FACULTE DE MEDECINE ET DE PHARMACIE -RABAT- ANNEE: 200 THESE N°:
L
L
e
e
s
s
c
c
o
o
m
m
p
p
l
l
i
i
c
c
a
a
t
t
i
i
o
o
n
n
s
s
g
g
r
r
a
a
v
v
e
e
s
s
d
d
e
e
l
l
a
a
d
d
r
r
é
é
p
p
a
a
n
n
o
o
c
c
y
y
t
t
o
o
s
s
e
e
THESE
Présentée et soutenue publiquement le :………..
PAR
Mlle. Ghizlane EL BADAOUI
Née le 11 Février 1984 à Rabat
Pour l'Obtention du Doctorat en
Médecine
MOTS CLES Drépanocytose – Syndrome thoracique aigu – Infections – Complications graves - Anesthésie
JURY
Mr. A. AZZOUZI PRESIDENT &
A mes très chers parents
Aucun mot, aucune dédicace ne saurait exprimer mon respect, ma considération , ma reconnaissance et l’amour éternel , pour les sacrifices que vous avez consentis pour mon instruction et mon bien être.
Ce travail vous est particulièrement dédié , tout le mérite vous revient. Que dieu , le tout puissant , vous garde et vous procure santé , bonheur et longue vie pour que vous demeuriez le flambeau illuminant notre chemin.
A mon cher frère khalid
En témoignage de la profonde affection et l’indéfectible attachement qui nous lie.
Je te souhaite une vie pleine de succès , santé et prospérité.
A ma chère sœur Milham Najdaa
Qu’il me soit permis de t’exprimer ici, toutes mes reconnaissances , mes remerciements et mon grand amour.
A mon cher frère Taha
Beaucoup de volonté et d’enthousiasme, c’est ainsi que tu pourras arriver à très plus hauts désirs.
A mes deux grand-mères.
A mes oncles et leurs épouses.
A mes tantes et leurs maris.
A mes cousins et cousines.
A toute la famille
Avec mes sentiments de respect et d’affection.
A tous mes amis
Que ce travail soit un gage d’amitié , en vous souhaitant le bonheur et la réussite.
A ma promotion de médecine
A tous ceux qui ont participé de près ou de loin
dans l’élaboration de ce travail.
A mon rapporteur et président de thèse
Monsieur le professeur ABDERRAHIM AZZOUZI
Professeur d'anesthésie réanimation
C’est un grand honneur pour nous de vous voir présider cette thèse. Nous vous prions de bien vouloir, cher maître , accepter le témoignage de notre profonde reconnaissance et notre grande estime.
A mon maître et juge de thèse
Monsieur le professeur IFRINE LAHSSAN
Professur de chirurgie générale
Je vous remercie de m’honorer en siégeant parmi le jury de cette thèse. Votre modestie et votre gentillesse nous particulièrement marqué. Veuillez trouver dans ce travail le témoignage de ma grande estime.
A mon maître et juge de thèse
Monsieur le professeur EL HIJRI AHMED
Professeur agrégé d'anesthésie réanimation
Vous nous avez fait le grand honneur d’être membre jury de cette thèse. Votre générosité, votre compréhension et votre modestie recueillent ma très grande estime.
A mon maître et juge de thèse
Monsieur le professeur HARMOUCHE HICHAM.
Professeur agrégé de médecine interne
Nous vous présentons nos remerciements pour l’intérêt que vous avez bien voulu porte à ce travail , en acceptant d’être parmi le jury de notre thèse.
ECG : électrocardiogramme
EFR : exploration fonctionnelle respiratoire
RPM : rupture prématurée de membrane
PDP : prélèvement distal protégé
ECBU : examen cytobactériologique des urines
Observations cliniques ... 3 Discussion ... 10 1er chapitre : la drépanocytose ... 11 Historique ... 12 Généralités- génétique ... 14 Epidémiologie ... 16 Physiopathologie ... 21 1-echelon moléculaire ... 21 2-echelon cellulaire ... 22 3-echelon vasculaire ... 23 Diagnostic biologique ... 26 A-moyens de diagnostic ... 26 B-diagnostic anténatal ... 28 C-diagnostic néonatal ... 29 Manifestations cliniques ... 30 A-drépanocytose hétérozygote ... 30 B-drépanocytose homozygote ... 30
1-de 03mois à cinq ans ... 30
2-de cinq ans à l’adolescence ... 32
3-Adolescence ... 33
4-Adulte ... 33
2ème chapitre : complications graves de la drépanocytose ... 36
Infection et drépanocytose ... 45 A- ... 46 1-physiopathologie ... 46 2-germes en cause ... 47 3-traitements préventifs ... 49 B-types d’infections ... 50 1-sépticémies ... 50 2-méningite ... 50 3-ostéomyélite ... 51 4-infections pulmonaires ... 51 5-infections urinaires ... 51
C- conduite à tenir devant une infection chez le drépanocytaire ... 53
1-principes généraux ... 53
2- devant une fièvre isolée ... 54
3- principaux antibiotiques : posologies, voies d’administration . 55 Atteinte neurologique centrale chez le drépanocytaire ... 56
A- Facteurs de risque et mortalité des accidents vasculaires cérébraux ... 56
B-Physiopathologie des accidents vasculaires cérébraux ... 57
C- clinique ... 57
1-Accidents ischémiques constitués ... 57
2- Accidents vasculaires cérébraux hémorragiques ... 58
D-traitement ... 58
E- Prévention des accidents vasculaires cérébraux chez le drépanocytaire ... 59
C-infarctus osseux ... 62
D-infections ostéo-articulaires ... 62
E-ostéonécrose aseptique ... 65
Autres complications ... 67
A-anémie aigue et aggravation de l’anémie chronique ... 67
B-priapisme ... 70
C-complications dermatologiques : les ulcères de jambe ... 72
D-atteinte rétinienne : la rétinopathie drépanocytaire ... 73
E-complications cardiaques ... 74
F-complications digestives et hépato-biliaires ... 74
G-complications rénales ... 75
3ème chapitre : drépanocytose et anesthésie ... 76
Introduction ... 77
Prise en charge préopératoire ... 78
A-évaluation pré-anesthésique ... 78
B-préparation à l’intervention ... 78
1-mesures générales ... 78
2-prise en charge transfusionnelle ... 79
Anesthésie chez le drépanocytaire ... 82
1-généralités ... 82
2-anesthésie générale ... 83
Prise en charge post-opératoire ... 90
Situation particulière : la cholécystectomie chez le drépanocytaire ... 92
Drépanocytose et grossesse. ... 93
Résumé ... 95
La drépanocytose est la maladie génétique humaine la plus fréquente. Elle correspond à la synthèse d'une hémoglobine (Hb) anormale, l'HbS, dont la polymérisation à l’état désoxygéné est à l’origine d’une anémie hémolytique chronique et de phénomènes vaso-occlusifs.
La maladie est surtout fréquente dans les populations d'origine africaine sub-saharienne. En raison des mouvements récents de population qui caractérisent notre époque, elle existe aujourd'hui sur tous les continents.
Les complications qui menacent de façon aiguë le sujet drépanocytaire sont nombreuses et graves : crises douloureuses intenses, syndrome thoracique aigu, infections graves à type de septicémie ; en outre de nombreuses complications chroniques, sources d’handicaps, peuvent survenir : rétinopathie, ulcères cutanés, nécroses osseuses….
Au cours des trois dernières décennies, la prise en charge thérapeutique s’est considérablement améliorée permettant ainsi une augmentation de l’espérance de vie, et par conséquent l’accroissement des adultes drépanocytaires.
Le traitement conventionnel est essentiel dans la drépanocytose, il comprend antibiothérapie et vaccination, antalgiques, transfusion sanguine, l’hydroxyurée et la transplantation médullaire sont l’objet de recherches en cours.
La drépanocytose constitue un défi en termes de santé publique, à la fois par des complications aigues, mais aussi et surtout par les handicaps prolongés qu’elle est susceptible d’entrainer.
Observation clinique 1(STA: Syndrome thoracique aigu)
K.Z. âgée de 28 ans, connue porteuse d’une drépanocytose homozygote S/S a été opérée pour lithiase vésiculaire pigmentaire simple à ciel ouvert. Le taux d’hémoglobine pré- opératoire était de 8,3g.dl-1
et les leucocytes à 12 000 élts.mm-3. Le reste du bilan était normal (fonction rénale, hémostase, exploration fonctionnelle respiratoire et échocardiographie). Les suites opératoires immédiates ont été marquées par l’apparition d’un ictère et de douleurs osseuses diffuses. Le traitement a consisté en une oxygénothérapie nasale, une analgésie par paracétamol 4 g/j et une hydratation par sérum salé (SS) et sérum glucosé (SG) isotonique 4 l/j. Au 3è jour post opératoire sont apparues une dyspnée intense et une fièvre motivant l’admission en réanimation. La patiente avait à son admission une polypnée à 40 cycles/min, une tachycardie à 140bpm et une température à 39,5°c. La pression artérielle était de 120/80mmHg. La gazométrie artérielle sous 6l d’02 nasal montrait : une PaO2 à 73mmHg, une
PaCO2 à 43mmHg, un pH à 7,45 et un taux de bicarbonates à 32mmol/l. Le taux
d’hémoglobine était de 6,9g.dl-1
, les leucocytes à 30 000 élts.mm-3, les plaquettes à 390 000 élts.mm-3, le taux de prothrombine à 42% et le temps de céphaline activée à 54’’/30’’. Les enzymes hépatiques ASAT et ALAT étaient normales mais le taux de la LDH était à 1638 UI.l-1 soit environ dix fois la normale. La radiographie thoracique montrait un infiltrat bilatéral diffus. Le traitement consistait en une ventilation artificielle contrôlée sous sédation continue par midazolam et fentanyl, à 60% de FiO2 et avec une Pression
expiratoire positive à 8 cmH20 ; une antibiothérapie probabiliste à large spectre
SG isotonique. L’évolution s’est faite lentement vers l’amélioration avec : obtention de l’apyrexie au 4è
jour de traitement antibiotique, amélioration du rapport PaO2/FiO2 devenu supérieur à 200 après 3 jours de ventilation et
supérieur à 300 après 5 jours. La dernière gazométrie artérielle avant le sevrage de la ventilation réalisé au 7è jour montrait à 40% de FiO2 : une PaO2 à 146
mmHg, une PaCO2 à 35 mmHg, un pH à 7,40 et un taux de bicarbonates
22mmol/l. Signalons l’apparition au 3è jour d’un emphysème sous cutané
bilatéral étendu jusqu’à l’abdomen ayant régressé après drainage pleural. Sur le plan biologique, la normalisation de l’hémostase s’est faite au 8è
jour et une hyperplaquettose s’est progressivement installée pour atteindre 934 000 élts. mm-3 à la sortie de réanimation au 14è jour. Les prélèvements bactériologiques pulmonaires, urinaires et sanguins sont tous revenus négatifs.
Observation clinique 2 (sepsis sévère/drépanocytose)
Mr. R.M âgé de 28ans est hospitalisé le 27 septembre 2000, dans le service de réanimation chirurgicale pour sepsis sévère sur drépanocytose.
Il est suivi depuis l’âge de 6ans pour drépanocytose homozygote S/S. Il a deux sœurs porteuses de la même hémoglobinopathie et dont l’une d’entre elles est décédée en 1982 dans les suites d’un syndrome infectieux.
Le début de la symptomatologie remonte à deux jours avant son admission en réanimation par la survenue de douleurs lombaires et abdominales quelques heures après un passage au bain maure, motivant ainsi l’hospitalisation initiale dans le service de médecine interne.
Au 2ème jour de son admission, le patient a présenté un tableau infectieux sévère avec une fièvre à 38,9°C, une polypnée à 43 cycles/min, une tachycardie à 100bpm et des sueurs profuses. La pression artérielle était à 160/50mmHg. Aucun point d’appel urinaire, respiratoire, digestif n’a été relevé.
Le bilan biologique montre une hyperleucocytose à 34000 élts/mm³, une anémie à 5g/dl d’Hb. L’ionogramme sanguin et le bilan de crase sanguine étaient corrects.
Le diagnostic de crise drépanocytaire est soulevé, et le patient est mis sous traitement symptomatique : réhydratation par SS 9%° et SG isotonique à raison de 4l/j, analgésie par du paracétamol 4g/j, transfusion de deux culots globulaires, et oxygénothérapie nasale à raison de 8l/min.
L’évolution était marquée par la survenue d’une détresse respiratoire et de troubles neurologiques motivant le transfert du patient au service de réanimation.
A son admission en réanimation l’examen clinique trouve un patient conscient avec un score de Glascow (GCS) à 15, sans déficit neurologique ou de focalisation, fébrile à 40°C, polypnéique à 45 cycles/min. La pression artérielle était à 90/50mmHg. L’auscultation pulmonaire révélait des râles crépitants diffus au niveau des deux champs pulmonaires, plus accentués à gauche. Le patient était par ailleurs oligurique et ictérique.
Le bilan biologique montrait une hyperleucocytose à 46000élts/mm³, une anémie à 6g/dl d’Hb et un taux de plaquettes à 189000élts/mm³, une insuffisance rénale avec urée sanguine à 25mmol/l, et une créatinémie à 260mmol/l. La natrémie était à 132mmol/l et la kaliémie à 6mmol/l.
La gazométrie artérielle montrait une acidose sévère mixte avec un pH à 7,05, une réserve alcaline à 16mEq/l, une PaO2 à 55mmHg et une PaCO2 à
60mmHg.
La radiographie thoracique montrait un aspect d’OAP.(œdème aigu du poumon).
Le traitement a consisté en une ventilation artificielle, un remplissage vasculaire, une transfusion par 4 culots globulaires, une alcalinisation et une
Le bilan infectieux est venu confirmer le diagnostic de septicémie avec isolement d’un staphylocoque aureus sur 2 hémocultures.
Le staphylocoque en question est méthicilline résistant, sensible aux phénicolés, tétracyclines, macrolides, sulfamides, quinolones et glycopeptides.
Les autres prélèvements bactériologiques (ECBU, PDP) sont revenus négatifs.
Observation 3:
Le patient L.M, âgé de 22ans, drépanocytaire depuis l’enfance, a été admis dans un tableau de troubles de conscience avec une détresse respiratoire sur OAP.
Les troubles se sont installés de façon brutale 24 heures auparavant. L’examen à l’admission aux urgences trouvait un patient comateux, avec un GCS à 9, et une hémiparésie droite. Le patient était apyrétique, polypneique à 30cycles/min avec signes de lute. L’auscultation retrouvait des râles ronflants et crépitant bilatéraux. La SpO2 était à 88% sous 6L/min d’oxygène. La pression artérielle était à 210/120mmHg et la fréquence cardiaque à 120batt/min.
Le patient a bénéficié d’une intubation et d’une ventilation artificielle. Le scanner cérébral a retrouvé de multiples petites lésions ischémiques bilatérales.
Le bilan biologique a montré une insuffisance rénale sévère avec une urée à 3,45g/l, une créatininémie à 191mg/l. La kaliémie était à 6,5mmol/l, sans signes électriques à l’ECG. La natrémie à 129mmol/l.
L’échographie rénale a montré des reins de taille normale avec un index corticomédullaire respecté.
Le patient a bénéficié de deux séances d’hémodialyse et la diurèse est relancée sous diurétiques.
1
ER
CHAPITRE :
LA DREPANOCYTOSE
Historique de la drépanocytose
On situe l’origine de la drépanocytose à – 3000 ans, en Afrique centrale et occidentale, et dans la région arabo-indienne. A partir de ces zones d’origine, les migrations et les métissages de populations, la maladie s’est répandue dans le monde entier avec des prévalences variables d’une région à l’autre.
Il s’agit de la première maladie génétique identifiée chez l’Homme. [1]
Le premier cas est décrit par Herrick en 1910 , (médecin à chicago), il concernait un jeune étudiant Jamaïcain dont le sang contenait des globules rouges (hématies) déformés en forme de croissant ou faucille. En 1929 Hahn et Gillepsie font une découverte intéressante, ils remarquent que la déformation des globules rouges n'a lieu que lorsque la pression en oxygène dans le sang est inférieure à 50 mm de mercure. Ceci est réversible lors de l'augmentation de la pression en oxygène.
Dès 1940, un étudiant en médecine, Sherman, suggérait même qu'un bas niveau en oxygène altérait la structure de l'hémoglobine dans la molécule. [2]
En 1949, James Neel [3] démontre que la transmission de cette maladie est mendélienne,Pauling, Itano, Singer et Wells font accomplir un progrès majeur à la recherche en effectuant l'électrophorèse des hémoglobines d'un patient possédant des hématies falciformes mais sans autre symptôme marqué de la maladie. Il possède non seulement l'hémoglobine A normale (notée HbA) de l'adulte mais aussi une autre
hémoglobine, notée HbS (le S venant de Sickle signifiant faucille en anglais). Un tel patient est dit porteur du trait drépanocytaire. Il est hétérozygote HbS/HbA. Les malades, eux, ne possèdent pas du tout d'HbA. Ils sont homozygotes HbS/HbS. C'est le premier trouble de santé reconnu causé par une protéine.
Ingram démontre en 1956 que la différence entre HbA et HbS est due à la substitution d'un seul acide aminé : l'acide glutamique 6 de la chaîne β de l'HbA est remplacé par une valine. [4]
Au début des années 70 des tests de dépistage sont lancés aux USA. La population américaine d'origine africaine est en effet très touchée . En 1978, Tom Maniatis isole le gène de la bêta globine.
En 1995, l'hydroxyurée devient le premier et le seul médicament permettant de prévenir les complications dues à la maladie.
Généralités – génétique [5]
La drépanocytose est une maladie génétique de l’hémoglobine (Hb) qui se transmet sur un mode autosomique récessif.
Elle résulte de la mutation sur le chromosome 11 du sixième codon de la chaîne β globine de l'Hb (GAG GTG), entraînant la substitution d'un acide glutamique par une valine (GLU VAL) sur la protéine.
Cette mutation provoque la synthèse d’une Hb anormale, l’HbS.
L'HbS est capable de polymériser dans certaines circonstances, provoquant la falciformation des globules rouges (GR) d'où le terme d'anémie à hématies falciformes ou sickle-cell anemia des Anglo-Saxons.
Les sujets homozygotes pour la mutation, et certains sujets hétérozygotes composites SC et Sβ+
, ont un syndrome drépanocytaire majeur .alors que les sujets hétérozygotes sont porteurs du trait drépanocytaire AS et ne développent pas la maladie.
Epidémiologie de la drépanocytose
La drépanocytose est la plus fréquente des hémoglobinopathies dans le monde avec 50 millions de personnes atteintes environ. Elle est présente en Inde (certaines régions), aux Antilles, en Amérique du Sud (surtout le Brésil), chez les Afro-américains, mais surtout en Afrique intertropicale (entre le 15ème parallèle Sud et le 20ème parallèle Nord). Les pays les plus touchés sont : Sénégal, Bénin, Zaïre et Angola. On peut y observer une corrélation avec la résistance au paludisme. [6]
Aux Antilles et en Guyane, un nouveau-né sur 260 est atteint de drépanocytose ; en Afrique intertropicale, 1 sur 100. Les porteurs du trait drépanocytaire sont 25% en Afrique centrale, 15 à 20% en Afrique de l’ouest (35% si on y associe les porteurs des traits C et beta thalassémie) ; 10 à 12% dans les DOM antillais ; 1 à 15% dans les régions méditerranéennes.
On peut distinguer parmi les populations touchées :
celles à très haut risque : Afrique intertropicale, Inde (certaines régions)
celles à haut risque : Antilles, Amérique du Sud (Brésil), Noirs américains
celles à moyen risque : Afrique du Nord, Sicile, Grèce celles à faible risque : Portugal, Turquie, Israël.
La drépanocytose est ainsi répandue parce qu'à l'état hétérozygote, la présence du gène drépanocytaire contribue à protéger son porteur du paludisme
Plasmodium - de pénétrer dans les globules rouges) et lui procure donc un avantage sélectif par rapport aux porteurs des gènes normaux AA, qui sont eux vulnérables au Plasmodium. Les anomalies de l’hémoglobine se limitaient pratiquement aux zones impaludées et aux pays qui ont connu, au cours des siècles derniers, un important afflux d’esclaves d’origine africaine. Dans ces pays, les sujets homozygotes mouraient dans la petite enfance, alors que les hétérozygotes survivaient tout en bénéficiant d’un avantage sélectif. Au cours des dernières décennies, la distribution de ces anomalies génétiques a été considérablement modifiée, à la fois par d’importants flux migratoires vers les pays industrialisés et par les progrès de la médecine, et tout spécialement par l’amélioration de la prise en charge de ces maladies. [7]
Selon les projections de l’Organisation mondiale de la santé (OMS), le nombre de porteurs d’anomalies de l’hémoglobine devrait au cours des prochaines décennies se stabiliser à environ 8 % de la population mondiale.
Cette estimation tient compte à la fois d’une croissance sélective des populations atteintes et des progrès de la médecine. [7]
LA DISTRIBUTION DES SYNDROMES DRÉPANOCYTAIRES MAJEURS [7]
En France,La drépanocytose est aujourd’hui la plus fréquente des
L’accès effectif au conseil génétique suppose la recherche des anomalies de l’hémoglobine chez toute personne en âge d’avoir des enfants. [7]
Les spécificités historiques des flux migratoires dans les divers pays de la CEE sont à l’origine d’une fréquence différente de la drépanocytose d’un pays à l’autre.
Au Royaume-Uni, une étude de 1999 portant sur l’Angleterre montre, par
an, 3 000 nouveaux porteurs du trait drépanocytaire (0,47 %) et 2 800 (0,44 %) porteurs d’un trait β-thalassémique ; il y aurait 140 à 175 naissances d’enfants atteints de syndromes drépanocytaires majeurs et 10 à 25 enfants thalassémiques majeurs ou intermédiaires.
L’incidence des thalassémies en Grande-Bretagne est largement supérieure à celle observée en France. [8]
En Allemagne, on ne compte au total que 300 patients atteints de
syndromes drépanocytaires majeurs répartis dans une centaine d’hôpitaux. [9]
En Belgique, la population migrante s’élève à 30 % dans l’agglomération
bruxelloise et un dépistage néonatal systématique a été mis en place. Sur 23 136 tests pratiqués de 1994 à 1998, 11 syndromes drépanocytaires majeurs et un cas de thalassémie majeure ont été détectés. Tous les porteurs d’HbS (au total 277) avaient au moins un parent originaire d’Afrique subsaharienne. [10]
Aux Pays-Bas, les études épidémiologiques sont encore partielles et une
première projection ferait état d’environ 800 cas de syndromes drépanocytaires majeurs répartis sur tout le pays (P. Giordano, communication personnelle).
En Espagne les cas de syndromes drépanocytaires majeurs sont encore
rares mais tendent à se multiplier avec l’accroissement récent de la population immigrée. Une étude d’un centre hospitalier de Barcelone rapporte 22 patients diagnostiqués entre 1985 et 2001. Les patients sont de différentes origines ethniques : subsaharienne mais également nord-africaine et afro-américaine. [11]
L’histoire coloniale du Portugal est plus ancienne et la mutation drépanocytaire y a été introduite à partir du bassin méditerranéen entre les VII et XIIIe siècles, puis de l’Afrique et s’est diluée dans l’ensemble de la population.[12]
La drépanocytose indigène est fréquente dans le sud de l’Italie (800 cas connus), la Grèce, et l’Albanie.
Le Maroc, à l’instar des autres pays du pourtour méditerranéen et le moyen
orient, est une région de faible endémie. La drépanocytose touche 1 à 2% de la
Physiopathologie de la drépanocytose (moléculaire, cellulaire, et vasculaire)
La drépanocytose est une hémoglobinopathie héréditaire dont la sévérité est tributaire de l’interaction de multiples mécanismes propres au déclenchement des événements vaso-occlusifs [15]. l’atteinte pouvant être micro ou macro vasculaire impliquant de nombreux facteurs :
o facteurs liés aux GR : polymères d'HbS, anomalies rhéologiques
globulaires (déshydratation, fragilité mécanique, baisse de
déformabilité, augmentation de la viscosité sanguine, présence de cellules denses) ;
o facteurs liés aux globules blancs (GB) ;
o facteurs extraérythrocytaires : hémostase, endothélium vasculaire ; Echelon moléculaire : la polymérisation.
En milieu désoxygéné,l’HbS se polymérise et se colle à la membrane du GR.
Cette polymérisation dépend de la concentration en Hb (concentration corpusculaire moyenne en Hb [CCMH]), de la composition de l'Hb (présence d'autres Hb : HbF et C notamment), de la saturation en oxygène (O2), de la
La polymérisation aura pour conséquences :
cristallisation de l’HbS et déformation caractéristique irréversible du GR en faucille : le drépanocyte.
augmentation de la rigidité des GR favorisant leur accumulation dans la microcirculation ;
augmentation de la viscosité sanguine ; rupture et fragmentation des érythrocytes ;
augmentation de la perméabilité cationique du GR induisant sa déshydratation
Echelon cellulaire 1-GR
La déshydratation, phénomène important dans la constitution de l'anémie et la diminution de durée de vie érythrocytaire est le résultat de l’augmentation de la perméabilité du GR aux cations(Na+
, K+, Mg2+, Ca2+) [ 16]
Altérations structurales et fonctionnelles de la membrane
érythrocytaire
La surface des GR SS est propice à une hyperfixation d'immunoglobuline (Ig)G, proportionnelle à la densité globulaire. Cela favoriserait leur séquestration et leur destruction par les macrophages du système réticuloendothélial [17].
Hyperviscosité [18 ; 19]
La rhéologie des GR SS dépend de multiples paramètres : viscosité sanguine, hématocrite, CCMH, propriétés mécaniques.
La viscosité sanguine moyenne est plus élevée chez un sujet drépanocytaire que chez un sujet normal.
La diminution de la déformabilité des GR,déjà présente en phase d’oxygénation, est aggravée par la désoxygénation, ce qui augmente la viscosité et l’incapacité des érythrocytes à traverser la microcirculation.
2-GB
il existe fréquemment une hyperleucocytose au cours des crises douloureuses :les patients ayant les leucocytes les plus élevés ont une mortalité plus élevée. [20]
Les monocytes sont impliqués dans le phénomène de vaso occlusion en activant l’endothélium par l’intermédiaire du facteur kappa nucléaire endothélial B. [21 ; 22]
Echelon vasculaire : rôle de l’endothélium vasculaire
Le premier mécanisme du complexe physiopathologique décrit depuis 1980 est souligné par l’adhérence anormale du globule rouge drépanocytaire aux cellules de l’endothélium vasculaire.[23]
Diagnostic biologique de la drépanocytose A- Moyens de diagnostic
1-Le diagnostic biologique s’effectue à l’aide d’une étude
électrophorétique de l’hémoglobine qui révèle la présence de l'HbS. Une confirmation est obligatoire par le test de falciformation (test d'Emmel qui fait
apparaître les drépanocytes parmi les GR incubés dans un milieu dépourvu d'O2)
ou le test de solubilité (test de précipitation de l'HbS en milieu réducteur). [27]
a- Mise en évidence de l’HbS :
par l’électrophorèse de l’Hb à pH alcalin, qui permet une séparation des HbS,HbA, et HbC en fonction de leur charge.
b- Confirmation de la présence de l’HbS :
Par la CLHP et le test de précipitation d’Itano.
La chromatographie liquide haute performance(CLHP) a l’avantage de fournir en même temps un dosage précis des différentes fractions de l’Hb.c’une éléctrophorèse sur agar à pH acide.
Le test d’Itano permet la caractérisation de l’HbS et de l’HbS Antilles par précipitation.
c- Dosage des différentes fractions.
2-Bien que le diagnostic de certitude de la drépanocytose repose sur la mise en évidence de l’HbS, la numération formule sanguine, le taux d’hémoglobine,le volume globulaire moyen,la numération des réticulocytes et l’examen du frottis sanguin sont des éléments essentiels pour l’orientation diagnostique. [28]
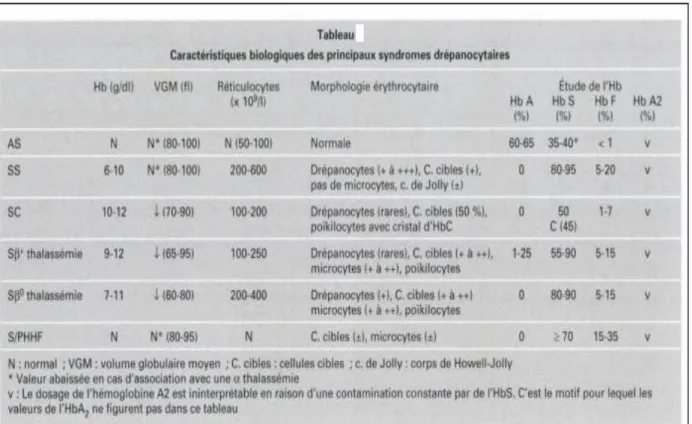
Le terme de syndrome drépanocytaire désigne , outre la drépanocytose homozygote (formeSS) qui est la forme la plus fréquemment rencontrée, les hétérozygoties composites associant l’HbS à une autre anomalie de l’Hb. De la nature de cette association dépendent les signes cliniques et biologiques. Certaines formes sont asymptomatiques, d’autres sont caractérisées par des signes cliniques et des anomalies hématologiques proches de la drépanocytose homozygote, on les désigne par le terme clinique de syndrome drépanocytaire majeur (SDM)
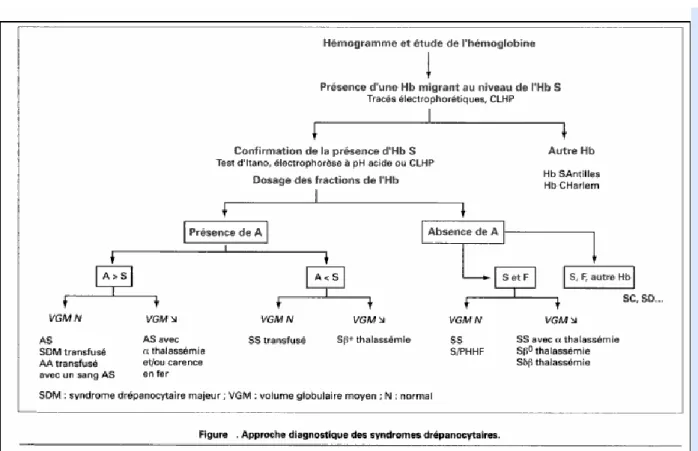
Tableau : approche diagnostique des syndromes drépanocytaires.
B- Diagnostic anténatal :
Proposé aux couples exposés au risque d'avoir un enfant atteint d'un syndrome drépanocytaire majeur.
Ainsi la naissance d’un enfant drépanocytaire peut être prévenue par l’interruption thérapeutique de la grossesse si les parents le souhaitent.
Il consiste en une étude génétique mettant en évidence la mutation caractéristique de la drépanocytose par prélèvement des villosités chorioniques ou fibroblastes par amniocenthèse.
C- Diagnostic néonatal : [29 ; 30]
Le dépistage néonatal est un dépistage de masse destiné à toucher tous les nouveau-nés d’un pays dans le but de détecter une ou plusieurs affections, le plus souvent héréditaires, à des fins de prévention secondaire. Le dépistage repose sur une technique d’isoélectrofocalisation. En cas d’anomalie, le diagnostic est confirmé par électrophorèse ou par chromatographie liquide à haute performance.
La prise en charge précoce de l’enfant permet de réduire la mortalité précoce par des vaccinations appropriées (antipneumococcique), des transfusions, le traitement par l’hydroxyurée pour augmenter le taux d’hémoglobine foetale, l’éducation des familles et le suivi régulier. Ces conditions de prise en charge ont permis de réduire la mortalité avant cinq ans à une fréquence de 2–5 %. (selon une étude faite par l’AFDPHE « Association Française pour le Dépistage et la Prévention des Handicaps de l’Enfant » en 2004.)
Manifestations cliniques de la drépanocytose [35]
Drépanocytose hétérozygote (trait drépanocytaire) [31 ; 32]
La grande majorité des patients drépanocytaires hétérozygotes se porte bien. Dans certains cas, cependant, on peut observer chez ces patients des infarctus spléniques au cours de situations d'hypoxémie sévère. On a également rapporté des hématuries macroscopiques en rapport avec des nécroses papillaires. La principale recommandation à faire aux drépanocytaires hétérozygotes est de ne pas se placer dans des situations à risque d'hypoxémie sérieuse (altitude élevée, plongée sous-marine). Ces patients peuvent subir des anesthésies générales comme tout sujet normal, sans préparation particulière.
Drépanocytose homozygote et autres syndromes drépanocytaires majeurs
L'expression clinique de la drépanocytose est large, avec des manifestations nombreuses et variées. Cette variabilité reflète principalement des influences génétiques et environnementales. Différents marqueurs hématologiques, qui dépendent eux-mêmes de ces précédentes influences, modifient la nature et la fréquence des complications de la drépanocytose.
De trois mois à cinq ans
Sous l'effet protecteur de l'HbF, les trois premiers mois de l'enfant HbSS sont asymptomatiques. Les premiers signes cliniques coïncident avec le remplacement progressif de HbF par l'Hb adulte porteuse de la mutation S. À partir du 4e mois vont apparaître les manifestations qui domineront durant les cinq premières années de vie : l'anémie, les infections et les CVO.
Anémie
Elle est la conséquence de la diminution de durée de vie des GR et apparaît vers 4 mois. Elle est normochrome, normocytaire et régénérative. Elle est absente dans les formes S/C, et microcytaire, dans les formes S/β . Parallèlement à cette évolution, la rate (site de l'hémolyse) augmente de volume, cette splénomégalie régresse en général vers l'âge de 6 ou 7 ans. La séquestration splénique aiguë, complication redoutée surtout entre 6 mois et 5 ans, réalise un
trapping brutal du sang dans la rate qui devient volumineuse et douloureuse,
l'anémie et l'hypovolémie aiguës conduisent alors rapidement au collapsus en l'absence de traitement adapté (transfusion). Face à la menace d'une récidive ou d'un passage à la chronicité, la splénectomie est souvent discutée ; elle semble de plus en plus conseillée malgré la menace du risque infectieux [33] .
La crise érythroblastopénique aiguë secondaire à une infection par le parvovirus B19 réalise un tableau comparable.
Risques infectieux
Ils sont majeurs dans la première enfance, avec notamment une vulnérabilité particulière face aux germes encapsulés. Ceci s'explique par un dysfonctionnement splénique précoce avec défaut d'opsonisation du sérum, déficit en tufsin, activation excessive de la voie alterne du complément, associé à une asplénie fonctionnelle rapidement progressive, secondaire aux infarcissements répétés [34] . Les sepsis sont donc fréquents et graves chez les
augmentés. Les salmonelles souvent en cause dans les ostéomyélites, et d'autres germes à multiplication intracellulaire ( Mycoplasma pneumoniae, Chlamydia
pneumoniae ) participent également à la morbidité. Par ailleurs l'infection,
source potentielle de fièvre, d'acidose métabolique voire de déshydratation, favorise la polymérisation de l'HbS et donc la survenue de CVO. Cette fragilité justifie des mesures préventives spécifiques essentiellement représentées par la prise continue de pénicilline et les vaccinations antipneumocoque et anti-hémophilus.
Crises vaso-occlusives
Elles sont les manifestations les plus fréquentes de la maladie drépanocytaire. Quasi pathognomoniques, elles sont la traduction clinique de l'obstruction des microvaisseaux par les GR rigidifiés lors de la polymérisation de l'HbS. Chez le jeune enfant, le tableau le plus typique, et souvent révélateur de la maladie, est la dactylite aiguë ou syndrome pied-main, qui est une atteinte inflammatoire des extrémités, souvent associée à un syndrome fébrile. La rate, les os longs et le parenchyme pulmonaire sont les sites privilégiés d'accidents vaso-occlusifs à cette période de la vie ; l'atteinte des ganglions mésentériques réalise une crise douloureuse abdominale et peut simuler un tableau pseudochirurgical. Le priapisme peut aussi se rencontrer chez le jeune enfant.
De l'âge de cinq ans à l'adolescence
À cet âge, l'asplénie fonctionnelle est constante. L'adaptation hémodynamique à l'anémie chronique se traduit par un état cardiaque hyperkinétique conduisant parfois à une myocardiopathie dilatée. L'ictère hémolytique est habituel, et les lithiases biliaires sont fréquentes. À cette période de la vie, les CVO hyperalgiques multifocales sont les premières complications.
La symptomatologie douloureuse prédomine souvent au niveau osseux, témoignant soit de lésions ischémiques soit de processus infectieux de type ostéomyélite. Les accidents vasculaires cérébraux ou pulmonaires peuvent mettre en jeu le pronostic vital. D'une façon générale, la fréquence et la sévérité des crises vaso-occlusives conditionnent le pronostic fonctionnel de chaque organe. C'est d'ailleurs à cette période que doit commencer le dépistage des premières atteintes dégénératives.
Adolescence
C'est souvent une période difficile, comme dans toutes les maladies chroniques, avec l'apparition d'un sentiment de découragement, voire de révolte qui rend la prise en charge délicate. La croissance staturopondérale est ralentie, le retard pubertaire est fréquent surtout chez le garçon. La fréquence des crises douloureuses et des hospitalisations varie selon l'expression de la maladie qui elle-même varie non seulement avec l'anomalie génétique initiale, mais également pour un même génotype en fonction d'éléments encore assez mal déterminés. Il semble, à l'heure actuelle qu'une concentration d'HbF relativement élevée soit associée à une évolution moins sévère de la maladie.
Adulte
La qualité de vie de l'adulte drépanocytaire dépend en grande partie de la façon dont s'est exprimée sa maladie antérieurement. En effet, pour chaque
aux complications osseuses, oculaires ou cutanées. Les crises anémiques s'espacent et les complications infectieuses diminuent à l'âge adulte. La morbidité est surtout liée aux syndromes pulmonaires aigus parfois mortels, aux ostéomyélites et aux infections urinaires fréquentes et redoutées chez la femme enceinte ; plus rarement à des méningites, des septicémies ou des infections intestinales.
La maladie dans sa phase intercritique
Il est de plus en plus fréquent que l’on découvre une drépanocytose chez un enfant à l’occasion d’un examen systématique clinique ou même biologique.
-l’anémie est la manifestation la plus connue, sans laquelle on ne peut évoquer le diagnostic. C’est une anémie hémolytique chronique, aux alentours de 7 à 9 g d’Hb/dl. Le degré d’anémie est différent d’un enfant à l’autre.
L’anémie chronique peut engendrer des signes propres : à l’examen un souffle systolique mésocardiaque , de type fonctionnel, est presque constant. Une cardiomégalie est retrouvée très précocement sur la radio du cœur et doit être différenciée d’une cardiomyopathie.
Chez l’enfant, une dyspnée d’effort, une asthénie, peuvent être remarquées. L’ictère cutanéo-muqueux n’est pas constant. Il est lié à l’hémolyse chronique. Selon les auteurs, on l’observe dans un peu plus de 10% des cas, mais il peut apparaître ou s’intensifier à l’occasion des crises, s’accompagnant alors d’une hyperhémolyse.
la splénomégalie est constante chez le nourrisson. Elle disparaît au fur et à mesure des années car les infarctus successifs entraînent une atrophie splénique.
l’hépatomégalie est fréquente dès le début de la maladie et persiste très longtemps dans l’enfance.
2
ème
CHAPITRE :
LES COMPLICATIONS GRAVES
DE LA DREPANOCYTOSE
I- Syndrome thoracique aigu [53 ;54 ;55 ;56 ;57]
Le STA est la principale cause de décès et la deuxième cause d'hospitalisation des patients drépanocytaires [36;37]. Son traitement optimal n'est pas codifié, surtout en raison de l'absence de causes précises reconnues [38].
1. Définition :
Toute complication pulmonaire aiguë associant des signes fonctionnels et physiques respiratoires et des signes radiologiques chez un drépanocytaire est un STA [39 ;40]. En pratique, il correspond à l'existence d'un nouvel infiltrat radiologique pulmonaire au moins segmentaire (syndrome alvéolaire), en dehors d'une atélectasie, associé à des signes respiratoires (tachypnée, wheezing, toux, hémoptysie...) ou des douleurs thoraciques 41 ;42 survenant parfois dans un contexte fébrile.
2. Epidémiologie, facteurs de risque :
Le STA est plus fréquent chez les patients homozygotes que chez les autres drépanocytaires (SS > Sβ° > SC > Sβ+
) [38]Son incidence varie en fonction des séries de 25 à plus de 80 % [43 ;44 ;39], avec un maximum entre 10 et 15 ans [45]. L'incidence est âge-dépendante [41 ;42] de 24,5/100 patients/an chez des enfants SS et 8,8/100 patients/an chez des adultes. Elle dépend aussi du taux d'HbF : ainsi, on l'observe moitié moins souvent chez les patients dont le taux d'HbF est de 15% par rapport à ceux dont le pourcentage est de 5 %.
Certains événements cliniques ont une influence défavorable sur le risque évolutif du STA : CVO douloureuses, syndrome fébrile, intervention chirurgicale récente, grossesse, ostéonécrose aseptique, anémie aiguë et atteinte pulmonaire antérieure.
3. Physiopathologie, étiologies
Le syndrome thoracique aigu est caractérisé par une occlusion vasculaire qui a lieu probablement au niveau de la veinule pulmonaire post-capillaire. Cette agression pulmonaire aiguë, qui peut évoluer vers le syndrome de détresse respiratoire aigu, est rarement primitive, elle est déclenchée par un certain nombre de causes dont les 4 principales sont : 1. les pneumopathies infectieuses bactériennes ou virales ; 2. les embolies graisseuses ou plus rarement fibrino-cruoriques ; 3. les thromboses in situ ; 4. les hypoventilations d’origine algique, ou dues à des infarctus osseux ou à des pathologies sous-diaphragmatiques, plus rarement secondaires à un surdosage en opiacés.
Le STA ressemble au tableau d'une pneumonie infectieuse avec fièvre, infiltrats radiologiques et détresse respiratoire. Sa physiopathologie est cependant multifactorielle. L'asplénie, la réduction de l'immunité humorale et de la phagocytose sont responsables d'un déficit immunitaire, tandis que l'ischémie pulmonaire locale et la diminution de la fonction alvéolaire favorisent la prolifération microbienne [40]. Ces éléments plaident en faveur d'une étiologie infectieuse comme principale cause du STA.Mais l'examen clinique et l'examen radiologique ne permettent pas de préjuger du germe causal, s'il en existe un, et les recherches bactériologiques sont trop longues pour être d'un intérêt quelconque à la phase aiguë. D'autre part, même lorsque des explorations
invasives sont effectuées (en particulier la fibroscopie bronchique associée au lavage bronchioloalvéolaire [LBA]), près d'un tiers des STA reste inexpliqué.
Une étiologie microbienne est retrouvée dans 20 à 50% des cas selon les séries. Dans la récente étude du National Acute Chest Syndrome Study Group (NACSSG) [38], les bactéries représentent 73% des germes isolés contre 27% pour les virus, tous âges confondus. Des germes habituellement peu pathogènes, tels que les mycoplasmes et Chlamydia, sont potentiellement graves sur ce terrain [38 ;46] La prédominance classique des pneumocoques n'est pas retrouvée dans l'étude du NACSSG où les germes atypiques représentent plus de 70 % des bactéries causales. Bactériémies et septicémies accompagnant le STA sont rares chez l'enfant et plus encore chez l'adulte (< 1-2 %), ce qui conduit peut-être à sous-estimer la fréquence des pneumonies bactériennes [39 ;47].
Le STA est parfois secondaire à une pneumonie virale, essentiellement chez l'enfant de moins de 10 ans, très rarement chez les adultes de plus de 19 ans. Les virus impliqués sont nombreux : virus respiratoire syncytial (VRS) surtout (39% ), parvovirus B19 (15%), rhinovirus (12%), cytomégalovirus (CMV), virus d'Epstein-Barr (EBV), Herpes simplex virus (HSV), grippe, adénovirus, virus para- influenza [38 ;42]
Les embolies graisseuses, recherchées par des techniques spéciales sur le produit du LBA, sont retrouvées dans 13 à 75% des cas [42]. Chez les
ambiant plus basse (89% versus 94% en cas d'infection et 91% en cas d'infarctus), et enfin, plus grande fréquence des CVO douloureuses (74% contre 54% en cas d'infection et 68% en cas d'infarctus).
Le taux sérique de phospholipase A2, un médiateur inflammatoire libérant
des acides gras (libres), est très augmenté au cours des STA avec des taux corrélés à la sévérité clinique [48 ;49]. Il pourrait constituer un marqueur précoce car il s'élève avant la constitution du STA.
Les infarctus pulmonaires secondaires à l'obstruction vasculaire de petits ou moyens vaisseaux sont rarement documentés (angiographie, scintigraphie pulmonaire), et constituent en quelque sorte un diagnostic d'élimination (16 % dans l'étude du NACSSG [38] )
L'hyperadhésion des GR drépanocytaires à l'endothélium qui a un rôle dans l'occlusion microvasculaire pourrait jouer un rôle dans ce type d'atteinte au niveau pulmonaire [50 ;51].
Enfin, les embolies pulmonaires fibrinocruoriques semblent être une complication aiguë inhabituelle chez le drépanocytaire, malgré l'état biologique d'hypercoagulabilité.
4. Clinique, biologie, examens morphologiques :
La présentation clinique typique chez l'adulte est celle d'une crise thoracique douloureuse, parfois sévère, habituellement sans fièvre, et dont l'aggravation progressive en quelques jours va provoquer l'hospitalisation. Mais dans près de 50 % des cas, l'admission est motivée par une CVO douloureuse des membres avec un examen pulmonaire normal. Ce n'est que 2 à 3 jours plus
fréquence du retard au diagnostic de STA [38 ;39 ;45]. Toute crise douloureuse doit donc être considérée comme un prodrome éventuel du STA. La surveillance doit être renforcée car l'état clinique du malade peut se détériorer très rapidement, en quelques jours, voire quelques heures.
Une CVO paraissant simple doit toujours faire l'objet d'une surveillance de la saturation du sang artériel en oxygène (SaO2) et du débit expiratoire de pointe
(DEP) (moyen à la phase aiguë à 53 % de la théorique) [38]. Le tableau est plus sévère s'il s'agit d'embolies graisseuses avec des douleurs osseuses intenses, thoraciques et/ou des membres, une toux avec expectoration ayant parfois l'aspect de beurre frais, et des modifications comportementales et/ou de la conscience.
La radiographie initiale est soit normale, insuffisante pour porter le diagnostic précoce, soit montre de discrets infiltrats qui s'étendent rapidement aux bases avec une atteinte pleurale dans 20 à 50 % des cas [38 ;39]. Des infiltrats multiples évoquent surtout des emboles graisseux.
L'Hb chute de 1 à 2 g/dL, plus encore en cas d'embolie graisseuse, la leucocytose augmente en moyenne de 70 % et une thrombopénie relative ou absolue apparaît [38 ;42]. La gazométrie artérielle montre une hypoxie modérée (70 mmHg) avec hypocapnie (35 mmHg). Chez près d'un patient sur quatre, l'hypoxie est inférieure à 60 mmHg, non corrélée à la gravité clinique.
CVO ou une CVO à l'entrée, des plaquettes inférieures à 200 000/mm3 à l'admission, de la fièvre, des anomalies radiologiques étendues, une insuffisance respiratoire aiguë (IRA), une atteinte neurologique ou un traitement transfusionnel. Une IRA survient chez 10 à 15 % des malades, surtout en cas d'atteinte radiologique d'emblée étendue, de plaquettes inférieures à 200 000/mm3 ou d'antécédents cardiaques [38]. La ventilation mécanique est maintenue habituellement 4 à 5 jours.
Des troubles neurologiques apparaissent chez plus de 20 % des adultes : troubles de conscience (> 50 %), convulsions (> 10 %), atteinte neuromusculaire (< 10 %) et parfois AVC hémorragique ou ischémique. [38]. La mortalité par STA chez l'adulte varie de 2 % à 10 %, très supérieure à celle observée chez l'enfant [38]. Les facteurs de risque de décès sont une atteinte multilobaire, une IRA précoce, un sepsis bactérien, une atteinte neurologique ou une baisse importante de l'Hb(≤ 5 g/dL). Les récidives augmentent le risque de mortalité précoce et de maladie pulmonaire chronique [41]. L'autopsie, quand elle est pratiquée, révèle fréquemment une embolie pulmonaire correspondant souvent à des emboles graisseux massifs.
6. Traitement :
En raison du risque imprévisible de décompensation brutale de l’état respiratoire, le syndrome thoracique justifie une hospitalisation systématique. Le transfert en unité de soins intensifs doit être envisagé dans les formes sévères ou lorsqu’une surveillance rigoureuse ne peut pas être correctement assurée.
Le traitement du syndrome thoracique aigu essaie donc d’agir sur ces différents mécanismes physiopathologiques.
Les modalités thérapeutiques sont proches de celles des crises osseuses : hydratation, supplémentation en folates, apport d’oxygène et antalgiques. Les morphiniques peuvent être utilisés, même en cas d’hypoxie, ce qui peut justifier une admission en soins intensifs afin de surveiller l’état respiratoire du patient.
Une antibiothérapie couvrant le pneumocoque est systématique si le malade est fébrile, même si une origine infectieuse n’est en cause que dans moins de 20% des cas.
La kinésithérapie respiratoire est utile pour améliorer l’ampliation thoracique et lutter contre les atélectasies qui majorent l’hypoxie et risquent ainsi d’aggraver la situation en favorisant la falciformation des hématies. D’autres approches thérapeutiques telles que la ventilation non invasive et l’utilisation du monoxyde d’azote (NO) sont en cours d’évaluation.
Une insuffisance respiratoire chronique et une hypertension artérielle pulmonaire peuvent se développer à la suite d’épisodes répétés de syndromes thoraciques. Le retentissement du syndrome thoracique aigu est évalué à distance de l’épisode par les épreuves fonctionnelles respiratoires.
Le traitement des récidives de STA est difficile. Il fait appel, soit aux programmes transfusionnels entraînant une disparition quasi totale des récidives
[52]
, soit à hydroxyurée, qui diminue d'environ 50 % la fréquence de STA chez l'adulte
II- L’ infection chez le drépanocytaire [58;59;60]
Les infections sont fréquentes dans la drépanocytose et souvent graves chez l'enfant de moins de 5 ans (c'est la première cause de mortalité avant 3 ans).
Toute infection peut aussi déclencher des crises vaso-occlusives car elle provoque fièvre, hypoxie et déshydratation, 3 facteurs favorisant la falciformation.
On sait aussi que la crise vaso-occlusive favorise l'infection locale du fait de la stase veineuse qu'elle entraîne (fréquence des ostéomyélites après crise osseuse par exemple). Il s'agit là d'un véritable cercle vicieux infection-crise
A.
1. physiopathologie:Immunité et drépanocytose
S'il est certain que le drépanocytaire fait souvent des infections parfois très graves, voire foudroyantes, et ceci dès l'âge de 2 mois et jusqu’à la fin de sa vie, cette susceptibilité aux infections est mal expliquée.
L’asplénisme fonctionnel : la rate véritable filtre à microbes fonctionne mal dès la naissance; ensuite ces fonctions se dégradent car les thromboses itératives de ses vaisseaux aboutissent à une véritable atrophie de celle-ci. On comprend qu’en cas de bactériémie celle-ci devient souvent une septicémie puisque le filtre splénique ne fonctionne pas.
Altération de la voie du complément : le complément est un ensemble de protéines stimulant ―en urgence‖ nos globules blancs qui vont se précipiter pour phagocyter les germes intrus, permettant à notre système immunitaire de synthétiser des anticorps en quelques jours. Chez le drépanocytaire le système est défaillant ce qui est surtout grave chez le jeune qui n’a pas encore eu le temps de faire des anticorps.
Nécroses intestinales source de brèches : les crises vaso-occlusives intestinales provoquent de minimes nécroses de la muqueuse intestinale expliquant le passage sanguin de bactéries souvent présentes dans le tube digestif (ceci explique la relative fréquence des bactériémies à salmonelles).
Crises vaso-occlusives causes de stase sanguine donc d’infection : c’est probablement ce qui se passe dans les poumons et dans l’os. Par contre, le drépanocytaire synthétise normalement des anticorps après vaccination et ses lymphocytes T (défaillants dans l’infection à VIH) sont ici normaux : ceci explique pourquoi l’adulte qui s’est immunisé contre de nombreux germes fait beaucoup moins d’infections.
2. Germes en cause
Le pneumocoque (Streptococcus pneumoniae) est responsable, chez le
sujet normal d’infections graves aux âges extrêmes de la vie : méningites, pneumonies, septicémie avant 2 ans, pneumonies après 65 ans. Chez le drépanocytaire de moins de 5 ans le risque de méningite et de septicémie est élevé et la mortalité de l’ordre de 25 % : c’est dire si la priorité doit être la prévention de l’infection pneumococcique par la Pénicilline V et la vaccination.
L’Haemophilus influenzae b, autre responsable de méningites et de
pneumonies avant 5 ans ; il est aussi plus fréquent chez le drépanocytaire : nous avons ici la chance de disposer d’un vaccin protégeant à près de 100 % contre ce germe redoutable.
Les salmonelles : il s’agit rarement de la typhoïde mais des bacilles para
typhi (S. Enteritidis) le vaccin antityphoïdique n’a donc pas d’indication particulière chez le drépanocytaire ; par contre, ce vaccin est utile pour tous du
Le paludisme : s’il est vrai qu’au cours des siècles les porteurs du trait
drépanocytaire ont été moins décimés par le paludisme que les sujets normaux, le drépanocytaire homozygote a les mêmes risques que la population : de plus l’hémolyse que le paludisme provoque va aggraver l’anémie et nécessiter une transfusion avec ses risques : il faut donc conseiller grillages antimoustiques aux fenêtres, moustiquaires et chloroquine chez le jeune enfant.
L’hépatite B: les risques sont ici encore augmentés du fait des transfusions
rarement évitables : c’est dire si tout enfant drépanocytaire doit être vacciné le plus tôt possible.
La grippe : sévissant dans le monde entier, il a été montré que la grippe
entrainait des complications pulmonaires chez le drépanocytaire justifiant la vaccination à partir de l’âge de un an dans les pays ou le vaccin est disponible, sans oublier la nécessité d’un renouvellement annuel.
Le rotavirus grand responsable de diarrhées graves chez tous les enfants,
peut provoquer chez le drépanocytaire une déshydratation déclenchant une crise. Il faut donc hydrater constamment et réhydrater le plus tôt possible en cas de diarrhée (un vaccin arrive !!). Nous ne parlerons pas du parvovirus B 19 et du
Mycoplasma pneumoniae aussi fréquents chez l’enfant drépanocytaire car il
n’existe pour l’instant pas de prévention.
Enfin, il ne faut pas oublier que le drépanocytaire peut faire des infections à germes banaux : staphylocoque, streptocoque etc. Mais aussi la tuberculose d’où la nécessité d’une hygiène générale, d’une vigilance alimentaire, cutanée, et dentaire entre autres.
3. Traitements préventifs
Antibiothérapie anti pneumococcique
On a fait la preuve de son efficacité : elle est indispensable à base de PénicillineV donnée à la dose de 50 000 Unités/kg en 2 prises par jour : elle est supérieure à la pénicilline injectable dont les taux sériques sont rapidement insuffisants et à l’amoxicilline qui favorise la résistance du pneumocoque aux antibiotiques.
Quand commencer? Dés l’âge de 3 mois, âge auquel les anticorps maternels transplacentaires commencent à disparaître.
Jusqu’à quand? Aucun consensus: certainement jusqu’à 5 ans, pour beaucoup 15 ans surtout s’il existe une infection à pneumocoque dans les antécédents.
Vaccin anti-pneumococcique
Le vaccin anti-pneumococcique à 23 valences n’est pleinement efficace qu’après 2 ans : certains le proposent dès 9 mois avec un rappel tous les 3 ans : il ne dispense en aucun cas du traitement préventif par la PénicillineV. L’arrivée d’un vaccin anti-pneumococcique à 7 valences conjugué à une protéine a l’im-mense avantage de protéger dès les premières semaines de la vie et d’entraîner une production plus élevée d’anticorps : malheureusement les 7 sérotypes qu’il contient sont adaptés à l’Amérique du nord et l’Europe mais peu à l’Afrique ;
Vaccin anti-haemophilus
Heureusement proposé à tous les enfants béninois de moins de 2 ans, il devra être effectué en priorité dès le deuxième mois chez le drépanocytaire.
Vaccin anti-grippal
La primo vaccination se fait dès l’âge de un an avec une 1/2 dose répétée à un mois d’intervalle ; un rappel devant être effectué chaque année avec une 1/2 dose jusqu’à 10 ans, une dose ensuite.
Les vaccins contre le méningocoque, A-C ou mieux ACW135Y sont recommandés après l’âge de 18 mois et indispensables en cas d’épidémie.
Règle d’or : apprendre aux parents à surveiller la température de leur enfant et à consulter pour toute fièvre > 38,5° C.
B. Les types d'infection 1. Septicémies
Elles sont fréquentes et redoutables car foudroyantes avec fièvre élevée, collapsus en quelques heures.
Chez le jeune enfant, il s'agit presque toujours d'un pneumocoque. Les septicémies à bacille Gram (- ), notamment à colibacille et à salmonelle (typhoïde) se voient à tout âge.
2. Méningites
Leur fréquence est connue depuis longtemps. Elles surviennent surtout chez le jeune enfant et là encore, le pneumocoque vient en tête devant l'hémophilus. Le risque de récidive est important.
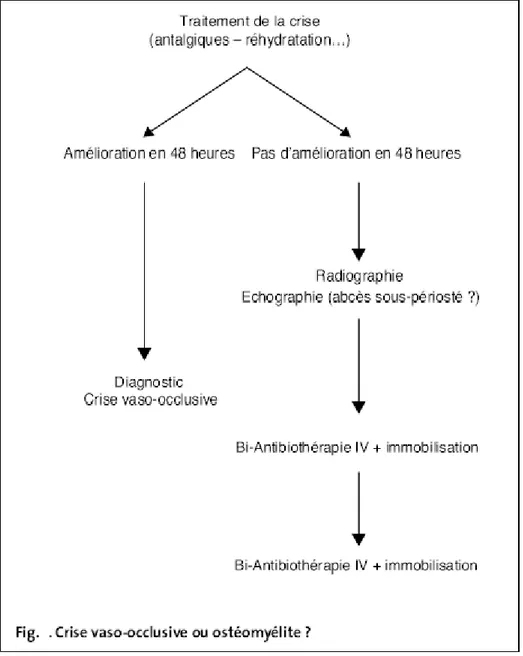
3. Ostéomyélites
Elles aussi très fréquentes, elles sont bien particulières :
• parce que les salmonelles sont très souvent en cause (ce qui est rare chez le sujet sain). Plus rarement il s'agit d'un staphylocoque ou d'un pneumocoque.
• parce qu'elles surviennent à tout âge (contrairement aux septicémies et aux méningites) :
• par la difficulté du diagnostic avec les thromboses osseuses: celles-ci entraînent douleur osseuse, oedème des parties molles mais en général pas de fièvre:
• enfin parce qu'il existe souvent plusieurs localisations et que les rechutes sont fréquentes.
4. Infections pulmonaires
Plus fréquentes que graves, elles sont provoquées par le pneumocoque et le mycoplasme. Il est quelquefois difficile de différencier infarctus pulmonaire et infection.
Infection et thrombose ont souvent associées !!! 5. Infections urinaires
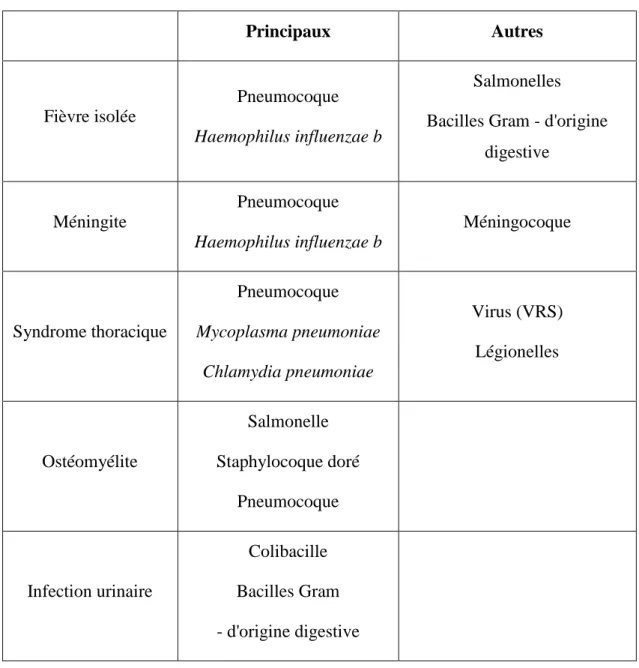
Tableau : principaux germes à évoquer en fonction du tableau clinique Principaux Autres Fièvre isolée Pneumocoque Haemophilus influenzae b Salmonelles Bacilles Gram - d'origine
digestive Méningite Pneumocoque Haemophilus influenzae b Méningocoque Syndrome thoracique Pneumocoque Mycoplasma pneumoniae Chlamydia pneumoniae Virus (VRS) Légionelles Ostéomyélite Salmonelle Staphylocoque doré Pneumocoque Infection urinaire Colibacille Bacilles Gram - d'origine digestive
C. Conduite à tenir devant une infection chez le drépanocytaire
1. Principes généraux
Dans la grande majorité des cas, devant une suspicion d'infection, le traitement antibiotique est démarré de manière empirique sans attendre les résultats des prélèvements.
En fonction du tableau clinique, il faut parier sur les germes (Tableau). L'antibiothérapie probabiliste doit être :
bactéricide et adaptée au site infectieux suspecté ou identifié : en cas de suspicion de méningite, il faudra choisir un antibiotique qui franchit la barrière méningée ;
active sur le Pneumocoque, même si l'enfant est correctement vacciné et si les parents certifient que le traitement par Oracilline est bien administré ;
large pour être aussi efficace sur Haemophilus b et les salmonelles. Les situations sont bien différentes suivant le tableau :
Fièvre isolée,
2-Devant une fièvre isolée
Toute fièvre >38,5°C chez un enfant drépanocytaire impose une consultation au dispensaire d'urgence et la réalisation d'une NFS, une radio pulmonaire, une bandelette urinaire : une ponction lombaire est indiquée s'il existe une altération de l'état général et le moindre doute sur un syndrome méningé. Idéalement, l'antibiothérapie d'urgence doit être parentérale : le Cefotaxime, la Ceftriaxone sont parfaitement adaptés à la situation car ils sont efficaces sur les principaux germes et ils traversent facilement la barrière méningée.
Ce traitement d'urgence est indispensable pour :
les enfants de moins de 3 ans avec une fièvre >38,5°C
tous les enfants avec une fièvre >39,5°C ou avec une atteinte de l'état général et/ou des troubles de la conscience,
tout enfant ayant un antécédent de septicémie et/ou présentant une radio pulmonaire anormale une hyperleucocytose >30 000 ou une leucopénie < 5 000 enfin une anémie avec Hg< 6gr/dl..
3-Tableau: principaux antibiotiques/posologie, voies d'administration
INDICATIONS
Amoxicilline 50à 100mgr/kg 3 prises/jour PO, IV
Infection pulmonaire
ORL
Ceftriaxone 50 à 100mgr/kg 1 par jour
IV IM
Infection sévère
Cefotaxime 75 à 200mgr/kg 3 par jour IV Infection sévère
Gentamycine 3 à 7 Mgr/kg 2 par jour
IV IM
Ostéite Infection urinaire
Erythromycine 30 à 50 Mgr/kg 2 par jour PO
Inf. pulmonaire (en association) -Amoxicilline -Acide clavulanique (Augmentin) 45 Mgr/kg 2 par jour PO Inf. Pulmonaires Urinaires
III- Atteinte neurologique centrale [65 ;66 ;67 ;68;69 ;70 ;71]
L'atteinte du système nerveux central est une cause majeure de morbidité de la drépanocytose, représentée principalement par les infarctus cérébraux et les hémorragies intracrâniennes. Les méningites bactériennes, avec ou sans septicémie, concernent surtout des enfants de moins de 5 ans.
L'âge médian au premier AVC est d'environ 13 ans chez les SS et 47 ans chez les SC [61]. La probabilité de survenue d'un premier AVC, ischémique ou hémorragique, augmente avec l'âge : 11 % à 20 ans jusqu'à 24 % à 45 ans chez le drépanocytaire homozygote SS, plus faible chez le drépanocytaire SC (de 2 % à 10 %) [62].
Les AVC sont principalement ischémiques (50-80 %), parfois associés à une hémorragie (≤ 2 %).
A. Facteurs de risque et mortalité des accidents vasculaires cérébraux
Les facteurs de risque des accidents ischémiques sont les antécédents de déficits neurologiques transitoires (DNT), de STA récent (≤ 15 jours), le nombre de STA, la pression artérielle systolique (corrélation positive) et le taux de base d'Hb (corrélation négative). [62 ;63]
Les AVC hémorragiques sont corrélés positivement au taux basal de leucocytes et négativement à celui de l'Hb.
La mortalité précoce (dans les 14 jours) liée aux AVC est variable selon la nature de l'accident et l'âge des patients. Elle est globalement proche de 10 %, plus élevée s'il s'agit d'une hémorragie (environ 25 %) ou d'un premier AVC.