HAL Id: hal-02113388
https://hal.archives-ouvertes.fr/hal-02113388
Submitted on 16 Jul 2019
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
Riccardo Piccardi, Serge Turcaud, Erica Benedetti, Laurent Micouin
To cite this version:
Riccardo Piccardi, Serge Turcaud, Erica Benedetti, Laurent Micouin. Synthesis and Reactivity of
Mixed Dimethylalkynylaluminum Reagents. SYNTHESIS, Georg Thieme Verlag, 2018, 51 (01),
pp.97-106. �10.1055/s-0037-1610392�. �hal-02113388�
97
R. Piccardi et al.
Short Review
Syn thesis
Synthesis and Reactivity of Mixed Dimethylalkynylaluminum
Reagents
Riccardo Piccardi Serge Turcaud Erica Benedetti Laurent Micouin* 0000-0003-1080-8591
Laboratoire de Chimie et Biochimie Pharmacologiques et Toxicologiques, UMR 8601 CNRS-Université Paris Descartes, Faculté des Sciences Fonda-mentales et Biomédicales, 45 rue des Saints-Pères, 75006 Paris, France Laurent.micouin@parisdescartes.fr
Published as part of the 50 Years SYNTHESIS – Golden Anniversary Issue
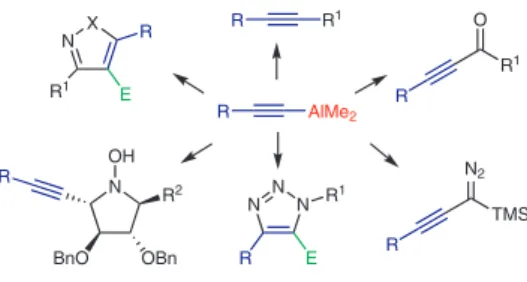
R AlMe2 R R1 N R2 R O R1 R N2 TMS R X N R1 R E N N N R 1 R E OH OBn BnO Received: 20.10.2018 Accepted: 29.10.2018 Published online: 20.11.2018
DOI: 10.1055/s-0037-1610392; Art ID: ss-2018-z0706-sr
License terms:
Abstract Organoaluminum derivatives are mostly appreciated for their Lewis acidity properties, but generally not considered as reagents of choice in synthetic transformations involving the creation of C–C bonds. Among these species, dimethylalkynylaluminum reagents rep-resent a special class of compounds, with, in many cases, unique reac-tivity. This review summarizes the preparation and reactivity of these organometallic reagents with a focus on their synthetic potential. 1 Introduction
2 Preparation of Dimethylalkynylaluminum Reagents
3 Reactivity of Dimethylalkynylaluminum Reagents
3.1 Reactions with Csp3 Electrophiles
3.2 Reactions with Csp2 Electrophiles
4 Transition-Metal-Catalyzed Reactions
4.1 Addition to ,-Unsaturated Enones
4.2 Coupling Reactions
5 Triple Bond Reactivity
6 Conclusion
Key words organometallics, alkynes, organoaluminum reagents, alkynylation, metalation
1 Introduction
Organoaluminum reagents have been known for more than 150 years since the first synthesis of ethylaluminum sesquiiodide by Hallwachs and Schafarik in 1859.1 However,
the chemistry of these organometallic species is still mostly restricted to the fields of olefin polymerization and thin-film fabrication by chemical vapor deposition techniques.2
The range of applications of organoaluminum reagents is
surprisingly limited, especially if one considers that alumi-num, the most abundant metal in the earth’s crust, is pro-duced at low price (a mole of aluminum is now cheaper than a mole of lithium). Trimethylaluminum is also a major, widely available inexpensive organometallic compound that can act not only as a cheap methyl donor, but also as metalating or transmetalating agent for the synthesis of more elaborated organoaluminum reagents.3
There are probably a few reasons why organoaluminum reagents are still less frequently used in organic synthesis than other main group organometallic reagents, such as or-ganolithium or organomagnesium reagents. One of the lim-itations is that generally only one of the 3 C–Al bonds will react in synthetic transformations.4 Furthermore,
competi-tive hydride transfer can occur with substituents bearing an sp3-C–H bond to the sp3-C–Al bond.5 Finally, the main
reason for the rather low popularity of organoaluminum as synthetic reagents has been highlighted by Eisch in his seminal review on the history of aluminum chemistry:
if ether solvents are essential in the reactivity of organolithi-um or magnesiorganolithi-um reagents, they dramatically reduce the re-activity of alkylaluminum reagents through the formation of Lewis acid-base adducts.6 However, these solvents are still
generally used for the preparation of organoaluminum re-agents by transmetalation, C–H activation, or direct inser-tion, leading to organometallic species with limited reactiv-ity.
Among the large variety of organoaluminum com-pounds, dimethylalkynylaluminum reagents (Figure 1) look quite attractive.
Figure 1 General properties of mixed dimethylalkynylaluminum
re-agents Me Al Me R no β C-H bond
selective alkynyl transfer reagent no disproportionation
high Lewis Acidity in non-polar solvents
SYNTHESIS0039-78811437-210X
Georg Thieme Verlag Stuttgart · New York 2019, 51, 97–106
short review
enThese species are indeed devoid of the major limitations described above. The three substituents on aluminum do not bear a C–H bond in the -position. The greater reactivi-ty of the sp-C–Al bond compared to that of sp3-C–Al bond
will enable selective transfer of the alkynyl moiety, the methyl group behaving as a nontransferable moiety. Finally, we were able to propose a new route to these reagents in non-polar solvents, providing compounds with enhanced reactivity.
In this short review, our goal is to focus on the prepara-tion and reactivity of mixed dimethylalkynylaluminum re-agents with a special focus on their peculiar reactivity and synthetic utility in selective organic transformations.7
2 Preparation
of
Dimethylalkynylaluminum
Reagents
A classical way to prepare dialkylalkynylaluminum compounds is by salt metathesis from the corresponding al-kali acetylides (Scheme 1). This is the method of choice starting from terminal acetylenes bearing tert-butyl8 or
tri-alkylsilyl groups,9 or showing weak acidity such as
alkoxy-acetylenes.10
This approach requires a first metalation step, generally conducted at low temperature, and careful control of the transmetalation experimental conditions to avoid the pres-ence of residual traces of dialkylaluminum halides. The equivalent of lithium or sodium chloride produced in the second step can be removed by filtration if a non-polar sol-vent is used, but can affect the reactivity of the resulting or-ganoaluminum reagents if the transmetalation step is con-ducted in Et2O.
Trimethylaluminum is not sufficiently basic to metalate a terminal alkyne at room temperature in non-polar sol-vents. The use of higher temperatures generally leads to a mixture of compounds resulting from competitive car-boalumination side reactions.11 Interestingly, clean
metala-tion occurs at room temperature in triethylamine, as re-ported by Binger in 1963.12 This observation led to the
de-velopment of a base-catalyzed alumination of terminal alkynes with trimethylaluminum.13 A mechanistic
investi-gation showed that several Lewis bases can catalyze this transformation, the most efficient being N,N-bis(trimethyl-silyl)methylamine (Scheme 2).14 Using this base, clean
ter-minal alumination can be obtained at room temperature with only 1 or 2 mol% of the catalyst. In 2018, the use of zwitterionic neodymium(III) heterobimetallic compounds was reported to catalyze a similar reaction.15
Dimethylalkynylaluminum reagents are typically pre-pared by simply adding MeN(SiMe3)2 and the alkyne to a 2
M commercial solution of AlMe3, leading to a stable 1.6 M
solution of the organometallic reagent. The prepared air-and moisture sensitive organoaluminum solutions can be stored under argon in the dark for several days at room temperature. Although commercially available heptane or Serge Turcaud (left) obtained his CNAM Engineer degree in 1995.
After working for 19 years in the laboratory of Prof. B. Roques at the University of Paris Descartes, he joined the group of Dr. J. Royer in 2003 and obtained his Ph.D. in 2009. He then joined the group of Dr. L. Micouin where he works on aluminum chemistry.
Laurent Micouin (2nd from left) obtained his Ph.D. in 1995 under the
supervision of Prof. H.-P. Husson and Dr. J.-C. Quirion. After a post-doctoral stay at the Philipps Universität Marburg in the group of Prof. P. Knochel, he was recruited as a CNRS researcher at Paris Descartes University. His main research interests are methodological develop-ments including organoaluminum chemistry, asymmetric synthesis, di-versity-oriented synthesis, and developments at the interface between chemistry and biology, such as the design of new RNA binders.
Erica Benedetti (2nd from right) obtained her Ph.D. in 2011 under the
supervision Prof. L. Fensterbank (UPMC-Sorbonne Universités) and Prof. A. Penoni (Università degli Studi dell’Insubria). In 2012, she joined the group of Prof. K. M. Brummond at the University of Pittsburgh as a postdoctoral fellow. After a second post-doctoral experience at the ESPCI ParisTech in the group of Prof. J. Cossy, she was recruited in 2014 as a CNRS researcher at Paris Descartes University in the team of Dr. L. Micouin. Her current research mainly involves the development of synthetic methodologies providing access to new chemical space.
Riccardo Piccardi (right) obtained his Ph.D in 2005 under the
supervi-sion of Prof. P. Renaud at the University of Bern. The same year, he moved to Germany for a post-doctoral stay at the Technical University of Munich in the group of Prof. T. Bach. He then joined the team of Prof. O. Baudoin at University Claude Bernard Lyon I for a second postdoctor-al experience. In 2008, he joined the group of Prof. M.-C. Scherrmann at University of Paris Sud. The next year he was recruited as a CNRS re-searcher in the same group. He finally joined the team of Dr. L. Micouin in 2014, where he is developing new methods combining flow chemis-try and organoaluminum reactivity.
Scheme 1 Preparation of dimethylalkynylaluminum reagents by
trans-metalation Me3Si Li hexane, 0 °C + LiCl Me3Si AlMe2 1) BuLi 2) Me2AlCl hexane/toluene
tBuO AlMe2 + LiCl
–40 °C Cl tBuO Cl Me2AlCl t-Bu Li cyclopentane + LiBr t-Bu AlMe2 Me2AlBr
99
R. Piccardi et al.
Short Review
Syn thesis
toluene solutions can both be used indiscriminately, di-methyl(phenylethynyl)aluminum tends to crystalize in al-kanes and is better prepared in toluene.
An alternative to this batch procedure based on flow chemistry has also been proposed. In this case, a resin-sup-ported tertiary amine is used to promote the metalation step. This procedure delivers dimethylalkynylaluminum re-agents without any residual traces of the catalyst with an increased reaction rate (Scheme 2).16
3 Reactivity of Dimethylalkynylaluminum
Reagents
Alkynylaluminum reagents exhibit a rather low nucleo-philic character. This reactivity can be explained by the low ionic character of the C–Al bond, and their bridged dimeric nature in non-coordinating solvents. As a result, most of the reactions involving the carbon–metal bond will be trig-gered by the complexation of the acidic aluminum center to the substrate, leading to activation of the nucleophile. As a consequence, the nature of the solvent will play a major role in the reactivity of dimethylalkynylaluminum reagents. This typical behavior is perfectly illustrated in the reaction with chiral oxazolopiperidines (Scheme 3). These polyfunc-tional compounds are known to react with organolithium, -cuprates,17 or -magnesium reagents18 at their
amino-nitrile moiety. Dimethylalkynylaluminum reagents react, in a complementary manner, selectively with the oxazolidine motif by coordinating the most Lewis basic part of the sub-strate; diastereoselective alkynylation, without methyl transfer, is obtained.14 The reaction proceeds at 0 °C in
tolu-ene. No reaction occurs if THF is used as a cosolvent, high-lighting the role of coordination in this transformation.
3.1 Reactions with Csp
3Electrophiles
3.1.1 Alkyl Halides
The reaction of dimethylalkynylaluminum reagents with alkyl halides takes place through a dissociative path-way and involves the coordination of the metal center to the leaving group. The exceeding high affinity of aluminum
for fluorine (663 kJ mol–1) has been exploited for the
selec-tive alkynylation of tertiary alkyl fluorides (Scheme 4).19
In-terestingly, no reaction was observed using the correspond-ing chloro analogues.
Scheme 4 Nucleophilic substitutions of alkyl fluorides
3.1.2 Propargylic Sulfonates
The Lewis base activation of dimethylalkynylaluminum reagents associated with their low basicity has been ex-ploited in a very elegant synthesis of skipped diynes from propargylic electrophiles (Scheme 5).20 The coordination of
the metallic nucleophile to the electrophile drives a clean SN2 reaction and avoids an undesired SN2′ substitution at
the triple bond. Particularly noteworthy is the reaction of terminal propargylic sulfonates, known to generally react predominately on the least substituted carbon and lead to allene intermediates. The corresponding chloro or iodo de-rivatives do not react under similar reaction conditions. A six-membered aluminum-coordinated transition state has been proposed to explain this difference in reactivity.
Scheme 5 Selective synthesis of skipped diynes Scheme 2 Preparation of dimethylalkynylaluminum reagents by
base-catalyzed terminal alumination
toluene or heptane r.t AlMe3 + 1–2 mol% MeN(SiMe3)2 R H R AlMe2 N AlMe3 + AlMe3 20 μL·min–1 R H R AlMe2
Scheme 3 Example of a selective Lewis acidity triggered reaction of
di-methylalkynylaluminum reagents N O NC Ph AlMe2 Pent AlMe2 Pent THF no reaction toluene 0 °C to r.t., 1 h N NC Ph OH Pent 71%, d.e. > 97% RLi or R2CuLi N O Ph R NH AgBF4 RMgX N O R Ph F Ph Ph AlMe2 toluene –78 °C, 30 min Ph Ph Cl Ph Ph AlMe2 toluene –78 °C, 30 min no reaction 70% Ph AlMe2
X = OSO2Me, R = Et, 92%
X = OSO2Me, R = H, 88%
X = OSO2pTol, R = Et, 96%
X = OSO2pTol, R = H, 85% X = Cl R = Et 0% + R X heptane/CH 2Cl2 0 °C R Ph
3.1.3 Thioacetals
Sulfones are excellent leaving groups for the synthesis of medium-sized -substituted cyclic ethers; the nucleo-philic substitution involves a reactive oxonium intermedi-ate. Interestingly, thioacetals are selectively activated in the presence of acetals (Scheme 6).21
Scheme 6 Alkynylation of lactone-derived sulfones
Such selective alkynylation was exploited in the synthe-sis of the C15–C38 fragment of okadaic acid (Scheme 7).22
This example is particularly illustrative of the great func-tional group tolerance of organoaluminum species as well as their excellent chemoselectivity.
Scheme 7 Selective alkynylation as a key step in the synthesis of the
C15–C38 fragment of okadaic acid
3.1.4 Hemiaminals
Like oxoniums, iminium precursors such as hemiami-nals can react with dimethylalkynylaluminum reagents in a similar manner. This reactivity enables a simple access to various substituted propargylamines (Scheme 8).23
Scheme 8 Synthesis of mono- and bis-propargylamines
3.1.5 -Lactones
Alkynylaluminum compounds are excellent partners in alkyne-propanoic acid homologation reactions based on the regioselective ring opening of -propiolactone.24 This
reaction was used as a key step in the synthesis of unsatu-rated fatty acids (Scheme 9); the same reaction with the corresponding organolithium was unsuccessful.25
Scheme 9 Homologation of alkynes with -propiolactone
3.1.6 Epoxides
Epoxides react readily with dimethylalkynylaluminum reagents (Scheme 10).26 As expected, the ring opening is
generally fully regioselective with terminal epoxides, the least substituted carbon being the most reactive one.
Scheme 10 Regioselective epoxide opening by
dimethylalkynylalumi-num reagents O SO2Ph AlMe2 Ph toluene/CH2Cl2 O Ph O O O Ph Ph H H H O O Ph H H H OBn O O Ph H H H OBn O Ph O H H H O Ph O O H H H Ph O O O S H OTIPS OBn OMe O O H + O O Me Me Me2Al CH2Cl2 O O O H OTIPS OBn H O O Me Me H 63% d.r. > 20:1 BnO BnO 0 °C to r.t. R1 AlMe 2 R1 = Ph R2 = Ph, 98% R1 = Pent R2 = Ph, 94% R1 = (CH 2)2Ph R2 = Ph, 98% R1 = cPr R2 = Ph, 90% R1 = Ph R2 = H, 98% N R2 tBuO toluene 0 °C to 60 °C N R2 R1 N OMe MeO N R1 R1 R1 = Ph 95% R1 = Pent 92% Bu Bu Cl 1) MeLi 2) 1) BuLi 2) Me2AlCl 3) O O Bu CO2H O O no reaction 62% OTBDPS O + TIPSO AlMe3 toluene 0 °C to r.t. OTIPS OH TBDPSO 92%
101
R. Piccardi et al.
Short Review
Syn thesis
The regioselectivity of the reaction is less pronounced with internal epoxides. It can however be controlled by neighboring coordination groups. This effect has been beautifully illustrated with fluorine-substituted substrates (Scheme 11). Thus, a -fluorinated epoxide (X = F) reacted in less than 10 minutes at –78 °C to give a homopropargyl alcohol I in a fully regioselective manner and 70% chemical yield. In marked contrast, the corresponding non-fluorinat-ed substrate (X = H) was less reactive, delivering an almost equimolar mixture of isomers I/II in only 40% yield. This fluorine assistance can be explained by the high affinity of aluminum to fluorine, leading to the formation of a tran-sient pentacoordinate complex that delivers its alkynyl group in a regioselective manner.27
Scheme 11 Fluorine-assisted epoxide opening
The formation of an acid-base complex prior to the alkynyl group delivery can lead to interesting rearrange-ments. The regioselective alkynylation of 2,3-epoxy sul-fides28 or selenides29 (Scheme 12) involves an episulfonium
or episelenium intermediate. In the case of sulfur deriva-tives, the C-2 alkynylation product III was preferentially ob-tained with a global retention of configuration, whereas alkynylation occurred at C-1 to give IV starting from the corresponding selenium precursor.
Scheme 12 Alkynylations of 2,3-epoxy sulfides or selenides
Another chemo- and regioselective alkynylation of an epoxide involving a neighboring group participation can be observed on bicyclic hydrazines (Scheme 13).30 This
behav-ior is a remarkable example of the synthetic potential of strong Lewis acidic, poor nucleophilic reagents such as or-ganoaluminum compounds.
Scheme 13 Neighboring group participation in bicyclic hydrazine
ep-oxide alkynylation 3.1.7 Aziridines
Dimethylalkynylaluminum compounds react with N-sulfonylaziridines. The reaction has to be conducted in CH2Cl2 and proceeds via coordination of the organometallic compound to the aziridine-protecting group with subse-quent intramolecular alkynylation.31 Interestingly, a
trans-1,2-disubstituted cyclohexene was obtained from an aziri-dine-fused cyclohexene whereas cis-1,4-disubstituted cy-clopentene was formed from an aziridine-fused cyclopen-tene. The same aziridine reacted with trimethylaluminum to give a cis-1,2-disubstituted cyclopentene (Scheme 14).
Scheme 14 Ring opening of cyclic vinyl aziridines
3.1.8 -Azido Enol Ethers
-Azido silyl enol ethers can be ionized by Lewis acids like AlMe3 and generate a reactive enonium that can
under-go a conjugate 1,4-addition. This original reactivity was used to prepare -alkynyl enol ethers in 70–98% yield (Scheme 15).32
Scheme 15 Alkynylation of -azido silyl enol ethers
Ph X O Ph AlMe2 Ph X OH Ph Ph X OH Ph + toluene I II X = F, –78 °C, 0.1 h, 70%, I/II = 99:1 X = H, –20 °C, 5 h, 40%, I/II = 1:1.2 Pr SiMe3 AlMe2 X = S 91% III/IV = 90:10 X = Se 87% III/IV = 0:100 O XPh Pr XPh OH SiMe3 Pr OH XPh SiMe3 + CH2Cl2 III IV AlMe2 R NCO2Bn NCO2Bn O NCO2Bn NCO2Bn O R Me2Al NCO2Bn NCO2Bn HO 59–63% – + + R Ph AlMe2 NTs 0 °C, CH2Cl2 NHTs Ph 45% NTs Ph AlMe2 0 °C, CH2Cl2 TsHN Ph 65% NTs 0 °C, CH2Cl2 NHTs 62% AlMe3 Me OTIPS N3 R AlMe2 hexane OTIPS R R = Ph 98% R = SiMe3 70% R = Bu 84%
3.2 Reactions with Csp
2Electrophiles
3.2.1 Carbonyl Compounds
Dimethylalkynylaluminum reagents react readily with carbonyl compounds by selectively transferring their alkynyl moiety (Scheme 16).33 The general order of
reactiv-ity is acyl chlorides > aldehydes > ketones >> esters. Of par-ticular interest is the reaction with acyl chlorides, which provides a very simple and convenient access to ynones.34
The use of 1,2-dichloroethane as a solvent is crucial in this transformation since no reaction occurs in THF. The prepa-ration of symmetrical diynones from oxalyl chloride is also noteworthy.34
Scheme 16 Reaction of dimethylalkynylaluminum reagents with
car-bonyl compounds
3.2.2 Amino Derivatives
The reaction of dimethylalkynylaluminum with nitriles requires a higher temperature than that used with acyl chlorides, but this is an alternative way to prepare ynones after hydrolysis of the ,-alkynylketimine.35 Interestingly,
in a one-pot procedure, treatment of the N-(dimethylalumi-no)-,-alkynylketimine addition product with a chlorofor-mate led to an ynimine carbachlorofor-mate; this straightforward re-action enabled a very efficient access to 1,2-dihydropyri-dines via an alkyne isomerization/electrocyclization sequence (Scheme 17).36
Scheme 17 Two-step synthesis of disubstituted dihydropyridines from
nitriles
Hydroximoyl chlorides and hydrazinoyl chlorides react-ed readily with dimethylalkynylaluminum reagents; the transient aluminate underwent an intramolecular cycliza-tion, leading to aluminated isoxazoles and pyrazoles. The stability of the C–Al bonds enables this transformation to be performed at 50 °C without degradation by -elimina-tion (Scheme 18). These aluminated heterocycles were then reacted with strong electrophilic reagents such as isocya-nates or N-halosuccinimides.37
Scheme 18 One-pot, two-step synthesis of trisubstituted oxazoles and
pyrazoles
The reaction of dimethylalkynylaluminum compounds with carbohydrate-derived nitrones was highly stereoselec-tive (Scheme 19);38 cyclization onto the triple bond was not
observed. In absence of any other competitive Lewis basic center, nitrones themselves catalyzed the terminal alumi-nation of alkynes, leading to a one-pot -addition from ter-minal alkynes in the presence of trimethylaluminum.39
Scheme 19 Reaction of alkynylaluminum reagents with cyclic nitrones
Activated imines, such as N-phosphinoyl40 or
N-sulfinylimines,41 react with dimethylalkynylaluminum
re-agents. Good to excellent diastereoselectivities were gener-ally obtained when performing the reaction in toluene (Scheme 20). No reaction was observed in THF or diethyl ether, highlighting the importance of coordination for this transformation. R1 R2 OH R1 R2 O R1 O R1 R1 R2 OH R3 R2COCl R2COR3 R2CHO Cl(CO) 2Cl Me2Al R1 R1 + R2CN AlMe3 Et3N (10 mol%) hexane/toluene 60 °C, 20 h R 1 R2 NAlMe2 R3O Cl O THF R1 R2 N OR3 O dppp (30 mol%) toluene, 110 °C 24 h N R1 R2 R3 O 44–88% N XH N X R1 "Al" R2 Cl R1 E+ N X R1 E R2 R2 AlMe2 AlMe3, toluene X = O, NR3 N O Cl Pr 91% Cl N O I Pr 57% N O Pr 65% Cl N O Pr 70% HON O NH2 Cl N Ph N Cl Pr 89% N Ph N Cl 71% N BnO OBn O + – + toluene 0 °C to r.t. 92% d.r. > 98:2 Ph AlMe2 BnO BnO N BnO OBn OH BnO BnO Ph
103
R. Piccardi et al.
Short Review
Syn thesis
Scheme 20 Diastereoselective synthesis of propargylamines
The reaction of dimethylalkynylaluminum reagents with diazo(trimethylsilyl)methane is particularly notewor-thy. Instead of attacking the metal center, as classically de-scribed with diazomethane and alkyl-, alkenyl-, or arylalu-minum compounds, diazo(trimethylsilyl)methane behaves as an electrophile with dimethylalkynylaluminum com-pounds. This unique reactivity enabled a simple access to -silylated alkynyl hydrazones.42 These species were readily
oxidized into the corresponding diazo derivatives, which served as useful precursors for geminal bis-propargylsi-lanes43 or -silylated propargyl esters (Scheme 21).44
Scheme 21 Reaction of dimethylalkynylaluminum reagents with
di-azo(trimethylsilyl)methane and subsequent transformations 3.2.3 Enones
Dimethylalkynylaluminum reagents add to conjugated enones provided they can adopt a chelated transition state, leading to an intramolecular delivery of the alkynyl moi-ety.45 As a consequence, enones devoid of a proximal
coor-dinating group can undergo a 1,4-addition only if they are able to adopt a cisoid conformation. This strategy was used for the two-step synthesis of -butyrolactones from a Meldrum’s acid derivative (Scheme 22).46 The low basicity
of organoaluminum reagents enabled a clean 1,4-addition whereas any attempts to add lithium alkynides resulted in nearly quantitative recovery of starting material, probably because of undesirable -deprotonation at the methyl posi-tion.
Scheme 22 1,4-Addition to a Meldrum’s acid derivative
The need for a coordinating group in 1,4-addition onto cyclic enones has been exploited in the diastereoselective desymmetrization of p-quinols by dimethylalkynylalumi-num reagents (Scheme 23).47
Scheme 23 1,4-Addition to p-quinols
4 Transition-Metal-Catalyzed
Reactions
4.1 Addition
to
,-Unsaturated Enones
Conjugated addition of dimethylalkynylaluminum re-agent to cyclic enones can be quite challenging due to the lack of a polar directing group allowing the delivery of the alkynyl group to the enone moiety. This problem can how-ever be circumvented by using transition-metal-catalyzed reactions. Indeed, the selective 1,4-addition of alkynyl groups to ,-unsaturated compounds can be observed in the presence of nickel catalysts (Scheme 24).48 In this case,
an excess of the dimethylaluminum reagent was necessary to prevent undesired side reaction of the final aluminum enolate with the unreacted enone.
Scheme 24 Nickel-catalyzed 1,4-alkynylations
Interestingly, enantioselective 1,4-additions can be achieved either in the presence of a Ni(II)–bisoxazolidine catalyst, or using nickel(II) complexes with chiral biphos-phine ligands (Scheme 25). In this last case, the alkynylating species proposed to be a bis(phosphine)nickel diacetylide,
toluene 60 °C 42–70% d.r. = 75:25 to 98:2 R1 AlMe2 N P R2 O Me Ph NH P R2 O Me Ph R1 toluene 0 °C N S R2 O pTol NH S R2 O R1 pTol 22–88% d.r. = 84:16 to 99:1 N2 Me3Si + R1 N SiMe3 NH2 AlMe3 (1 equiv) 45–92% Cl(CH2)2Cl 60 °C R1 AlMe2 MnO2 R1 N2 SiMe3 R1 SiR2 3 Me3Si R1 OCOR3 Me3Si O O Ph Me Ph O O O O Ph O O Ph AlMe2 toluene 0 °C to r.t. 77% Ag2CO3 10 mol% PhH/H2O O O Ph Ph Me 92% + O R1 R1 HO S O pTol CH2Cl2 0 °C O R1 HO S O pTol R3 56–87% R2 R2 R2 AlMe2 R3 R2 O R AlMe2 Ni(acac)2/DIBAL-H (0.2 equiv) O R Et2O + n n n = 0, 1 15–85%
while the dimethylaluminum chloride formed during the metathesis activates the enone by coordinating to the car-bonyl group.49
Scheme 25 Asymmetric nickel-catalyzed 1,4-alkynylations
4.2 Coupling
Reactions
Compared to terminal alkynes or other metallated alkynes, dimethylalkynylaluminum reagents are rarely used in transition-metal-catalyzed coupling reactions. These compounds can nonetheless be employed as valuable re-agents in palladium-catalyzed cross-couplings for the selec-tive alkynylation of aromatic halides or triflates (Scheme 26).50 This approach prevents the formation of undesired
homodimeric byproducts frequently observed in Glaser-type reactions.
Scheme 26 Palladium-catalyzed cross-coupling reactions
Dimethylalkynylaluminum reagents can also react with alkynyl bromides in the presence of nickel(0) catalysts to generate unsymmetrical diynes in good yields (Scheme 27).51
Scheme 27 Synthesis of unsymmetrical diynes
An interesting palladium-catalyzed coupling between -tosylvinylsulfoximines and alkynylaluminum reagents has been reported for the stereospecific synthesis of enyn-ylsulfoximines (Scheme 28).52
Scheme 28 Stereospecific synthesis of enynylsulfoximines
5 Triple Bond Reactivity
Although dimethylalkynylaluminum reagents behave mostly as -nucleophilic alkynides, their triple bond can also be engaged in several reactions. This reactivity, com-bined with the stability of the C–Al bond, can be exploited in the synthesis of various organoaluminum compounds. Thus, dimethylalkynylaluminum reagents undergo carbom-etalation reactions in the presence of titanium- or zirconi-um-based catalysts to form geminal dimetallic species (Scheme 29). Such compounds can also react easily with different electrophiles, such as aldehydes, ketones, halo-gens, or acyl chlorides. Remarkably, while for the Al/Ti bis-metallic compound a slow E/Z isomerization can occur at room temperature, the zirconium derivative has proven to be stereochemically stable.53
Scheme 29 Methylmetalation of alkynylaluminum reagents
Zirconium-catalyzed reactions can also be used to con-vert -halogenated alkynylaluminum derivatives into iodo-cycloalkenes via the formation of cyclic organometallic in-termediate species (Scheme 30).54
Scheme 30 Intramolecular reaction of bis-metalated reagents
Another remarkable transformation is the copper-cata-lyzed 3+2 cycloaddition of azides. This reaction delivered various aluminated triazoles in a regioselective manner. The new organometallic species were then reacted with various electrophiles (Scheme 31). This synthetic route en-ables a straightforward access to trisubstituted triazoles.55 O R AlMe2 O R toluene + 61–74% ee = 85–90% P(Ar)2 P(Ar)2 NiCl2 R AlMe2 + ArX 2.5 mol% Pd2(dba)3•CHCl3 5 mol% dppf R Ar heptane/DME X = I, Br, Cl, OTf 58–100% Ph AlMe2 Tol Br 5 mol% Ni(OAc)2 10 mol% P(o-furyl)3 Et2O, rt, 3 h + Tol Ph 68% S OTs O TBDMSN Ph + Me2Al Bu Pd(PPh3)4 pentane/PhH/toluene 1:1:1 50 °C, 15 min S O TBDMSN Ph Bu 52% Pr AlMe2 Cl(Me)MCp2 CH2Cl2 Pr MCp2Cl AlMe2 Pr R2 R1 R1 R2 O M = Ti 67–83% AcCl AlCl3 M = Zr O Pr 61% Z/E 92:8 AlMe2 X n Cl2ZrCp2 AlMe3 M I2 I n n n = 1, 2 X = I, Br M = Al, Zr
105
R. Piccardi et al.
Short Review
Syn thesis
Scheme 31 Copper-catalyzed 3+2 cycloaddition of azides with
di-methylalkynylaluminum reagents and subsequent transformations
6 Conclusion
As shown in this short review, the reactivity of dimeth-ylalkynylaluminum reagents is, in many aspects, unique and complementary to classical reactivities observed with polar organometallic reagents. The combination of a metal atom acting as strong Lewis and a rather stable carbon– metal bond with significant covalent character leads to in-teresting regio- and chemoselectivities, which can general-ly be observed at room temperature or under refluxing sol-vent conditions. In all the cases the alkynyl group is selec-tively transferred, while the methyl groups remain spectator substituents. This reactivity, associated with the low price and wide availability of trimethylaluminum, make these compounds valuable reagents for many syn-thetic applications.
Funding Information
A part of the work reported in this review and conducted in our lab has been finance by ANR (TRIBAL and AluMeth grants) ()
Acknowledgment
CNRS and Paris Descartes University are acknowledged.
References
(1) Hallwachs, W.; Schafarik, A. Justus Liebigs Ann. Chem. 1859, 109, 207.
(2) Atwood, D. A.; Yearwood, B. C. J. Organomet. Chem. 2000, 600, 186.
(3) (a) Suzuki, K.; Nagasawa, T.; Saito, S. e-EROS Encycl. Reagents Org. Synth. 2007, DOI: 10.1002/9780470842898.rt216.pub2. (b) The Chemistry of Organoaluminum Compounds; Micouin, L.; Marek, I.; Rappoport, Z., Ed.; Wiley: Chichester, 2017.
(4) Saito, S. In Comprehensive Organometallic Chemistry III;Vol. 9 Mingos, D. M. P.; Crabtree, R. H., Ed.; Elsevier: Oxford, 2007, 245. (5) Oishi, M. In Science of Synthesis;Vol. 7 Yamamoto, H., Ed.; Thieme:
Stuttgart, 2004, 261.
(6) Eisch, J. J. In Comprehensive Organometallic Chemistry;Vol. 1 Wilkinson, G.; Stone, F. G. A.; Abel, E. W., Ed.; Pergamon: Oxford, 1982, 555.
(7) For a more general review on alkynylaluminum species see: Piccardi, R.; Micouin, L.; Jackowski, O. In The Chemistry of Orga-noaluminum Compounds; Micouin, L.; Marek, I.; Rappoport, Z., Ed.; Wiley: Chichester, 2017, 157.
(8) Starowieyski, K. B.; Chwojnowski, A.; Kusmierek, Z. J. Organomet. Chem. 1980, 192, 147.
(9) Woo, S. H.; Parker, M. H.; Ploypradith, P.; Northrop, J.; Posner, G. H. Tetrahedron Lett. 1998, 39, 1533.
(10) (a) Danishefsky, S.; Kitahara, T.; Tsai, M.; Dynak, J. J. Org. Chem.
1976, 41, 1669. (b) Verrier, C.; Carret, S.; Poisson, J.-F. Org.
Biomol. Chem. 2014, 12, 1875.
(11) Reinäcker, R.; Schwengers, D. Justus Liebigs Ann. Chem. 1970, 737, 182.
(12) Binger, P. Angew. Chem., Int. Ed. Engl. 1963, 2, 686.
(13) Feuvrie, C.; Blanchet, J.; Bonin, M.; Micouin, L. Org. Lett. 2004, 6, 2333.
(14) Zhou, Y.; Lecourt, T.; Micouin, L. Adv. Synth. Catal. 2009, 351, 2595.
(15) Kanbur, U.; Ellern, A.; Sadow, A. D. Organometallics DOI: 10.1021/acs.organomet.8b00374.
(16) Piccardi, R.; Coffinet, A.; Benedetti, E.; Turcaud, S.; Micouin, L. Synthesis 2016, 48, 3272.
(17) (a) Zhu, J.; Quirion, J.-C.; Husson, H.-P. Tetrahedron Lett. 1989, 30, 5137. (b) Froelich, O.; Desos, P.; Bonin, M.; Quirion, J.-C.; Husson, H.-P. J. Org. Chem. 1996, 61, 6700. (c) Cutri, S.; Chiaroni, A.; Bonin, M.; Micouin, L.; Husson, H.-P. J. Org. Chem. 2003, 68, 2645.
(18) Guerrier, L.; Royer, J.; Grierson, D. S.; Husson, H.-P. J. Am. Chem. Soc. 1983, 105, 7754.
(19) Ooi, T.; Uraguchi, D.; Kagoshima, N.; Maruoka, K. Tetrahedron Lett. 1997, 38, 5679.
(20) (a) Kessabi, J.; Beaudegnies, R.; Jung, P. M. J.; Martin, B.; Montel, F.; Wendeborn, S. Org. Lett. 2006, 8, 5629. (b) Kessabi, J.; Beaudegnies, R.; Jung, P. M. J.; Martin, B.; Montel, F.; Wendeborn, S. Synthesis 2008, 655. (c) Hamada, N.; Yoshida, Y.; Oishi, S.; Ohno, H. Org. Lett. 2017, 19, 1875.
(21) Suga, Y.; Fuwa, H.; Sasaki, M. J. Org. Chem. 2014, 79, 1656. (22) (a) Ley, S. V.; Humphries, A. C.; Eick, H.; Downham, R.; Ross, A.
R.; Boyce, R. J.; Pavey, J. B. J.; Pietruszka, J. J. Chem. Soc., Perkin Trans. 1 1998, 3907. (b) Fuwa, H.; Sakamoto, K.; Muto, T.; Sasaki, M. Tetrahedron 2015, 71, 6369.
(23) Korbad, B. L.; Lee, S.-H. Eur. J. Org. Chem. 2014, 5089.
(24) Shinoda, M.; Iseki, K.; Oguri, T.; Hayasi, Y.; Yamada, S.-I.; Shibasaki, M. Tetrahedron Lett. 1986, 27, 87.
(25) Charoenying, P.; Davie, D. H.; McKerrecher, D.; Taylor, R. J. K. Tetrahedron Lett. 1996, 37, 1913.
(26) Bijoy, P.; Avery, M. A. Tetrahedron Lett. 1998, 39, 209.
(27) Ooi, T.; Kagoshima, N.; Maruoka, K. J. Am. Chem. Soc. 1997, 119, 5754.
(28) Sasaki, M.; Tanino, K.; Miyashita, M. J. Org. Chem. 2001, 66, 5388.
(29) Sasaki, M.; Hatta, M.; Tanino, K.; Miyashita, M. Tetrahedron Lett.
2004, 45, 1911.
(30) Bournaud, C.; Bonin, M.; Micouin, L. Org. Lett. 2006, 8, 3041. (31) Bertolini, F.; Woodward, S.; Crott, S.; Pineschi, M. Tetrahedron
Lett. 2009, 50, 2515.
(32) Magnus, P.; Lacour, J.; Evans, P. E.; Rigollier, P.; Tobler, H. J. Am. Chem. Soc. 1998, 120, 12486.
AlMe2
R1
+ R2-N 3
CuI (10 mol%), THF (1 equiv) [Me2N(CH2)2]2NMe(10%) N N N R2 R1 AlMe2 E+ toluene, rt, 48 h N N N R2 E R1 E = D, CO2Me, CO2Bn, Cl, Br, I 52–98%
(33) (a) Lehmkuhl, H.; Ziegler, K.; Gellert, H.-G. In Houben-Weyl; Thieme: Stuttgart, 1970, 4th ed, Vol. 13 158. (b) Zweifel, G.; Miller, J. A. Org. React. 1984, 32, 375.
(34) Wang, B.; Bonin, M.; Micouin, L. J. Org. Chem. 2005, 70, 6126. (35) Korbad, B. L.; Lee, S.-H. Synlett 2013, 24, 1953.
(36) Trost, B. M.; Biannic, B. Org. Lett. 2015, 17, 1433.
(37) Jackowski, O.; Lecourt, T.; Micouin, L. Org. Lett. 2011, 13, 5664. (38) Pillard, C.; Desvergnes, V.; Py, S. Tetrahedron Lett. 2007, 48,
1457.
(39) Bunlaksananusorn, T.; Lecourt, T.; Micouin, L. Tetrahedron Lett.
2007, 48, 6209.
(40) Benamer, M.; Turcaud, S.; Royer, J. Tetrahedron Lett. 2010, 51, 645.
(41) Turcaud, S.; Berhal, F.; Royer, J. J. Org. Chem. 2007, 72, 7893. (42) Kumar, R.; Turcaud, S.; Micouin, L. Org. Lett. 2014, 16, 6192. (43) Courant, T.; Kumar, R.; Turcaud, S.; Micouin, L. Org. Lett. 2016,
18, 4818.
(44) Zhao, T.; Piccardi, R.; Micouin, L. Org. Lett. 2018, 20, 5015.
(45) Gampe, C. M.; Carreira, E. M. Chem. Eur. J. 2012, 18, 15761. (46) Ahmar, S.; Fillion, E. Org. Lett. 2014, 16, 5748.
(47) (a) Carreño, M. C.; González, M. P.; Ribagorda, M.; Houk, K. N. J. Org. Chem. 1998, 63, 3687. (b) Carreño, M. C.; González, M. P.; Ribagorda, M.; Fischer, J. J. Org. Chem. 1996, 61, 6758.
(48) Hansen, R. T.; Carr, D. B.; Schwartz, J. J. Am. Chem. Soc. 1978, 100, 2244.
(49) (a) Larionov, O. V.; Corey, E. J. Org. Lett. 2010, 12, 300. (b) Kwak, Y.-S.; Corey, E. J. Org. Lett. 2004, 6, 3385.
(50) Wang, B.; Bonin, M.; Micouin, L. Org. Lett. 2004, 6, 3481. (51) Mo, S.; Shao, X.-B.; Zhang, G.; Li, Q.-H. RSC Adv. 2017, 7, 27243. (52) Paley, R. S.; Snow, S. R. Tetrahedron Lett. 1990, 31, 5853. (53) Yoshida, T.; Negishi, E. J. Am. Chem. Soc. 1981, 103, 1276. (54) (a) Boardman, L. D.; Bagheri, V.; Sawada, H.; Negishi, E. J. Am.
Chem. Soc. 1984, 106, 6105. (b) Negishi, E.; Sawada, H.; Tour, J. M. J. Org. Chem. 1988, 53, 913.
(55) Zhou, Y.; Lecourt, T.; Micouin, L. Angew. Chem. Int. Ed. 2010, 49, 2607.