RNA binding and assembly of human influenza A virus polymerases
Texte intégral
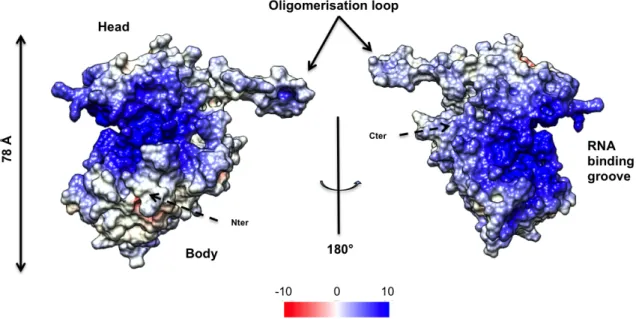
Figure
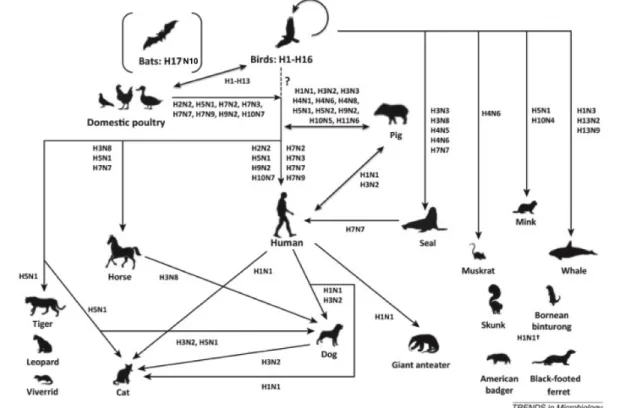
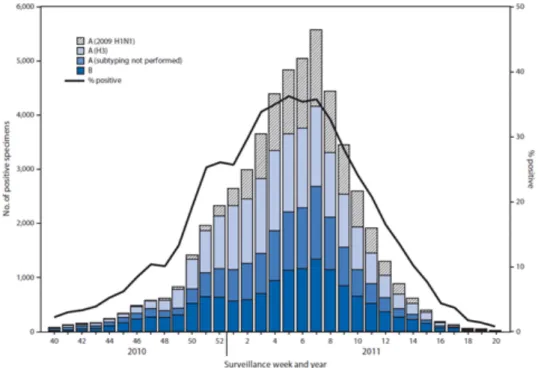

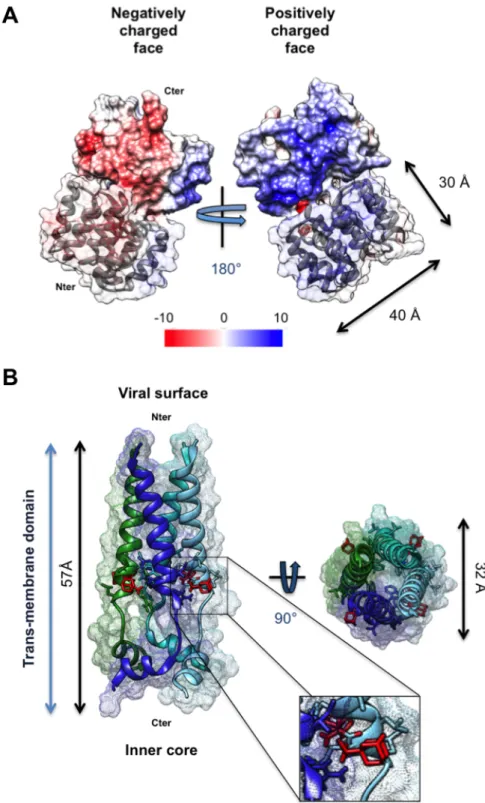
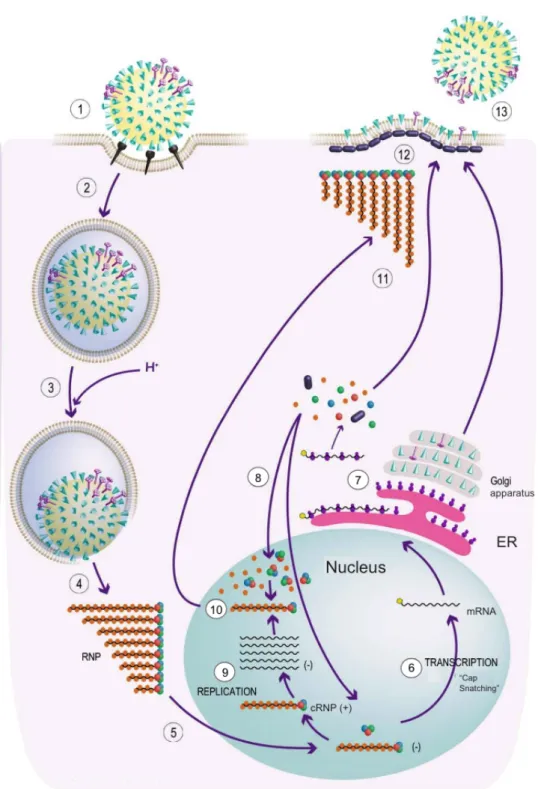


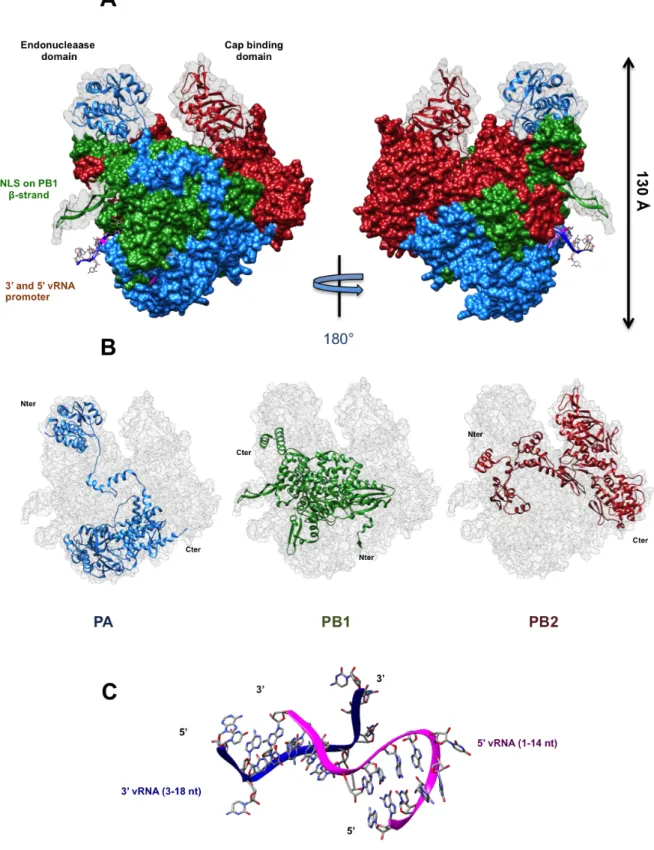
Documents relatifs
Results: The double substitution R38A-K41A in the RBD dramatically reduced the pathogenicity and replication potential of the virus, whereas the substitution A149V that was
However, the growth of the deacetylated mutant virus WSN-NS1-108R was sig- nificantly impaired compared with that of the other two viruses in MDCK and A549 cells at 36 and 48
Here, we analyzed the mRNA and protein expression of RNA-Binding Protein with Multiple Splicing-2 (RBPMS2), an early marker of gastrointestinal SMC precursors, in human GISTs (n=23)
Spread and pathway modelling to support pest risk assessment under global change.. Christelle Robinet, Wopke van den Werf, Jc Bob Douma, Lia Hemerik, Monique C
Cette diversité est Cherchant à favoriser une meilleure reconnais- même souhaitée, vue comme une richesse à sance de la diversité des pratiques sans sacrifier préserver,
In December 2009, a 43-year-old, mechanically ventilated patient with acute respiratory distress syndrome (ARDS) due to A(H1N1) influenza infection was transferred to our intensive
In the present study, novel H1N1 haemaglutinin was isolated from a confirmed clinical sample and cloned into the baculovirus expression system to produce re- combinant HA1
Twenty-four hours after transfection, about 30% of the transfected cells displayed a few micrometric fluorescent condensates dispersed throughout their cytoplasm, whereas the other
