Characterization of Influenza A Virus Evolution in
Laboratory Hosts.
by
Kimberly Ryan Davis
B.S., University of California Santa Cruz (2011)
Biochemistry and Molecular Biology
Submitted to the Graduate Program in Microbiology
in partial fulfillment of the requirements for the degree of
Doctor of Philosophy
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
February 2018
©Kimberly Ryan Davis, 2018. All rights reserved.
Author . . . .
Graduate Program in Microbiology
December 1, 2017
Certified by . . . .
James G. Fox
Professor of Biological Engineering
Thesis Supervisor
Accepted by . . . .
Martin F. Polz
Professor of Civil and Environmental Engineering
Co-Director, Microbiology Graduate Program
Characterization of Influenza A Virus Evolution in
Laboratory Hosts.
by
Kimberly Ryan Davis
Submitted to the Graduate Program in Microbiology on December 1, 2017, in partial fulfillment of the
requirements for the degree of Doctor of Philosophy
Abstract
Surveillance of influenza A virus (IAV) is conducted for many different hosts including humans, swine, poultry, and wild birds. The surveillance samples are screened by var-ious methods, but ultimately the isolates that are positive for IAV are propagated in a laboratory host prior to genome sequencing and characterization. Previous research has shown that passaging influenza viruses in laboratory hosts results in changes in viral sequence and receptor binding preferences. These studies have been limited to human IAV strains, and it remains unclear how propagation in laboratory hosts alters viruses isolated from animals. This thesis explores the evolutionary dynamics of IAV in a laboratory host environment.
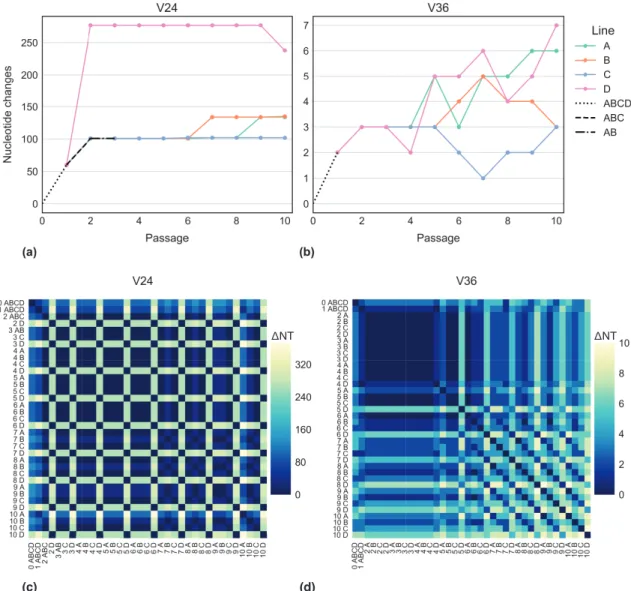
Chapter 2 of the thesis examines how the genomes of avian IAV strains change during a single infection in ECEs and MDCKs, and during ten serial infections in ECEs. The results from these experiments indicate that there is a wide range in the number of sequence differences between pre- and post-passaged viruses, and that sequencing viruses prior to passage results in better identification of mixed infections. The sequencing results from the serial passaged viruses show that egg adaptation is not limited to a single set of predictable changes. The results also suggest that the hemagglutinin gene is important for adaptation to a novel host environment.
The aim of this thesis is to identify the changes associated with propagation of IAV in different laboratory host environments. This data can be incorporated into phylodynamic studies that track global IAV transmission through different hosts. In combination with surveillance efforts, these experiments will augment our ability to predict influenza evolution and our pandemic preparedness.
Thesis Supervisor: James G. Fox
Acknowledgments
I want to thank all members of the Runstadler lab. Thank you Jon for the guidance you provided me while I was a student in your lab. I sincerely appreciate the freedom you gave me to explore new topics and experimental techniques. Thank you Wendy for your scientific and personal support. Thanks for helping me handle the many egg orders that I needed for this thesis, and for keeping our lab running. Thank you Nichola for being such a wonderful lab mate. I have learned so much from you about science and life. Thank you Islam for teaching me about all things virology and for being a great lab mate. Thank you to Eric for helping me learn python and for telling me about Jupyter. This thesis would not have been completed otherwise. Special thanks to all of the UROPs in the Runstadler lab, Annie, Lizy, Ellie, Andrea, Justin, Kyle, Nana, Laila. You all made the lab an exceptionally fun place to be. Thank you Darcy for for being such a great mentee. It was great to have your help developing the project in this thesis. Thank you to our lab admin Kathy for endless support over the years.
I want to thank my thesis committee, Martin Polz, Michael Birnbaum, and James Fox, for all of the guidance you have provided me during my time in graduate school. Thank you to Marta Gaglia for serving on my defense committee.
Thank you to my Maseeh family. Being a GRT has been one of the best parts of graduate school and my life. To the members of the house team, thank you for always being there for me. Special thanks to Becky,Yasmin, and Sebastian for helping me through all of the tough times. Thank you to all of my students over the last four years. I have learned so much from all of you, and I can’t wait to see what great things you accomplish in life.
Thank you to all my fellow Micro classmates. Thank you to Bonnielee and Susan for your support during my time at MIT.
To all of my friends, I could have not gotten through this experience without you. Jen, thank you for being my friend since we first me at MIT. It is not possible to express how grateful I am to have met you. Thank you to Chris. I am so happy
that we joined the same lab. All of the coffee breaks, TC jam sessions, and weekend adventures made the grad school grind much more bearable.
Finally, thank you to my parents and brother. You have supported me through all of the ups and downs of grad school. Thank you for always encouraging me to learn new things, and for letting me take over your house while I wrote this thesis.
Contents
1 Introduction 11
1.1 Influenza surveillance methods . . . 13
1.1.1 Detection . . . 13
1.1.2 Isolation . . . 14
1.1.3 Sequencing . . . 15
1.2 Influenza ecology . . . 17
1.2.1 Wild aquatic birds . . . 17
1.2.2 Swine . . . 19
1.2.3 Domestic poultry . . . 20
1.2.4 Humans . . . 22
1.3 Influenza evolution . . . 23
1.3.1 RNA-dependent, RNA polymerase . . . 23
1.3.2 Reassortment . . . 25
1.4 Influenza experimental evolution . . . 28
1.4.1 Airborne transmission . . . 28
1.4.2 Novel host adaptation . . . 29
1.5 Conclusion . . . 31
2 Influenza A virus adaptation during laboratory host passage impacts viral subtype identification and phylogenetic reconstruction. 53 2.1 Abstract . . . 53
2.2 Background . . . 54
2.3 Results . . . 56
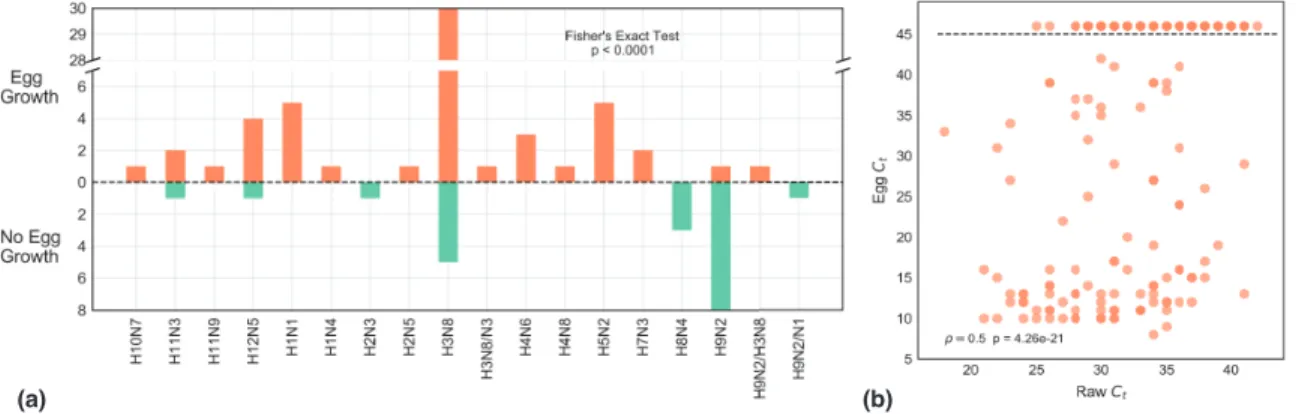
2.3.1 The subtype of IAV is predicative of growth in ECEs. . . 56
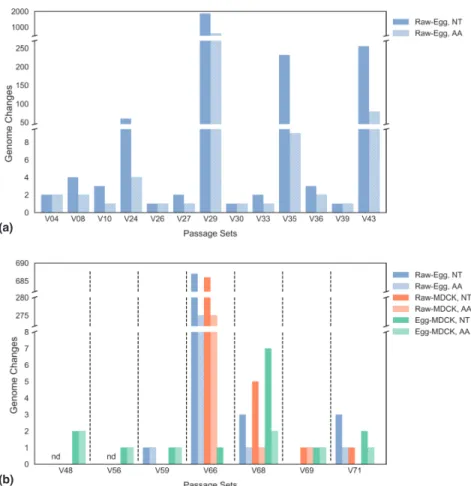
2.3.2 Passage in ECEs and MDCKs results in a range of changes in viral genome sequence. . . 58
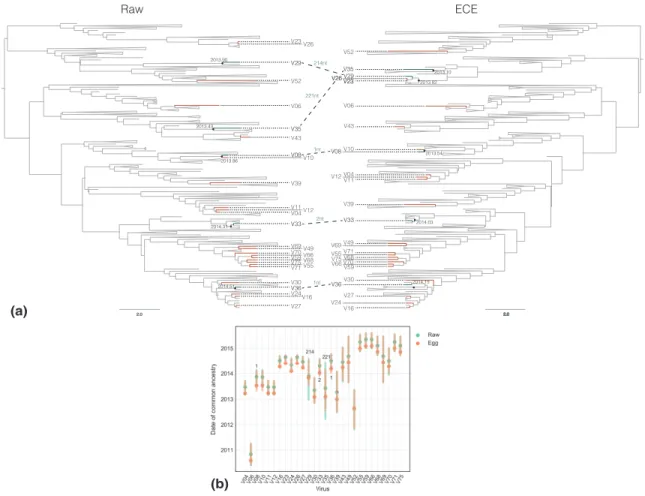
2.3.3 Passage-associated sequence changes impact phylogenetic tree construction and molecular dating analysis. . . 58
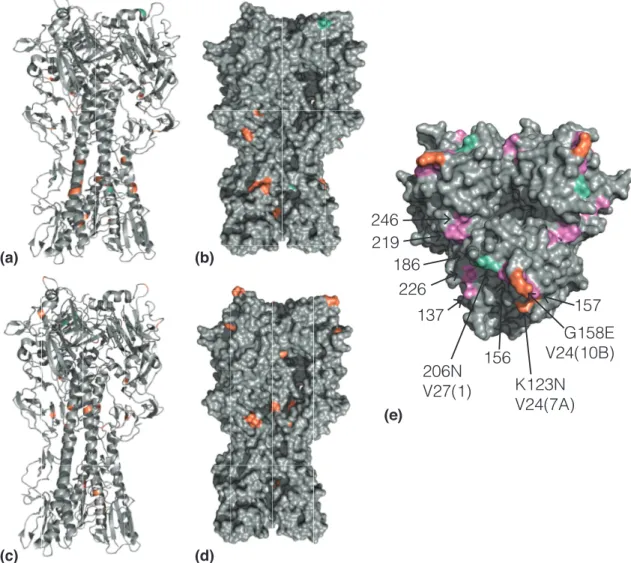
2.3.4 Changes in the hemagglutinin stem domain are common during passage in ECEs. . . 61
2.3.5 ECE grown samples have lower levels of intra-host population diversity than the raw samples. . . 61
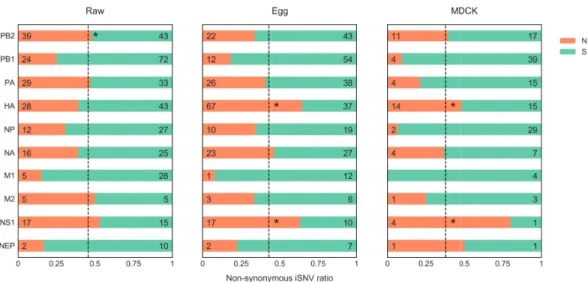
2.3.6 Non-synonymous intra-host single nucleotide variations are over-represented in HA and NS1 in passaged samples. . . 63
2.3.7 Serial passage replicates exhibit unique evolutionary trajectories. 63 2.3.8 HA mutations that occur during serial passaging are concen-trated in the head region. . . 66
2.3.9 Isolates with mixed infections have lower intra-host population diversity. . . 66
2.4 Discussion . . . 66
2.5 Materials and Methods . . . 71
2.5.1 Sampling of wild birds. . . 71
2.5.2 Viral RNA isolation. . . 71
2.5.3 IAV matrix qRTPCR . . . 72
2.5.4 ECEs infections . . . 72
2.5.5 MDCK infections . . . 72
2.5.6 Serial ECE passage . . . 73
2.5.7 RNA amplification . . . 73
2.5.9 Genome assembly and iSNV identification. . . 74
2.5.10 Sequence alignments of passaged viruses. . . 75
2.5.11 Bayesian phylogenetics and molecular dating. . . 75
2.5.12 HA mutation mapping. . . 76
2.5.13 Statistical analyses. . . 76
2.6 Supplemental figures . . . 77
2.7 Supplemental tables . . . 86
3 Conclusions and future directions 93 3.1 Conclusions . . . 93
Chapter 1
Introduction
Influenza A virus is a member of the Orthomyxoviridae family of viruses. This family is made up of the four types of influenza (A,B,C, and D), isavirus, thogotovirus, and quaranjavirus. The viruses in this family are characterized by segmented negative-sense RNA genomes and lipid envelope membranes[1]. The four influenza genera cause infections in vertebrates. Influenza B and C exclusively infect humans, and influenza D, a newly identified virus, has been isolated from cattle and swine[1, 2]. Isaviruses infect salmon, and both thogotoviruses and quarajaviruses are arboviruses and are transmitted by arthropod vectors[1]. Influenza A viruses (IAV) infect a range of vertebrate species, and are the most well known of the Orthomyxoviridae.
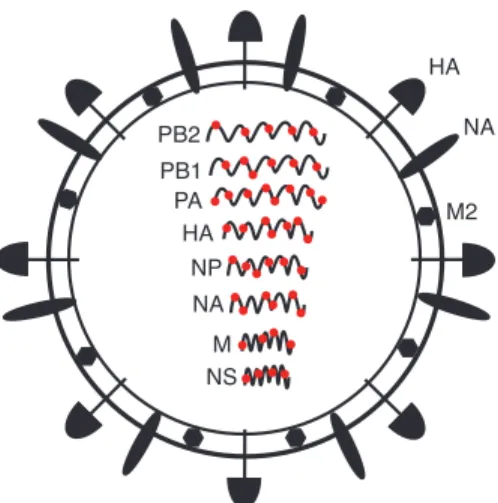
The genome of IAV is made up of eight individual negative-sense, single-stranded
RNA segments and encodes at least 11 proteins (Fig. 1)[3]. Three viral proteins–hemagglutinin (HA), neuraminidase (NA), and M2–are embedded in the lipid envelope. The HA
pro-tein binds to receptors on host cells that contain a terminally linked sialic acid. Once the virion is internalized, HA facilitates endosomal fusion. Upon assembly and bud-ding of the virions at the cell surface, NA is responsible for cleaving the terminal sialic acids on the host cell membrane to release the progeny virions[4].
Influenza-like illnesses have been documented since at least the Middle Ages. Ret-rospective studies suggest that since that time there have been 13 or more influenza pandemics. In the last 150 years, there have been 6 human influenza pandemics[5].
PB2 PB1 PA HA HA NP NA NA M M2 NS
Figure 1 IAV particle. Each of the RNA genome segments has been labeled with the common name. The hemagglutinin(HA), neuraminidase (NA), and M2 proteins have been labeled on the outside of the virion. The red circles represent the NP protein that coats the viral RNA.
The 1918 Spanish Flu pandemic was the most severe of these pandemics, and was estimated to have killed 50 million people worldwide[6]. Today, influenza viruses are endemic in several hosts, including humans, birds, swine, horses, and dogs. IAV is estimated to infect 5-10% of adults and 20-30% of children annually[7]. In the US, the economic cost of seasonal influenza infections is greater than $87 billion dollars per year[8, 9]. In addition, IAV infections in the livestock and poultry industry are a major economic concern. From 2014-2015, nearly 45 million poultry were killed, and $3.3 billion dollars was lost in revenue due to the highly pathogenic IAV that swept through the US[10].
In this chapter I review what is currently known about the evolution of IAV at both the ecological and molecular scale. In the following chapter I discuss how IAV evolves when grown in a laboratory host setting. The findings in chapter 2 illustrate that infecting IAV in a laboratory host can result in changes in the viral genome sequence. These changes impact our ability to correctly identify viral subtype and to determine patterns of IAV evolution and emergence in natural hosts. Finally in chapter 3, I discuss the future directions for the research projects.
1.1
Influenza surveillance methods
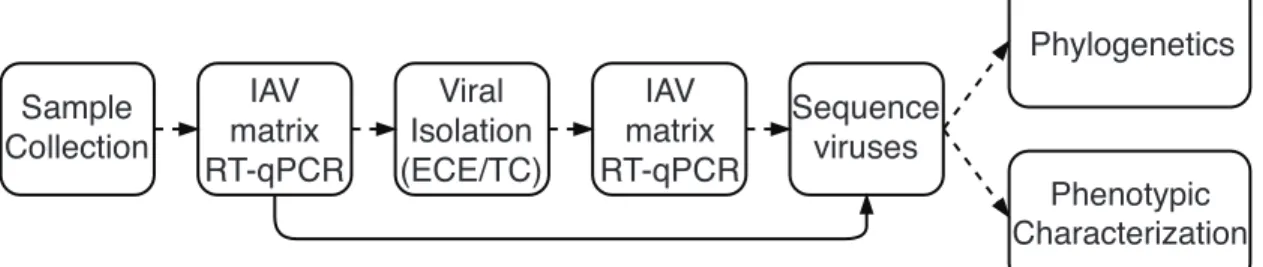
In order to study the viruses that circulate in wild animals and humans, several meth-ods have been developed for sample collection, virus detection, virus isolation, and sequencing. A typical IAV surveillance workflow in depicted in Figure 2. Depend-ing on the particular host of interest, there are particular methods and techniques for obtaining samples. Examples of these techniques are net launchers, rocket nets, and mist nets for the capture of birds, and specialized nets for the capture of ma-rine mammals[11, 12]. Once the animals or humans have been obtained, the samples (usually nasal, oral, throat, rectal, or cloacal swabs) are collected and preserved and transported in viral transport media[13].
Sample Collection IAV matrix RT-qPCR IAV matrix RT-qPCR Viral Isolation (ECE/TC) Sequence viruses Phylogenetics Phenotypic Characterization
Figure 2. A typical IAV surveillance workflow. The dashed lines represent the most commonly used workflow for IAV surveillance. The solid line represents an alternative workflow to bypass viral isolation. RT-qPCR = quantitative reverse transcription PCR, ECE = embryonated chicken egg, TC = tissue culture.
1.1.1
Detection
After sample collection, the next step is to determine whether the samples contain IAV. In a human clinical setting many rapid diagnostic procedures have been devel-oped and have been reviewed elsewhere[14]. The most commonly used method for detecting influenza in an laboratory setting is quantitative reverse transcriptase poly-merase chain reaction (RT-qPCR), also known as real-time PCR[15]. This RT-qPCR uses a probe that is specific for a highly conserved region of the IAV matrix gene. It has been used to detect many different subtypes of viruses from essentially all known
hosts of IAV[15, 16, 17]. There is also a mammalian IAV-specific probe that has been utilized to increase the assay sensitivity for samples collected from mammalian hosts[15]. Similar methods have been designed to specifically detect highly pathogenic H5 and H7 viruses[18]. Once a sample has tested positive for the presence of IAV, virus isolation is attempted.
1.1.2
Isolation
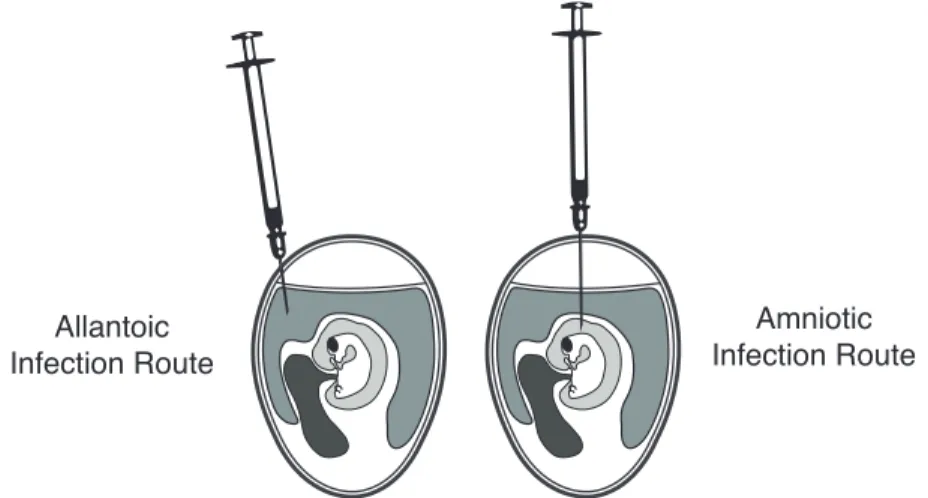
The first method used for isolating IAV was developed in 1940 prior to the advent of tissue culture[19]. This method involves infecting IAV in an embryonated chicken egg (ECE), and is still in wide use today. Currently most influenza vaccines are produced in ECEs, although recently there has been a significant effort to transition the production of vaccines to modern tissue culture methods[20]. Influenza viruses are predominately grown in either the amniotic cavity or the allantoic cavity of an ECE(Fig. 3)[21]. The allantoic cavity contains cells with receptors that are preferred by avian viruses, while the amniotic cavity contains mammalian IAV receptors (ref. section 1.2.2). With the advent of tissue culture, many immortalized cell lines have also been utilized for growing IAV. Madin Darby canine kidney (MDCK) and modified MDCK (MDCK-SIAT1) cell lines have been shown to be shown to be better for isolating swine and human viruses than ECEs[22, 23, 24]. Although many studies report ECEs as the best host for isolating avian origin virus, a study by Munster et al. showed that virus was only obtained from 32.5% of the 1,483 IAV positive samples isolated from wild birds[17].
More recently, researchers have been developing fully-differentiated, primary cell lines for growing IAV. These cells are isolated directly from the host tissue of interest, initially expanded in two-dimensional culture, and then finally allowed to differentiate and grow in three-dimensional culture at the air liquid interface (ALI)[25]. Airway epithelial cell cultures grown using this method exhibit morphological features (ex. mucus and cilia production) that are more physiologically similar to the native host tissue[25]. These systems have mostly been used for studying viral tropism and
Allantoic Infection Route
Amniotic Infection Route
Figure 3. Allantoic and amniotic routes of ECE infection.
replication dynamics, but there is potential for these cells to be useful for isolating viruses from clinical and animal samples[26, 27]. A study by Farsani et al. tested the growth of respiratory clinical samples in fully-differentiated pseudo-stratified tra-cheobronchial human airway epithelial (HAE) cultures, and found that IAV could be grown to a level sufficient for genome sequencing[28].
1.1.3
Sequencing
Before the development of genome sequencing techniques, scientists characterized in-fluenza viruses using a number of different techniques including the hemagglutination assay. This assay is based on the ability of the IAV HA protein to bind and agglu-tinate red blood cells[29]. The ability of a virus to aggluagglu-tinate red blood cells from different hosts (eg. chicken, ferret, horse) was used to characterize the viral type[30]. Now a virus is characterized by its genome sequence.
The segmented nature of the IAV genome is well suited for amplicon-based genome sequencing. Prior to 2009, each IAV segment was amplified individually with one or more sets of primers using PCR[31]. In 2009, Zhou et al. showed that all 8 segments of the IAV genome could be amplified in a single PCR reaction with primers that are specific for the untranslated region (UTR) of the genome[32]. The 5’ and 3’ ends of each genome segment contain a universally conserved sequence of 13 and
12 nucleotides respectively. Initially, each segment was cloned into a plasmid and sequenced using Sanger sequencing, but nearly all new IAV genomes are sequenced using next generation sequencing (NGS)[32, 33, 34]. Despite the ease and convenience of using an amplicon-based method to sequence IAV genomes, this method has been shown to have extensive biases towards certain sequence motifs[35, 36].
Recent developments in library preparation reagents and sequencing technology have helped circumvent the biases associated with amplicon methods and the errors that are inherent to NGS. Malboeuf et al. showed that the need to amplify the viral genome could be avoided by developing a sample preparation method to sequence very low titer HIV, RSV, and WNV clinical samples. In a recent study of Zika virus, Metsky et al. used a hybrid capture method to specifically capture and sequence the Zika virus genome[36]. This is particularly useful for Zika virus since it is present in very low titers in the human and mosquito hosts[37]. To address the errors due to NGS, Acevedo et al. developed a technique known as CirSeq that utilizes rolling circle amplification. By using this method to sequence the Polio virus genome, they were able to reduce the rate of sequencing error from 10−4 to 10−12per base sequenced[38,
39]. The influenza field would benefit by adapting these novel protocols. The ability to sequence viruses with low viral titers, would allow viruses to be directly sequenced from clinical and animal isolates. This would bypass the need for isolation in ECEs or cell culture, and eliminate biases associated with culture.
The information gained through routine surveillance activities is used for many purposes. The collection of human strains during the annual influenza season pro-vides the information necessary to design the vaccine for the following year. Regular surveillance of agricultural animals helps to identify highly pathogenic IAV infections before further spread of the virus and concomitant culling of animals. Additionally, surveillance identifies IAV strains with novel genetic mutations and functions that can be studied in the lab. Improving the surveillance workflow and identifying potential biases created by the methods in use is essential for understanding the biology of IAV.
1.2
Influenza ecology
All of the viruses responsible for the pandemics in the last 150 years have originated from viruses circulating in the avian population, a reservoir host of IAV[40, 41]. IAV is classified by its subtype, which is determined by the two main antigenic pro-teins: hemagglutinin (HA, 18 types) and neuraminidase (NA, 11 types). The primary reservoir, wild aquatic birds, host the greatest diversity of viral subtypes. Over fifty percent of all possible combinations of HA and NA types have been confirmed in these hosts[42, 43]. In addition to infecting wild birds, IAV is able to infect a wide range of other hosts (Fig. 4). The infections in the reservoir hosts are predominately asymptomatic, but avian IAV can transmit to other species and cause symptomatic, and periodically severe, disease[3]. The occurrence of these infections is generally limited and there is little or no onward transmission[44]. In the rare event when a virus becomes established in a new species, this can result in epidemic or pandemic spread. Through the accumulation of genomic mutations viruses can move from the avian host into the human population and sustain transmission, resulting in height-ened morbidity and mortality[3]. Retrospective studies of pandemic IAV strains have shown that mammalian host adaptation is polygenic in nature[5]. To improve our knowledge of global IAV circulation, and in an attempt to predict outbreaks and pandemic, surveillance is conducted across many of the IAV hosts. Here I will review what is known about the evolutionary dynamics and global circulation of IAV in four major hosts: wild aquatic birds, swine, domestic poultry, and humans.
1.2.1
Wild aquatic birds
Wild aquatic birds are considered to be the reservoir for IAV because of the genetic diversity of viruses that circulate in this population, and because of the asymptomatic nature of the infections[40]. Aquatic birds can be classified into either the order Anseriformes (ducks, swans, and geese) or the order Charadriiformes (Shorebirds, gull, auks). Among these birds, the dabbling ducks (family: Anatidae) are known to have high rates of IAV prevalence, and are thought to be the primary IAV reservoir[45,
Figure 4. IAV transmission. Lines with a single arrow illustrate the flow of IAV transmission. Lines with two arrows indicate bi-directional transmission between hosts. Hosts shown in this figure (from top-left to bottom-right): domestic poultry, Anseriformes, Charadriiformes, marine mammal, swine, human, and canine.
46, 43, 47, 48]. Within the dabbling duck family, mallards have been found to be robust hosts. This is in part because they are the most abundant and wide-ranging duck species, but also because they have been shown to shed high levels of virus for extended time periods[49, 50]. Within the Anseriformes, transmission has been shown to occur through the fecal-oral route[51, 52].
Charadriiformes are also considered to be an important reservoir for IAV, but IAV surveillance in these hosts is infrequent compared to that of Anseriformes[53]. It is known that Charadriiformes harbor a distinct pool of viruses. H9, H13, and H16 HA types are considered unique to Charadriiformes, while the H3, H6, and H7 types are considered to be more promiscuous and can be found in both hosts[54, 55, 42, 56]. As true long distance migrants, Charadriiformes are thought to play an important role in the spread of IAV between hemispheres[57, 58]. Globally avian IAV can be divided into two main lineages that are defined by the hemisphere that the viruses circulate in. Viruses and the individual genome segments are usually classified as being from the North American lineage, or the Eurasian lineage[59, 60]. Alaska and western Canada are considered to be important inter-hemispheric mixing grounds because
many species of birds congregate there for breeding and nesting[61, 59, 62]. Several analyses have indicated that Charadriiformes are critical for the mixing of viruses from the two lineages[63, 64, 65, 66]. On the intra-continental level, viral gene flow is shaped by the avian flyway[67, 68]. In the US, there are four major flyways—Pacific, Central, Mississippi, and Atlantic—that approximately describe the patterns of bird migration[69, 70, 68, 71]. Broadly, viral gene flow has been shown to move from north to south along the avian migratory flyways. In addition, studies indicate that the rates of IAV migration are higher between flyways than within flyways[71]. As a consequence of paired viral and avian migration, wild birds have been linked to the global distribution of IAV, and have been shown to be important for the origin and distribution of highly pathogenic H5 viruses[72, 73, 74, 75, 76].
1.2.2
Swine
Influenza-like illnesses have been documented in swine since the emergence of the 1918 Spanish flu pandemic in humans[77]. Unlike the asymptomatic infections of wild birds, IAV in swine is a major cause of viral pneumonia and contributor to the porcine respiratory disease complex[77]. Given the large number of swine in the world (it is estimated that China had 476.2 million pigs in 2010), IAV infections and outbreaks are of a global economic importance[77].
The subtype diversity of endemic swine viruses is relatively limited compared to that of wild avian hosts. H1N1, H1N2, and H3N2 are the predominant global subtypes, although distinct lineages of these viruses have been found to circulate in North America, Europe, and Asia[78, 79, 80]. Swine are often considered to be mixing vessels for human and avian IAV strains. Evolutionary analyses of circulating strains have provided ample evidence for infections of swine by both human and avian IAV. In China, the seasonal human H3N2 virus was transmitted to pigs several times[81]. Wholly avian strains have also been detected in swine populations in Europe, and Asia[77, 82]. In addition, other subtypes of IAV–H9N2, H5N1, H4N8, and H6N6–have been transmitted from the avian population to the swine population
in Asia[83, 84, 85, 86]. Swine viruses have also reassorted with human and avian lineages, and gone on to circulate in the swine population. This particular dynamic preceded the emergence of the 2009 H1N1 pandemic[87, 88, 89].
At a molecular level there is also support for the hypothesis of swine as virus mix-ing vessels. An important determinant of virus host range is the sialic acid receptor binding preferences of the HA protein. The receptor preferences of the virus can be partially explained by the distribution of these sialic acids on the host cells. Avian strains of the virus have been shown to preferentially bind sialic acids with an alpha-2,3 linkage (α-alpha-2,3-SA). In contrast, human strains show a preference for sialic acid receptors with an alpha-2,6 linkage (α-2,6-SA)[90, 91, 92]. The α-2,3-SA receptors are prevalent in the gut of birds, the predominant site for IAV infection[90]. In contrast, α-2,6-SA receptors are predominately found in the upper respiratory tract of humans which is the primary site for IAV infection in humans[93, 94]. Swine are thought to be mixing vessels for avian and human influenza viruses because both α-2,3 and α-2,6 SA linkages are present on tracheal epithelial cells[90, 94].
In recent years, global surveillance efforts have expanded in an attempt to better understand the circulation of swine IAV. With the increased movement of pigs be-tween countries, and the nature of swine to be infected by avian and human viruses, the risk of zoonotic infections from swine is high[3, 77]. Monitoring and controlling IAV in swine is critical for both the global economy and human health.
1.2.3
Domestic poultry
Like swine, domesticated birds of the order Galiformes (chickens, turkeys, quail) are susceptible to IAV infection and are of global economic importance. IAV are usually introduced into the domestic poultry population by either direct or indirect contact with wild birds[95, 96]. Stable transmission of novel IAV strains from wild birds to poultry tends to be limited. Exceptions to this exist for H9N2 and H6 subtype viruses which are endemic in chickens in parts of China, and for H5 and H7 viruses which are endemic in many regions of the eastern hemisphere[97, 98, 99]. Turkeys
have also been shown to be an important host because of their permissivity to many subtypes of IAV. Nearly all subtypes (H1-H11) of IAV have been documented in turkeys. H5, H7, and H9 viruses tend to be isolated most frequently[99]. Quails, and other minor poultry species, have also been found to be infected with IAV. Quails are particularly interesting because they are permissive to most subtypes of IAV, and exhibit asymptomatic infections[100]. For this reason, quails are thought to be an intermediate host that allows for the emergence of wild bird IAV in domestic poultry[99, 100].
The genomic and phenotypic changes that allow a virus from the wild bird pop-ulation to adapt to domestic poultry have not been fully characterized. In a few studies, mutations in HA, and NA have been shown to be important for adaptation to poultry. In Campitelli et al., the sequence of a mallard H7N3 virus was compared to the sequence of the same virus isolated after an outbreak in turkeys, and was found to have six changes in HA, indicating that adaptation to the turkeys required a change in receptor binding[101]. In many poultry-isolated IAV strains, the NA pro-tein contains an in-frame deletion of approximately twenty amino acids[102]. A recent study showed that this deletion, referred to as a NA stalk deletion, was responsible for increased transmissibility and lethality of an highly pathogenic H5N1 virus[103].
It it thought that poultry are responsible for the genesis of highly pathogenic (HPAI) viruses from the low pathogenic (LPAI) versions that circulate in the wild bird population[99]. The mechanisms of this conversion are discussed in detail in 1.3.1. The first outbreak of HPAI in poultry occurred in Guangdong, China in 1996. The H5N1 virus first circulated in domestic geese, but later went on to cause an outbreak in live-poultry markets, and 6 human fatalities[104]. Due to the close proximity of humans and poultry in live-bird markets, these areas represent an increased risk for zoonotic transmission. Since the the 1996 outbreak HPAI has spread globally, causing more than 600 human cases and killing several million poultry[10, 105, 106]. Recently in the US, the 2014-2015 HPAI H5 outbreak is estimated to have infected more than 48 million poultry[10]. Unfortunately, the factors involved in the emergence of HPAI from LPAI are not well understood.
1.2.4
Humans
During the last 100 years, three stable lineages of IAV have circulated in the human population: H1N1, H2N2, and H3N2[51]. Influenza infections prior to this time were documented, but little is known about the genetic makeup of the viruses. Retrospec-tive studies have shown that H1N1 was introduced into the population during the 1918 Spanish influenza pandemic[107]. By 1920 the pandemic H1N1 strain, known as the classical H1N1 strain, was circulating seasonally[3]. In 1957, the H1N1 virus reassorted with an avian virus (HA, NA, and PB1 segments) to produce an H2N2 virus. This strain replaced the H1N1 virus as the dominant circulating strain[108]. Again in 1968, an avian virus (HA and PB1 segments) reassorted with the circu-lating H2N2 strain to generate an H3N2 virus[108]. Following the 1968 pandemic, this H3N2 strain went on to become endemic in the human population. This virus continues to circulate within the population today[109]. In 1977 the classical H1N1 strain reemerged. Eventually it was shown that the reemergence of the virus was due to the release of a frozen strain from 1950[110]. Since the lab release, this viral lineage has continued to circulate within the population[111]. In 2009 a novel H1N1 strain emerged from a triple reassortment swine virus (see section 1.3.2). In the first few weeks after the initial detection, the virus had spread to 30 countries[88]. To-day, this 2009 pandemic H1N1 strain has replaced the classical H1N1 strain as the predominant strain in circulation[51].
The replacement of IAV strains over time is especially pronounced in the human population. It is thought that these dynamics are partly due to herd immunity and the seasonal nature of IAV infections[51]. Using a 12-year dataset of 1,032 H1N1 and H3N2 isolates, Rambaut et al. characterized the levels of genetic diversity in influenza populations at different locations and times of the year[111]. They found that there is an annual series of peaks in viral diversity interspersed by periods of low sequence diversity. The troughs occur at the end of influenza seasons and corre-spond to strong genetic bottlenecks. The authors refer to this as the "source-sink" model. In this model, the tropics are the source of IAV epidemics (periods of high
genetic diversity), and the sink is the other areas of the world where a virus from the source spreads (periods of low genetic diversity). This model has been supported by another study that also found that tropical regions of the globe serve as sources for annual epidemics[112]. In contrast to this finding, a study of the distribution of H3N2 viruses showed that viruses from many different geographic origins could seed IAV epidemics. The authors of this study suggest that the success of a given lineage is the result of competitive exclusion and stochastic factors, rather than geographic location[113]. Since the 2009 H1N1 pandemic, H3N2 is infrequently detected in the human population. There is plenty of speculation about which strain will cause the next pandemic, but without a better understanding of the viruses that circulate in other hosts, and the molecular mechanisms that allow for adaptation, our ability to predict the next pandemic strain is limited.
1.3
Influenza evolution
The wide ecological distribution of IAV is due in large part to the virus’ capability to rapidly adapt to new host environments and to evade the host immune system[3, 44]. This rapid evolution of IAV is facilitated by an error-prone RNA polymerase and genome reassortment.
1.3.1
RNA-dependent, RNA polymerase
The IAV replication machinery is a heterotrimer composed of the PB2, PB1, and PA polypeptides, which are encoded for by the first three viral segments, respec-tively. As an RNA-dependent, RNA polymerase, the replication machinery lacks proofreading capacity[114]. The error rate of the polymerase has been estimated to be 2.3 × 10−5 substitutions per nucleotide per cell infection when studied in the
lab[115, 116, 117, 118, 119, 120]. When the rate of substitution was estimated using viral sequences from wild and domestic birds, the observed rate was > 10−3
viruses), a virus population within a host exists as a collection of similar, but not identical genotypes[122, 123, 124]. This collection of genotypes is often referred to as a mutant spectra. The genome changes that distinguish one virus from the average sequence of the population are referred to in this thesis as intra-host single nucleotide variations (iSNVs). Previous research on iSNVs of other RNA viruses have found that they are important for antiviral resistance and escape from immune pressure[125, 124]. It is thought that natural selection has favored the evolution of high mutation rates because of rapidly changing host environments, resulting from host defense mecha-nisms, and from heterogeneous environments (ex. a virus that infects multiple host species or tissue types)[126]. In the case of IAV, it is hypothesized that iSNVs are important for the ability to stably transmit and infect a novel host[3, 119]. When an iSNV becomes the majority sequence (or reaches fixation) at a given position, it is referred to as a single nucleotide polymorphisms (SNPs). SNPs have been shown to give rise to a variety of improved IAV functions.
Several studies have investigated the SNPs associated with increased viral pathogenic-ity. One of the most prominent changes is the acquisition of a polybasic cleavage site within the HA protein that results in the conversion of a low pathogenic virus to a highly pathogenic virus. If the sequence of the HA cleavage site is R-x-K/R-R-1, the
protein can be cleaved by a family of widespread intracellular proteases[127, 128]. This leads to systemic spread of the virus and concomitant increase in pathogenicity[129, 130]. Mutations in other IAV genes have also been known to convey increased pathogenicity. A retrospective analysis of the 2009 H1N1 pandemic virus identi-fied four specific mutations, NP-V1001, NP-I133L, NP-1373T, and HA-S202T, that all led to increased virulence of the virus[131].
Replication generated mutations have also been shown to be important for evad-ing the host immune system. An extensively studied class of mutations are those in the antigenic regions of HA that allow the virus to evade the host humoral immune response, a process known as antigenic drift. These mutations are the result of strong selective pressure by the antibodies present in the host, and are the cause of sea-sonal influenza infections[132, 133, 111]. IAV is also known to evade recognition by
virus-specific T cells. The NP protein has an overrepresentation of non-synonymous mutations in the region of the cytotoxic T cell (CTL) epitopes suggesting this region is under strong selective pressure[134]. Two specific NP mutations, R384K and R384G, were identified in circulating seasonal H3N2 strains and were shown to abolish MHC class I presentation. Both mutations allowed for the virus to escape specific CTL recognition[135].
A third class of mutations allows IAV to alter its host and/or tissue tropism. The most well known of the IAV mutations that is a major determinant of host range is the PB2 627 mutation. PB2 segments derived from avian viruses predominately contain a glutamic acid at this position, whereas human derived PB2 segments almost exclusively contain a lysine. The E627K mutation has been shown to be correlated with increased polymerase activity, virus replication, and transmission in mammalian hosts[136, 137, 138, 139, 140, 141]. Tissue tropism has also been shown to be altered by SNPs. One study showed that the S227N HA change was sufficient to reduce the lethality and systemic spread of a circulating H5N1 strain. The authors of this study propose that the serine at position 227 is responsible for the increased infectivity of this virus in mammals[142]. In summary, the nature of the IAV polymerase allows the virus to evolve rapidly to improve protein function, avoid the host immune system, and to infect novel host and cell types.
1.3.2
Reassortment
The second major IAV evolutionary mechanism is reassortment. This phenomenon is characteristic of segmented RNA viruses and involves the exchange of intact gene segments between viruses that coinfect the same cell[143]. An infection containing more than one virus (>8 unique genome segments) is known as a mixed infection. Reassortment can potentially confer fitness advantages to resulting progeny, but can also result in progeny with reduced fitness. The segmented nature of the IAV genome results in rapid genetic diversification and has been shown to be important in the formation of pandemic strains. The most prominent example is the process by which
reassortment produces progeny virions with new combinations of the antigenic pro-teins, HA and NA. This phenomenon is known as antigenic shift, and was responsible for both the 1957 and 1968 IAV pandemics[143]. In the 1957 H2N2 pandemic, the HA, NA, and PB1 segments of an avian origin virus combined with five of the segments of the H1N1 virus circulating at the time[144, 145, 146, 147]. Similarly, the 1968 H3N2 pandemic was the result of a reassortant virus with HA and PB1 genes derived from an avian virus, and the remaining segments from the seasonal circulating H2N2 strain[144, 145, 147]. In both cases, reassortment introduced avian origin HA seg-ments, generating the antigenic novelty required for pandemic spread of the viruses. Reassortment not resulting in antigenic shift has also resulted in pandemics. The 2009 H1N1 pandemic arose from an IAV virus that emerged from swine[87, 88, 89]. The virus was the combination of a triple reassortment between a human, avian, and swine origin virus, and resulted in an increase in genetic diversity within the swine IAV pool[148]. The novel diversity was a necessary factor in the emergence of this pandemic[87]. It is thought that reassortment plays a critical role in the ability of a virus to stably transmission to a new host. In a recent study, Ma et al. used a network analysis to show that reassortment dominates over mutational drift in transmission between different host species[149].
Despite the abundance of examples of reassortment in nature, the exact mech-anisms for this process remain elusive. It is known that the process of reassort-ment requires conserved packaging signals and the maintenance of RNA and protein interactions[151]. Two mechanisms have been proposed for the packaging of the eight viral ribonucleoproteins (vRNP) into the budding virions: a selective process in which only one of each vRNP is packaged into a single virion, and a random process, in which vRNPs are selected randomly for packaging[150]. There is stronger evidence for the former mechanism. In support of the specific packaging hypothesis, a study by Noda et al. used microscopy to show that most virions contain 8 vRNPs arranged in what is known as a ’7+1’ arrangement[152]. In this conformation, there is one central vRNP and the other seven are arranged in a circle around it (Fig. 5a). Although the exact mechanism by which each of the specific genome segments is packaged
into a virion is unknown, it is known that each of the eight segments contains a conserved packaging signal that is located at the terminal ends of the segment. In addition, each segment also has an internal coding region that has a unique length (Fig 5b)[153, 154, 155, 156, 157, 158, 159, 160, 161].
Reassortment between two strains of IAV with unique genomes can create up to 254 novel genome constellations. Potentially, linkage between the segments could occur because of functional incompatibility or because of signals that link specific segments together during passaging. Studies that have assessed whether all possible reassortants are produced during a coinfection have found varying levels of linkage between the segments[63, 162, 163]. Among IAV strains that circulate in wild bird, there has been little to no evidence for linkage between any of the genome segments[63, 162]. In contrast to this finding, a study of human viruses found that positive selection was responsible for the linkage between HA and NA segments during the experimental coinfection of a H1N1 and H3N2 virus[163]. It is difficult to study reassortment
PB2 PB1 PA HA NP NA NS M Conserved genomic ends (non-coding) Segment-specific regions (coding) 120 60 12 9 60 183 222 35 35 220 157 120 80 21 60 120 (a) ‘7+1’ arrangement (b) vRNP Virus membrane
vRNA packaging signals
NT NT
Figure 5. (a) ’7+1’ arrangement of vRNPs. (b) Influenza- and segment-specific packaging signals. The number of nucleotides (NT) within each segment-specific re-gion is depicted. Adapted from Eisfeld et al. with permission from Nature Publishing Group[150].
dynamics because currently it is not possible to sequence an individual virion. Single virion sequencing has been conducted for DNA viruses, but the segmented and RNA genome of IAV has presented many technical challenges[164]. Once we have this capability, it will be possible to better characterize the molecular dynamics controlling reassortment.
ADD SUMMARY STATEMENT HERE
1.4
Influenza experimental evolution
Laboratory-based evolution experiments have provided insight into the mechanisms responsible for several viral functions. These types of experiments have been partic-ularly useful for understanding airborne transmissibility and novel host adaptation.
1.4.1
Airborne transmission
In hosts where IAV infects the respiratory tract, the virus can be transmitted via three mechanisms: contact, droplet, and aerosol transmission[165]. Contact trans-mission involves both indirect (passive transtrans-mission via contaminated objects) and direct contact (direct transfer of IAV between two susceptible hosts) modes. Droplet based transmission occurs via droplets (≥5 µm diameter) that are generated from the respiratory tract when an infected host coughs, sneezes, or talks. These droplets travel a maximum distance of 1m through the air. The transmission of aerosols oc-curs when IAV is contained in a droplet less than 5µm that eventually comes into contact with a susceptible host. Aerosolized droplets can stay suspended in the air for long periods of time and can be widely dispersed by air currents. Even though large droplets are not truly aerosolized, airborne transmission often refers to both droplet and aerosol modes.
There have been a substantial number of studies that have characterized the genome changes that are necessary for the airborne transmission of IAV. The ma-jority of these studies have focused on identifying the amino acid changes that allow
highly pathogenic viruses (HPAI) (H5 and H7) to transmit via the airborne route. In a 2012 study, Imai et al. tested the ability of pandemic H1N1 and HPAI H5 lab-generated reassortant viruses (HA from H5, all other segments were from the H1N1) to transmit between ferrets. The experimental setup eliminated the possibility of contact transmission between ferrets, allowing only for airborne transmission. They found that a virus containing both HA N224K and Q226L mutations was efficiently transmitted via the airborne route. None of the ferrets died when infected with the virus, indicating that for this particular virus, the ability to transmit via the airborne route involves a loss of virulence[166]. In a similar study, Herfst et al. serially pas-saged an HPAI H5N1 virus in ferrets for 10 passages. During passages 6-10, sneezing was induced in the ferrets and the resulting fluid was used to infect the next ferret. Following the 10 passages, they tested whether any of the progeny viruses could trans-mit to naïve recipient ferrets via the airborne route. They identified five mutations that correlated with the ability to transmit: PB1 - E627K, and HA - Q222L, G224S, H103Y, T156A. As in the case with the study by Imai et al., the ability to transmit via airborne transmission was accompanied by a loss of virulence[167]. In a more recent study that used a very similar experimental design to Herfst et al., but with HPAI H7 viruses, identified four amino acid mutations (PB2-T81I, NP-V284M, and M1-R59K, Q211K) that conferred the ability to transmit via an airborne route. Notably, a loss of virulence did not accompany airborne transmissibility in this study[168]. The re-sults from these and other similar studies indicate that the ability to transmit via the airborne route is polygenic in nature, and that particular amnio acid changes in one subtype of IAV does not confer the same effects in a different subtype of IAV.
1.4.2
Novel host adaptation
A major goal of IAV research is to understand how viruses are able to stably transmit and infect a novel host. Much of this research has been focused on studying the past pandemic strains. Because there have been so few pandemics, there is not one mutation, or set of mutations, that has been identified that completely explains why
the pandemic viruses were able to sustain transmission in humans. A smaller set of studies have attempted to answer this question by subjecting viruses to new host environments and monitoring the genotypic and phenotypic changes.
The experimental evolution studies can be classified into two groups: adaptation of a wild bird strain in a novel host, and adaptation of a human strain in a novel host. In a study from the former group, Cilloni et al. serially infected chickens, turkeys, and quails with an H7N3 virus isolated from a mallard. The virus was able to infect the turkeys and quails for all ten passages, but the virus went extinct after just two passages in chickens. Interestingly, the progeny viruses resulting from the 10th quail and turkey passages were both able to cause infections in chickens, although only the quail-adapted virus caused symptomatic illness[169]. A follow up study found 4 different amino acid mutations in both the HA sequence of the turkey and quail adapted viruses. These mutations were all located in or near the receptor binding domain of HA. Notably, the quail-adapted virus also had the characteristic poultry NA stalk deletion (a 23-amino acid deletion in the NA protein)[170]. This could explain the difference between the virulence of the turkey and quail-adapted strains, although the deletion of this region in the absence of the other mutations was not tested. The results from this study also indicate that turkeys and quail have a both wild bird-type receptors and chicken-type receptors, thus allowing them to serve as an intermediate host between wild birds and chickens. The HPAI poultry virus-ferret studies discussed in 1.4.1 can also be classified in this group. Comparing all of these studies indicates that the adaptation of avian strains to new hosts involves changes in or around the HA receptor binding domain, albeit the HA mutations cannot completely explain the adaptation process.
The importance of HA and other genes has also been described for the adaptation of human strains to novel hosts. A study that examined the adaptation of a human H3N2 virus to mice identified four amino acid positions in the two chains of the HA protein (HA1 - 162, 210, 218, HA2 - 154). These mutations were shown to enhance viral replication and infection in mice[171]. Another study in mice elucidated the role of NS1 in adaptation of a human H3N2 virus. Twelve unique mutations were
identified in NS1 after the virus was passaged in mice (20 and 21 passages). All but one of the mutations was associated with increased virulence, and ten of the twelve enhanced IFN-β antagonism[172]. The role of the viral polymerase has also been shown to be important for host adaptation in a number of different hosts (ref 1.3.1). One such study showed that the adaptation of a human H1N1 strain in bat cells (bats were recently identified as a potential IAV reservoir) involved the previously undocumented PA M258K mutation[173]. When the polymerase genes (PB2, PB1, and PA) of this virus were replaced with avian polymerase genes, serial passage in bat cells resulted in the well known PB2 E627K mutation that is known to alter host tropism[174]. The results from experimental evolution of human strains are more varied, and indicate that adaptation is polygenic.
One missing class of relevant experiments is the adaptation of wild bird IAV to mammalian hosts. A major reason for the dearth of these types of experiments is the recent gain-of-function ban. In October of 2014, the White House Office of Science and Technology Policy (OSTP) instituted a pause on research that "increases the ability of [influenza, SARS, MERS viruses] to cause disease by enhancing its pathogenicity or by increasing its transmissibility among mammals by respiratory droplets"[175]. During the pause which lasted for more than two years, the government investigated the risks and benefits of gain-of-function research. In January of 2017, OSTP issued a policy stating that any new research that has the potential to be considered gain-of-function must be approved by an advisory committee[176]. Presently, the individual funding agencies are responsible for evaluating if the research meets the criteria for gain-of-function research of concern. If the research meets these eight criteria it can be conducted.
1.5
Conclusion
IAV is a fast evolving organism with a very wide host range. Due in part to these factors influenza outbreaks and pandemics have been very difficult, if not impossible, to predict[5]. A substantial amount of effort and money is devoted to increasing our
understanding of IAV evolution and distribution every year. In chapter 1 of this thesis I examine the ecology and evolutionary dynamics of IAV.
In chapter 2 I examine the dynamics of IAV evolution during laboratory host infection. More specifically, I have characterized the changes in the viral sequence that are associated with infecting IAV in embryonated chicken eggs and a canine cell culture model. I show that passaging IAV in a laboratory host prior to genome sequencing results in biases that hinder the ability to detect mixed infections, and impact downstream phylogenetic analyses. Furthermore, I show that the evolution of two very similar viruses does not proceed identically over repeated infections in embryonated chicken eggs.
Bibliography
[1] International Committee on Taxonomy of Viruses. Orthomyxoviri-dae. https://talk.ictvonline.org/ictv-reports/ictv_9th_report/negative-sense-rna-viruses-2011/w/negrna_viruses/209/orthomyxoviridae, 2011.
[2] Ben M. Hause, Emily A. Collin, Runxia Liu, Bing Huang, Zizhang Sheng, Wuxun Lu, Dan Wang, Eric A. Nelson, and Feng Li. Characterization of a novel influenza virus in cattle and swine: Proposal for a new genus in the orthomyxoviridae family. mBio, 5(2), 2014.
[3] Jeffery K Taubenberger and John C Kash. Influenza virus evolution, host adaptation, and pandemic formation. Cell host & microbe, 7(6):440–51, jun 2010.
[4] Tasleem Samji. Influenza A: understanding the viral life cycle. The Yale journal of biology and medicine, 82(4):153–9, dec 2009.
[5] J K Taubenberger and D M Morens. Pandemic influenza–including a risk as-sessment of H5N1. Revue scientifique et technique (International Office of Epi-zootics), 28(1):187–202, 2009.
[6] Jeffery K. Taubenberger and David M. Morens. 1918 Influenza: The mother of all pandemics. Emerging Infectious Diseases, 12(1):15–22, 2006.
[7] WHO. Influenza. http://www.who.int/biologicals/vaccines/influenza/en/. [8] Noelle-Angelique M. Molinari, Ismael R. Ortega-Sanchez, Mark L. Messonnier,
William W. Thompson, Pascale M. Wortley, Eric Weintraub, and Carolyn B. Bridges. The annual impact of seasonal influenza in the us: Measuring disease burden and costs. Vaccine, 25(27):5086 – 5096, 2007.
[9] Liang Mao, Yang Yang, Youliang Qiu, and Yan Yang. Annual economic im-pacts of seasonal influenza on us counties: Spatial heterogeneity and patterns. International Journal of Health Geographics, 11(1):16, May 2012.
[10] Susan A. Shriner, J. Jeffrey Root, Mark W. Lutman, Jason M. Kloft, Kaci K. VanDalen, Heather J. Sullivan, Timothy S. White, Michael P. Milleson, Jerry L. Hairston, Shannon C. Chandler, Paul C. Wolf, Clinton T. Turnage, Brian J. Mc-Cluskey, Amy L. Vincent, Mia K. Torchetti, Thomas Gidlewski, and Thomas J.
DeLiberto. Surveillance for highly pathogenic H5 avian influenza virus in synan-thropic wildlife associated with poultry farms during an acute outbreak. Sci-entific Reports, 6(1):36237, 2016.
[11] Darrell Whitworth, Scott Newman, Taej Mundkur, Phil Harris, editor. Wild bird capture techniques. FAO, 2007.
[12] Steven J. Jeffries, Robin F. Brown, and James T. Harvey. Techniques for cap-turing, handling and marking harbour seals. Aquatic Mammals, 19:21–25, 1993. [13] F. B. Johnson. Transport of viral specimens. Clinical Microbiology Reviews,
3(2):120–131, 1990.
[14] Dae-Ki Kim and Barun Poudel. Tools to detect influenza virus. Yonsei Medical Journal, 54(3):560–566, 05 2013.
[15] Erica Spackman and David L. Suarez. Type A Influenza Virus Detection and Quantitation by Real-Time RT-PCR, pages 19–26. Humana Press, Totowa, NJ, 2008.
[16] J. A. Runstadler, G. M. Happ, R. D. Slemons, Z. M. Sheng, N. Gundlach, M. Petrula, D. Senne, J. Nolting, D. L. Evers, A. Modrell, H. Huson, S. Hills, T. Rothe, T. Marr, and J. K. Taubenberger. Using RRT-PCR analysis and virus isolation to determine the prevalence of avian influenza virus infections in ducks at Minto Flats State Game Refuge, Alaska, during August 2005. Archives of Virology, 152(10):1901–1910, 2007.
[17] Vincent J Munster, Chantal Baas, Pascal Lexmond, Theo M Bestebroer, Judith Guldemeester, Walter E P Beyer, Emmie de Wit, Martin Schutten, Guus F Rimmelzwaan, Albert D M E Osterhaus, and Ron a M Fouchier. Practical considerations for high-throughput influenza A virus surveillance studies of wild birds by use of molecular diagnostic tests. Journal of clinical microbiology, 47(3):666–73, 2009.
[18] Erica Spackman, Dennis A. Senne, T. J. Myers, Leslie L. Bulaga, Lindsey P. Garber, Michael L. Perdue, Kenton Lohman, Luke T. Daum, and David L. Suarez. Development of a real-time reverse transcriptase pcr assay for type a influenza virus and the avian h5 and h7 hemagglutinin subtypes. Journal of Clinical Microbiology, 40(9):3256–3260, 2002.
[19] F M Burnet. Influenza virus infection of the chick embryo lung. Br J Exp Pathol, 21:147–153, 1940.
[20] Peter C. Soema, Ronald Kompier, Jean-Pierre Amorij, and Gideon F.A. Ker-sten. Current and next generation influenza vaccines: Formulation and pro-duction strategies. European Journal of Pharmaceutics and Biopharmaceutics, 94(Supplement C):251 – 263, 2015.
[21] Peter R Woolcock. Avian Influenza Virus Isolation and Propagation in Chicken Eggs, chapter 6, pages 35–46. Humana Press, Totowa, NJ, 2008.
[22] Tina Lombardo, Silvia Dotti, Sabrina Renzi, and Maura Ferrari. Susceptibility of different cell lines to Avian and Swine Influenza viruses. Journal of Virological Methods, 185(1):82–88, 2012.
[23] Ruben O Donis. Performance characteristics of qualified cell lines for isolation and propagation of influenza viruses for vaccine manufacturing. Vaccine, 32:1–8, 2014.
[24] Yuan Oh Ding, Ian G. Barr, Jenny a. Mosse, and Karen L. Laurie. MDCK-SIAT1 cells show improved isolation rates for recent human influenza viruses compared to conventional MDCK cells. Journal of Clinical Microbiology, 46(7):2189–2194, 2008.
[25] Loretta Müller, Luisa E Brighton, Johnny L Carson, William a Fischer, and Ilona Jaspers. Culturing of human nasal epithelial cells at the air liquid inter-face. Journal of visualized experiments : JoVE, pages 1–7, October 2013. [26] Allen C. Bateman, Alexander I. Karasin, and Christopher W. Olsen.
Differ-entiated swine airway epithelial cell cultures for the investigation of influenza A virus infection and replication. Influenza and other Respiratory Viruses, 7(2):139–150, 2013.
[27] Jianqiang Zhang and Phillip C. Gauger. Isolation of Swine Influenza Virus in Cell Cultures and Embryonated Chicken Eggs, pages 265–276. Springer New York, New York, NY, 2014.
[28] Seyed Mohammad Jazaeri Farsani, Martin Deijs, Ronald Dijkman, Richard Molenkamp, Rienk E. Jeeninga, Margareta Ieven, Herman Goossens, and Lia van der Hoek. Culturing of respiratory viruses in well-differentiated pseudostrat-ified human airway epithelium as a tool to detect unknown viruses. Influenza and Other Respiratory Viruses, 9:51–57, 2015.
[29] Amie J Eisfeld, Gabriele Neumann, and Yoshihiro Kawaoka. Influenza a virus isolation, culture and identification. Nature Protocols, 9:2663 EP –, 10 2014. [30] F M Burnet and Diana H Bull. Changes In Influenza Virus Associated With
Adaptation To Passage In Chick Embryos. Aust J Exp Biol Med, 21(2):55–69, jun 1943.
[31] E. Hoffmann, J. Stech, Y. Guan, R. G. Webster, and D. R. Perez. Universal primer set for the full-length amplification of all influenza A viruses. Archives of Virology, 146(12):2275–2289, 2001.
[32] Bin Zhou, Matthew E Donnelly, Derek T Scholes, Kirsten St George, Masato Hatta, Yoshihiro Kawaoka, and David E Wentworth. Single-reaction genomic
amplification accelerates sequencing and vaccine production for classical and Swine origin human influenza a viruses. Journal of virology, 83(19):10309–13, oct 2009.
[33] Dirk Höper, Bernd Hoffmann, and Martin Beer. A comprehensive deep sequenc-ing strategy for full-length genomes of influenza A. PloS one, 6(4):e19075, jan 2011.
[34] Wiriya Rutvisuttinunt, Piyawan Chinnawirotpisan, Sriluck Simasathien, San-jaya K Shrestha, In-Kyu Yoon, Chonticha Klungthong, and Stefan Fernandez. Simultaneous and Complete Genome Sequencing of Influenza A and B with High Coverage by Illumina MiSeq Platform. Journal of virological methods, jul 2013.
[35] Melanie Schirmer, Umer Z. Ijaz, Rosalinda D’Amore, Neil Hall, William T. Sloan, and Christopher Quince. Insight into biases and sequencing errors for amplicon sequencing with the illumina miseq platform. Nucleic Acids Research, 43(6):e37, 2015.
[36] Hayden C Metsky, Christian B Matranga, Shirlee Wohl, Stephen F Schaffner, Catherine A Freije, Sarah M Winnicki, Kendra West, James Qu, Mary Lynn Baniecki, Adrianne Gladden-Young, Aaron E Lin, Christopher H Tomkins-Tinch, Daniel J Park, Cynthia Y Luo, Kayla G Barnes, Bridget Chak, Giselle Barbosa-Lima, Edson Delatorre, Yasmine R Vieira, Lauren M Paul, Amanda L Tan, Mario C Porcelli, Chalmers Vasquez, Andrew C Cannons, Marshall R Cone, Kelly N Hogan, Edgar W Kopp, Joshua J Anzinger, Kimberly F Gar-cia, Leda A Parham, Rosa Margarita Gelvez Ramirez, Maria Conseulo Mi-randa Montoya, Diana P Rojas, Catherine M Brown, Scott Hennigan, Bran-don Sabina, Sarah Scotland, Karthik Gangavarapu, Nathan D Grubaugh, Glenn Oliveira, Refugio Robles-Sikisaka, Andrew Rambaut, Lee Gehrke, San-dra Smole, M. Elizabeth Halloran, Luis Angel Villar Centeno, Salim Mattar, Ivette Lorenzana, Jose Cerbino-Neto, Wim Degrave, Patricia T Bozza, An-dreas Gnirke, Kristian G Andersen, Sharon Isern, Scott F Michael, Fernando A Bozza, Thiago ML L Souza, Irene Bosch, Nathan L Yozwiak, Bronwyn L MacIn-nis, Pardis C Sabeti, Simon H Ye, Daniel J Park, Cynthia Y Luo, Kayla G Barnes, Rickey R Shah, Bridget Chak, Giselle Barbosa-Lima, Edson Delatorre, Yasmine R Vieira, Lauren M Paul, Amanda L Tan, Carolyn M Barcellona, Mario C Porcelli, Chalmers Vasquez, Andrew C Cannons, Marshall R Cone, Kelly N Hogan, Edgar W Kopp, Joshua J Anzinger, Kimberly F Garcia, Leda A Parham, Rosa M Gélvez Ramírez, Maria C Miranda Montoya, Diana P Rojas, Catherine M Brown, Scott Hennigan, Brandon Sabina, Sarah Scotland, Karthik Gangavarapu, Nathan D Grubaugh, Glenn Oliveira, Refugio Robles-Sikisaka, Andrew Rambaut, Lee Gehrke, Sandra Smole, M. Elizabeth Halloran, Luis Vil-lar, Salim Mattar, Ivette Lorenzana, Jose Cerbino-Neto, Clarissa Valim, Wim Degrave, Patricia T Bozza, Andreas Gnirke, Kristian G Andersen, Sharon Is-ern, Scott F Michael, Fernando A Bozza, Thiago ML L Souza, Irene Bosch,
Nathan L Yozwiak, Bronwyn L MacInnis, and Pardis C Sabeti. Zika virus evolution and spread in the Americas. Nature, 546(7658):411–415, jun 2017. [37] Silvia I. Sardi, Sneha Somasekar, Samia N. Naccache, Antonio C. Bandeira,
Laura B. Tauro, Gubio S. Campos, and Charles Y. Chiu. Coinfections of zika and chikungunya viruses in bahia, brazil, identified by metagenomic next-generation sequencing. Journal of Clinical Microbiology, 54(9):2348–2353, 2016. [38] Ashley Acevedo, Leonid Brodsky, and Raul Andino. Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature, 505(7485):686–690, nov 2013.
[39] Ashley Acevedo and Raul Andino. Library preparation for highly accurate population sequencing of RNA viruses. Nature protocols, 9(7):1760–9, 2014. [40] R G Webster, W J Bean, O T Gorman, T M Chambers, and Y Kawaoka.
Evolution and ecology of influenza A viruses. Microbiological reviews, 56(1):152– 79, 1992.
[41] Hans Dieter Klenk. Influenza Viruses En Route from Birds to Man. Cell host & microbe, 15(6):653–654, jun 2014.
[42] Scott Krauss, David Walker, S Paul Pryor, Larry Niles, Li Chenghong, Vir-ginia S Hinshaw, and Robert G Webster. Influenza A viruses of migrating wild aquatic birds in North America. Vector borne and zoonotic diseases (Larchmont, N.Y.), 4(3):177–189, 2004.
[43] Vincent J. Munster, Chantal Baas, Pascal Lexmond, Jonas Waldenström, An-ders Wallensten, Thord Fransson, Guus F. Rimmelzwaan, Walter E P Beyer, Martin Schutten, Björn Olsen, Albert D M E Osterhaus, and Ron A M Fouch-ier. Spatial, temporal, and species variation in prevalence of influenza a viruses in wild migratory birds. PLoS Pathogens, 3(5):0630–0638, 2007.
[44] Martha I Nelson and Edward C Holmes. The evolution of epidemic influenza. Nature reviews. Genetics, 8(3):196–205, mar 2007.
[45] V. S. Hinshaw, R. G. Webster, and B. Turner. The perpetuation of orthomyx-oviruses and paramyxorthomyx-oviruses in canadian waterfowl. Canadian Journal of Mi-crobiology, 26(5):622–629, 1980. PMID: 7397605.
[46] C P Alfonso, B S Cowen, and H van Campen. Influenza A viruses isolated from waterfowl in two wildlife management areas of Pennsylvania. J Wildl Dis, 31(2):179–185, 1995.
[47] Anders Wallensten, Vincent J. Munster, Neus Latorre-Margalef, Mia Brytting, Johan Elmberg, Ron A.M. Fouchier, Thord Fransson, Paul D. Haemig, Malin Karlsson, Åke Lundkvist, Albert D.M.E. Osterhaus, Martin Stervander, Jonas Waldenström, and Björn Olsen. Surveillance of influenza A virus in migratory
waterfowl in northern Europe. Emerging Infectious Diseases, 13(3):404–411, 2007.
[48] Hon S Ip, Paul L Flint, J Christian Franson, Robert J Dusek, Dirk V Derksen, Robert E Gill, Craig R Ely, John M Pearce, Richard B Lanctot, Steven M Mat-suoka, David B Irons, Julian B Fischer, Russell M Oates, Margaret R Petersen, Thomas F Fondell, Deborah A Rocque, Janice C Pedersen, and Thomas C Rothe. Prevalence of Influenza A viruses in wild migratory birds in Alaska: Patterns of variation in detection at a crossroads of intercontinental flyways. Virology Journal, 5(1):71, 2008.
[49] Elsa Jourdain, Gunnar Gunnarsson, John Wahlgren, Neus Latorre-Margalef, Caroline Bröjer, Sofie Sahlin, Lovisa Svensson, Jonas Waldenström, Åke Lund-kvist, and Björn Olsen. Influenza virus in a natural host, the mallard: Experi-mental infection data. PLoS ONE, 5(1), 2010.
[50] Juthatip Keawcharoen, Debby Van Riel, Geert Van Amerongen, Theo Beste-broer, Walter E. Beyer, Rob Van Lavieren, Albert D M E Osterhaus, Ron A M Fouchier, and Thijs Kuiken. Wild ducks as long-distance vectors of highly pathogenic avian influenza virus (H5N1). Emerging Infectious Diseases, 14(4):600–607, 2008.
[51] Sun-Woo Yoon, Richard J Webby, and Robert G Webster. Evolution and ecol-ogy of influenza A viruses. Current topics in microbiolecol-ogy and immunolecol-ogy, 385:359–375, 2014.
[52] Robert G. Webster, Maya Yakhno, Virginia S. Hinshaw, William J. Bean, and K. Copal Murti. Intestinal influenza: Replication and characterization of in-fluenza viruses in ducks. Virology, 84(2):268–278, 1978.
[53] Jonathan Runstadler, Nichola Hill, Islam T.M. Hussein, Wendy Puryear, and Mandy Keogh. Connecting the study of wild influenza with the potential for pandemic disease. Infection, Genetics and Evolution, 17:162–187, mar 2013. [54] Yoshihiro Kawaoka, Thomas M. Chambers, William L. Sladen, and Robert
Gwebster. Is the gene pool of influenza viruses in shorebirds and gulls different from that in wild ducks? Virology, 163(1):247–250, 1988.
[55] G B Sharp, Y Kawaoka, S M Wright, B Turner, V Hinshaw, and R G Webster. Wild ducks are the reservoir for only a limited number of influenza A subtypes. Epidemiology and infection, 110(1):161–76, 1993.
[56] Scott Krauss and Robert G Webster. Avian Influenza Virus Surveillance and Wild Birds: Past and Present. Avian Diseases, 54(s1):394–398, mar 2010. [57] R. E Gill, T. L. Tibbitts, D. C Douglas, C. M Handel, D. M Mulcahy, J. C
Gottschalck, N. Warnock, B. J McCaffery, P. F Battley, and T. Piersma. Ex-treme endurance flights by landbirds crossing the Pacific Ocean: ecological
corridor rather than barrier? Proceedings of the Royal Society B: Biological Sciences, 276(1656):447–457, 2009.
[58] Nicolas Gaidet, Ahmed B. Ould El Mamy, Julien Cappelle, Alexandre Caron, Graeme S. Cumming, Vladimir Grosbois, Patricia Gil, Saliha Hammoumi, Re-nata Servan de Almeida, Sasan R. Fereidouni, Giovanni Cattoli, Celia Abolnik, Josphine Mundava, Bouba Fofana, Mduduzi Ndlovu, Yelli Diawara, Renata Hurtado, Scott H. Newman, Tim Dodman, and Gilles Balança. Investigating Avian Influenza Infection Hotspots in Old-World Shorebirds. PLoS ONE, 7(9), 2012.
[59] Andrew M. Ramey, John M. Pearce, Andrew B. Reeves, J. Christian Franson, Margaret R. Petersen, and Hon S. Ip. Evidence for limited exchange of avian influenza viruses between seaducks and dabbling ducks at Alaska Peninsula coastal lagoons. Archives of Virology, 156(10):1813–1821, 2011.
[60] John M. Pearce, Andrew M. Ramey, Hon S. Ip, and Robert E. Gill. Limited evidence of trans-hemispheric movement of avian influenza viruses among con-temporary north american shorebird isolates. Virus Research, 148(1):44 – 50, 2010.
[61] Paul L. Flint, Kiyoaki Ozaki, John M. Pearce, Brian Guzzetti, Hiroyoshi Higuchi, Joseph P. Fleskes, Tetsuo Shimada, and Dirk V. Derksen. Breeding-season sympatry facilitates genetic exchange among allopatric wintering popula-tions of Northern Pintails in Japan and California. The Condor, 111(4):591–598, 2009.
[62] Andrew B. Reeves, John M. Pearce, Andrew M. Ramey, Brandt W. Meixell, and Jonathan A. Runstadler. Interspecies transmission and limited persistence of low pathogenic avian influenza genomes among Alaska dabbling ducks. In-fection, Genetics and Evolution, 11(8):2004–2010, 2011.
[63] Vivien G Dugan, Rubing Chen, David J Spiro, Naomi Sengamalay, Jennifer Zaborsky, Elodie Ghedin, Jacqueline Nolting, David E Swayne, Jonathan a Runstadler, George M Happ, Dennis a Senne, Ruixue Wang, Richard D Sle-mons, Edward C Holmes, and Jeffery K Taubenberger. The evolutionary ge-netics and emergence of avian influenza viruses in wild birds. PLoS pathogens, 4(5):e1000076, may 2008.
[64] Justin Bahl, Dhanasekaran Vijaykrishna, EC Holmes, GJD Smith, and Yi Guan. Gene flow and competitive exclusion of avian influenza A virus in natural reservoir hosts. Virology, 390(2):289–297, 2009.
[65] Andrew M. Ramey, John M. Pearce, Craig R. Ely, Lisa M. Sheffield Guy, David B. Irons, Dirk V. Derksen, and Hon S. Ip. Transmission and reassort-ment of avian influenza viruses at the Asian-North American interface. Virology, 406(2):352–359, 2010.
[66] Linda Widjaja, SL Krauss, and RJ Webby. Matrix Gene of Influenza A Viruses Isolated from Wild Aquatic Birds : Ecology and Emergence of Influenza A Viruses. Journal of virology, 2004.
[67] Douglas Causey and Scott V. Edwards. Ecology of Avian Influenza Virus in Birds. The Journal of Infectious Diseases, 197(s1):S29–S33, 2008.
[68] Tommy Tsan Yuk Lam, Hon S. Ip, Elodie Ghedin, David E. Wentworth, Re-becca A. Halpin, Timothy B. Stockwell, David J. Spiro, Robert J. Dusek, James B. Bortner, Jenny Hoskins, Bradley D. Bales, Dan R. Yparraguirre, and Edward C. Holmes. Migratory flyway and geographical distance are barriers to the gene flow of influenza virus among North American birds. Ecology Letters, 15(1):24–33, 2012.
[69] John M. Pearce, Andrew M. Ramey, Paul L. Flint, Anson V. Koehler, Joseph P. Fleskes, J. Christian Franson, Jeffrey S. Hall, Dirk V. Derksen, and Hon S. Ip. Avian influenza at both ends of a migratory flyway: Characterizing viral ge-nomic diversity to optimize surveillance plans for North America. Evolutionary Applications, 2(4):457–468, 2009.
[70] Nichola J. Hill, John Y. Takekawa, Carol J. Cardona, Brandt W. Meixell, Joshua T. Ackerman, Jonathan A. Runstadler, and Walter M. Boyce. Cross-Seasonal Patterns of Avian Influenza Virus in Breeding and Wintering Mi-gratory Birds: A Flyway Perspective. Vector-Borne and Zoonotic Diseases, 12(3):243–253, 2012.
[71] Mathieu Fourment, Aaron E. Darling, and Edward C. Holmes. The impact of migratory flyways on the spread of avian influenza virus in North America. BMC Evolutionary Biology, 17(1):118, 2017.
[72] Robert G Wallace, Hoangminh Hodac, Richard H Lathrop, and Walter M Fitch. A statistical phylogeography of influenza A H5N1 The geographic diffusion of highly pathogenic influenza A H5N1 has largely been traced from the perspec-tive of the virus ’ s victims . Birds of a variety of avian orders have been sampled across localities. PNAS, 104(11):4473–4478, 2007.
[73] Dhanasekaran Vijaykrishna, Justin Bahl, Steven Riley, Lian Duan, Xia Zhang Jin, Honglin Chen, J. S.Malik Peiris, Gavin J.D. Smith, and Yi Guan. Evo-lutionary dynamics and emergence of panzootic H5N1 influenza viruses. PLoS Pathogens, 4(9):1–10, 2008.
[74] Nicola S. Lewis, Josanne H. Verhagen, Zurab Javakhishvili, Colin A. Russell, Pascal Lexmond, Kim B. Westgeest, Theo M. Bestebroer, Rebecca A. Halpin, Xudong Lin, Amy Ransier, Nadia B. Fedorova, Timothy B. Stockwell, Neus Latorre-Margalef, Björn Olsen, Gavin Smith, Justin Bahl, David E. Wentworth, Jonas Waldenström, Ron A.M. Fouchier, and Miranda de Graaf. Influenza A