–408.
17. Paupy C, Brengues C, Kamgang B, Hervé J-P, Fontenille D, Simard F. Gene flow between domestic and sylvan populations of Aedes aegypti (Diptera: Culicidae) in North Cameroon. J Med Entomol. 2014;45:391–400. 18. Paupy C, Brengues C, Ndiath O, Toty C, Hervé J-P, Simard F. Morphological
and genetic variability within Aedes aegypti in Niakhar. Senegal. Infect Genet Evol. 2010;10:473–80.
19. Sylla M, Bosio C, Urdaneta-Marquez L, Ndiaye M, Black WC IV. Gene flow, subspecies composition, and dengue virus-2 susceptibility among Aedes
aegypti collections in Senegal. PLoS Negl Trop Dis. 2009;3:e408.
20. Nosil P, Funk DJ, Ortiz-Barrientos D. Divergent selection and heterogene-ous genomic divergence. Mol Ecol. 2009;18:375–402.
21. Rose NH, Sylla M, Badolo A, Lutomiah J, Ayala D, Aribodor OB, et al. Climate and urbanization drive mosquito preference for humans. BioRxiv. 2020. https ://doi.org/10.1101/2020.02.12.93904 1.
22. Yan G, Chadee DD, Severson DW. Evidence for genetic hitchhiking effect associated with insecticide resistance in Aedes aegypti. Genetics. 1998;148:793–800.
23. Matthews BJ, Dudchenko O, Kingan SB, Koren S, Antoshechkin I, Crawford JE, et al. Improved reference genome of Aedes aegypti informs arbovirus vector control. Nature. 2018;563:501–7.
24. Marcombe S, Paris M, Paupy C, Bringuier C, Yebakima A, Chandre F, et al. Insecticide-driven patterns of genetic variation in the dengue vector
Aedes aegypti in Martinique Island. PLoS One. 2013;8:e77857.
25. Endersby-Harshman NM, Schmidt TL, Chung J, van Rooyen A, Weeks AR, Hoffmann AA. Heterogeneous genetic invasions of three insecticide resistance mutations in Indo-Pacific populations of Aedes aegypti (L.). Mol Ecol. 2020;29:1628–41.
26. Petersen JL. Behavioral differences in two subspecies of Aedes aegypti (L.) (Diptera: Culicidae) in East Africa. Notre Dame: PhD Thesis, University of Notre Dame; 1977.
27. Tabachnick WJ, Powell JR. Genetic structure of the East African domestic populations of Aedes aegypti. Nature. 1978;272:535.
28. Trpis M, Hausermann W. Genetics of house-entering behaviour in East African populations of Aedes aegypti (L.) (Diptera: Culicidae) and its relevance to speciation. Bull Entomol Res. 1978;68:521–32.
29. Evans BR, Gloria-Soria A, Hou L, McBride C, Bonizzoni M, Zhao H, et al. A multipurpose, high-throughput single-nucleotide polymorphism chip for the dengue and yellow fever mosquito, Aedes aegypti. G3. 2015;5:711–8. 30. Brown JE, Scholte E-J, Dik M, Den Hartog W, Beeuwkes J, Powell JR. Aedes
aegypti mosquitoes imported into the Netherlands, 2010. Emerging
Infect Dis. 2011;17:2335.
31. Gloria-Soria A, Lima A, Lovin DD, Cunningham JM, Severson DW, Powell JR. Origin of a high-latitude population of Aedes aegypti in Washington. DC. Am J Trop Med Hyg. 2018;98:445–52.
32. Pless E, Gloria-Soria A, Evans BR, Kramer V, Bolling BG, Tabachnick WJ, et al. Multiple introductions of the dengue vector, Aedes aegypti, into California. PLoS Negl Trop Dis. 2017;11:e0005718.
33. Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7.
34. Purcell S, Chang C. PLINK 1.9. www.cog-genom ics.org/plink /1.9/. 35. Abreu FVS, Morais MM, Ribeiro SP, Eiras ÁE. Influence of breeding site
availability on the oviposition behaviour of Aedes aegypti. Mem Inst Oswaldo Cruz. 2015;110:669–76.
36. Loiselle BA, Sork VL, Nason J, Graham C. Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am J Bot. 1995;82:1420–5.
37. Hardy OJ, Vekemans X. SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes. 2002;2:618–20.
38. Schmidt TL, Filipović I, Hoffmann AA, Rašić G. Fine-scale landscape genomics helps explain the slow spatial spread of Wolbachia through the
Aedes aegypti population in Cairns. Australia. Heredity. 2018;120:386.
39. Tabachnick WJ, Munstermann LE, Powell JR. Genetic distinctness of sympatric forms of Aedes aegypti in East Africa. Evolution. 1979:287–95. 40. Nazareno AG, Bemmels JB, Dick CW, Lohmann LG. Minimum sample sizes
for population genomics: an empirical study from an Amazonian plant species. Mol Ecol Resour. 2017;17:1136–47.
41. Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59. 42. Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of
individuals using the software STRU CTU RE: a simulation study. Mol Ecol. 2005;14:2611–20.
43. Earl DA. STRU CTU RE HARVESTER: a website and program for visualizing STRU CTU RE output and implementing the Evanno method. Conserv Genet Resour. 2012;4:359–61.
44. Jakobsson M, Rosenberg NA. CLUMPP: a cluster matching and permuta-tion program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23:1801–6. 45. Francis RM. pophelper: an R package and web app to analyse and
visual-ize population structure. Mol Ecol Resour. 2017;17:27–32.
46. R Development Core Team: R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2018. 47. Jombart T. adegenet: a R package for the multivariate analysis of genetic
markers. Bioinformatics. 2008;24:1403–5.
48. Jombart T, Ahmed I. Adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics. 2011;27:3070–1.
49. Frichot E, François O. LEA: an R package for landscape and ecological association studies. Methods Ecol Evol. 2015;6:925–9.
50. Raymond M. GENEPOP (version 12): population genetics software for exact tests and ecumenicism. J Hered. 1995;86:248–9.
51. Rousset F. genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour. 2008;8:103–6. 52. Kalinowski ST. hp-rare 10: a computer program for performing rarefaction
on measures of allelic richness. Mol Ecol Notes. 2005;5:187–9. 53. Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and
effective stochastic algorithm for estimating maximum-likelihood phy-logenies. Mol Biol Evol. 2014;32:268–74.
54. Kalyaanamoorthy S, Minh BQ, Wong TK, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587.
55. Lewis PO. A likelihood approach to estimating phylogeny from discrete morphological character data. Syst Biol. 2001;50:913–25.
56. Hoang DT, Chernomor O, Von Haeseler A, Minh BQ, Vinh LS. UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol. 2017;35:518–22.
57. Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolu-tion in R language. Bioinformatics. 2004;20:289–90.
58. Popescu A-A, Huber KT, Paradis E. ape 3.0: New tools for distance-based phylogenetics and evolutionary analysis in R. Bioinformatics. 2012;28:1536–7.
59. Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8. 60. Giraldo-Calderón GI, Emrich SJ, MacCallum RM, Maslen G, Dialynas
E, Topalis P, et al. VectorBase: an updated bioinformatics resource for invertebrate vectors and other organisms related with human diseases. Nucleic Acids Res. 2014;43:D707–13.
61. Trpis M, Hausermann W. Demonstration of differential domesticity of
Aedes aegypti (L.) (Diptera, Culicidae) in Africa by mark-release-recapture.
Bull Entomol Res. 1975;65:199–208.
62. Reid W. Genomics of permthrin resistance in the Southern house mos-quito, Culex quinquefasciatus Say. Auburn: PhD Thesis, Auburn University; 2014.
63. Bariami V, Jones CM, Poupardin R, Vontas J, Ranson H. Gene amplification, ABC transporters and cytochrome P450s: unraveling the molecular basis of pyrethroid resistance in the dengue vector, Aedes aegypti. PLoS Negl Trop Dis. 2012;6:e1692.
64. Stevenson BJ, Pignatelli P, Nikou D, Paine MJI. Pinpointing P450s associ-ated with pyrethroid metabolism in the dengue vector, Aedes aegypti: developing new tools to combat insecticide resistance. PLoS Negl Trop Dis. 2012;6:e1595.
65. David J-P, Ismail HM, Chandor-Proust A, Paine MJI. Role of cytochrome P450s in insecticide resistance: impact on the control of mosquito-borne diseases and use of insecticides on Earth. Philos Trans R Soc Lond, Ser B: Biol Sci. 2013;368:20120429.
66. Bergé J, Feyereisen R, Amichot M. Cytochrome P450 monooxygenases and insecticide resistance in insects. Philos Trans R Soc Lond, Ser B: Biol Sci. 1998;353:1701–5.
67. Scott JG, Liu N, Wen Z. Insect cytochromes P450: diversity, insecticide resistance and tolerance to plant toxins. Comp Biochem Physiol C, Phar-macol Toxicol Endocrinol. 1998;121:147–55.
68. Ayala D, Fontaine MC, Cohuet A, Fontenille D, Vitalis R, Simard F. Chromo-somal inversions, natural selection and adaptation in the malaria vector
Anopheles funestus. Mol Biol Evol. 2011;28:745–58.
69. Kirkpatrick M, Barton N. Chromosome inversions, local adaptation and speciation. Genetics. 2006;173:419–34.
70. Redmond SN, Sharma A, Sharakhov I, Tu Z, Sharakhova M, Neafsey DE. Linked-read sequencing identifies abundant microinversions and intro-gression in the arboviral vector Aedes aegypti. BMC Biol. 2020;18:26. 71. Kotsakiozi P, Gloria-Soria A, Schaffner F, Robert V, Powell JR. Aedes aegypti
in the Black Sea: recent introduction or ancient remnant? Parasit Vectors. 2018;11:396.
72. Jasper M, Schmidt TL, Ahmad NW, Sinkins SP, Hoffmann AA. A genomic approach to inferring kinship reveals limited intergenerational dispersal in the yellow fever mosquito. Mol Ecol Resour. 2019;19:1254–64. 73. Chadee DD, Ward RA, Novak RJ. Natural habitats of Aedes aegypti in the
Caribbean - a review. J Am Mosq Control Assoc. 1998;14:5–11.
74. Mangudo C, Aparicio JP, Gleiser RM. Tree holes as larval habitats for Aedes
aegypti in urban, suburban and forest habitats in a dengue affected area.
Bull Entomol Res. 2015;105:679–84. Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in pub-lished maps and institutional affiliations.