A Bayesian framework for high-throughput T cell receptor pairing
Texte intégral
Figure
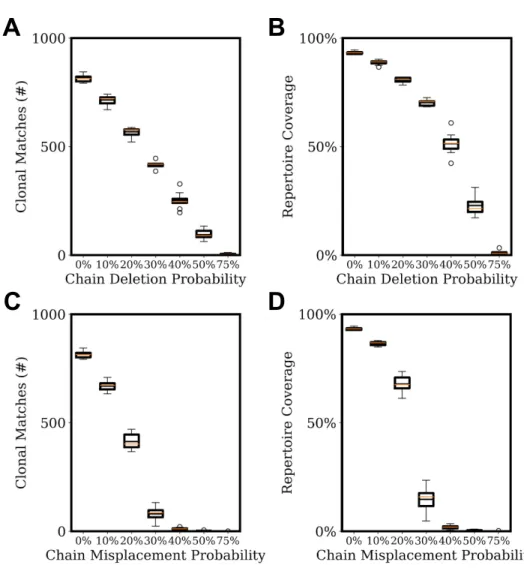

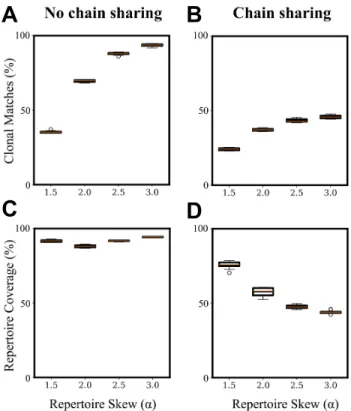


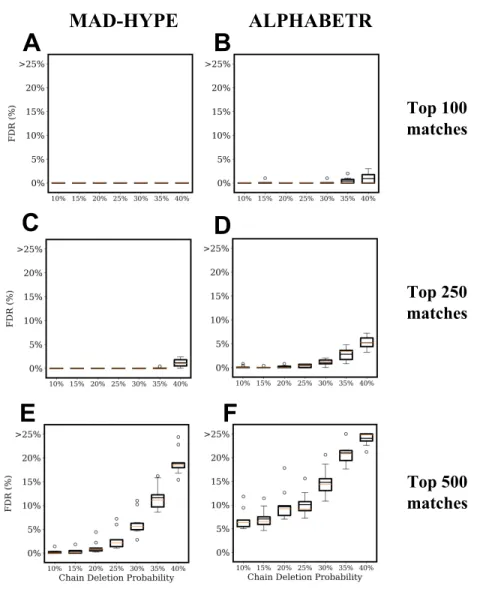


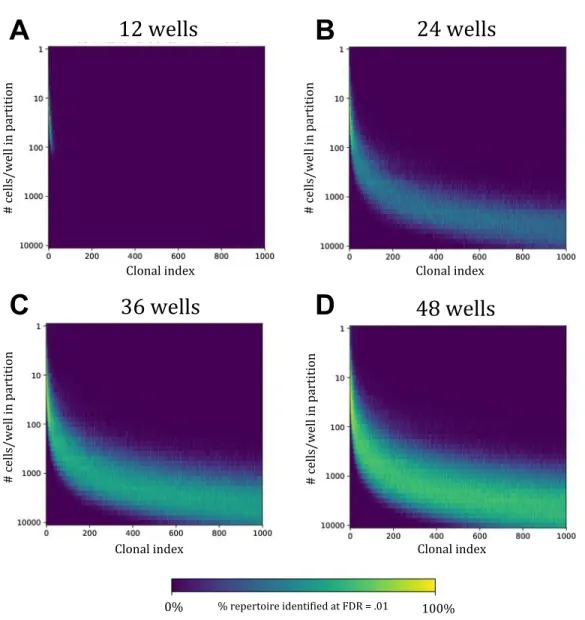

Documents relatifs
A two step approach is used: first the lithofacies proportions in well-test investigation areas are estimated using a new kriging algorithm called KISCA, then these estimates
Figure 3 compares well flow rate on production wells given by three simulations: the fine grid simulation and two coarse grid simulations using “linear flow” upscaling procedure
Absolute electroluminescence and photoluminescence measurements were carried out on strain-balanced quantum well solar cells Over a range of bias, a reduced radiative
Again, it can be surmised that the partial oxidation of ytterbium upon grinding in air before sintering leads to smaller effective ytterbium concentration in the Yb y Co 4 Sb 12
Les expériences fondées sur des modèles de déficiences génétiques en autophagie ont permis de démontrer l’importance de ce processus dans l’homéostasie des lymphocytes B et T
We describe the restriction of the Dehornoy ordering of braids to the dual braid monoids introduced by Birman, Ko and Lee: we give an inductive characterization of the ordering of
Responses to four phases will be conducted during this research on ASD participants and aged-matched controls: (1) 24 h pre-experimental recording for baseline, (2) a 2 h
During the past several years, sharp inequalities involving trigonometric and hyperbolic functions have received a lot of attention.. Thanks to their use- fulness in all areas
