SOMMAIRE
INTRODUCTION 1. Définition………..1 2. Intérêt de l’étude………...2 3. Buts du travail………..2 GENERALITES I. Epidémiologie……….3 1. Fréquence………3 2. Sex-ratio………3 3. Répartition géographique………..4 II. Histogenèse……….4III. Caractéristiques des tumeurs bénignes……….5
IV. Classification………...8
V. Etude anatomopathologique……….12
1. Les tumeurs cutanées……….12
1.1. D’origine épithéliale………12
1.1.1. Les verrues………..12
1.1.2. Le kérato-acanthome………12
1.1.3. L’acanthome à cellules claires………13
1.1.4. L’hamartome épidermique………..13
1.2. D’origine conjonctive……….13
1.2.1. Histiocytofibrome………13
1.2.2. Chéloïdes………13
1.2.4. Léiomyomes………..16
1.2.5. Les tumeurs vasculaires………..16
1.2.6. Xanthomes………16
1.2.7. Le naevus………17
1.2.8. La tumeur à cellules géantes des gaines et des tendons………..17
2. Les tumeurs conjonctives d’origine musculo- squelettiques……….18
2.1. Les tumeurs bénignes du tissu graisseux: les lipomes………18
2.1.1. Caractéristiques du tissu adipeux………...18
2.1.2. Siège des lipomes………19
2.1.3. Constitution………19
2.1.4. Variétés particulières de lipomes cutanés et sous cutanés………..20
2.2. Les tumeurs bénignes du tissu musculaire………23
2.2.1. Les léiomyomes………23
a. Caractéristiques des muscles lisses……….23
b. Aspect macroscopique……….24
c. Histogénèse………25
d. Constitution………26
e. Accidents évolutifs………..26
2.2.2. Le rhabdomyome………27
2.3. Les tumeurs bénignes du tissu vasculaire sanguin: les hémangiomes……….27
2.3.1. Structure du tissu vasculaire………..27
2.3.2. Types de tumeurs vasculaires………...28
2.4. Les tumeurs bénignes du tissu lymphatique: les lymphangiomes………..30
2.5. Les tumeurs bénignes nerveuses primitives………..31
2.5.1. Les tumeurs primitives solitaires des nerfs périphériques………32
a. Les tumeurs extirpables………..33
a.1. Le schwannome……….33
a.2. Le lipome intra-nerveux………35
b. Les tumeurs inextirpables………..35
b.1. Les neurofibromes solitaires………36
b.2. L’hémangiome de la gaine de schwann………38
b.3. Le neurofibrolipome……….39
2.5.2. Les tumeurs bénignes primitives multiples ou neurofibromatoses………..40
ETUDE CLINIQUE I. Interrogatoire……….41
1. Quelle est la durée présumée de l’évolution ? Y a-t-il eu une augmentation récente de volume ?...41
2. La tumeur est elle douloureuse ? existe-t-il un syndrome d’aval ?...41
3. Y a-t-il des antécédents traumatiques ?...42
4. Y a-t-il des antécédents néoplasiques ?...43
ETUDE PARACLINIQUE I. Imagerie………46 1. La radiographie standard……….46 2. L’échographie……….46 3. L’angiographie………48 4. La tomodensitométrie (TDM)………48
5. L’imagerie par résonance magnétique (IRM)………49
II. Biopsie………..50 1. La biopsie à l’aiguille………..50 2. La biopsie chirurgicale………...51 3. La biopsie-exérèse……….53 DONNEES ANATOMOPATHOLOGIQUES I. Fragment biopsique………54
II. Pièce d’exérèse………...55
PRISE EN CHARGE THERAPEUTIQUE I. Buts……….59
II. Principe………..59
III. Traitement spécifique de quelques tumeurs………..60
PARTIE PRATIQUE I. INTRODUCTION……….63
II. MATERIELS ET METHODES………65
1. Type d’étude………65
2. Durée de l’étude……….65
3. Sélection des patients………65
4. Critères d’inclusion ………65
III. RESULTATS………..69
1. Conduite à tenir pratique……….69
2. Présentation des résultats selon les critères cliniques, paracliniques, chirurgicaux et anatomopathologiques…………69
3. Présentation des résultats selon les critères Epidémiologiques………91
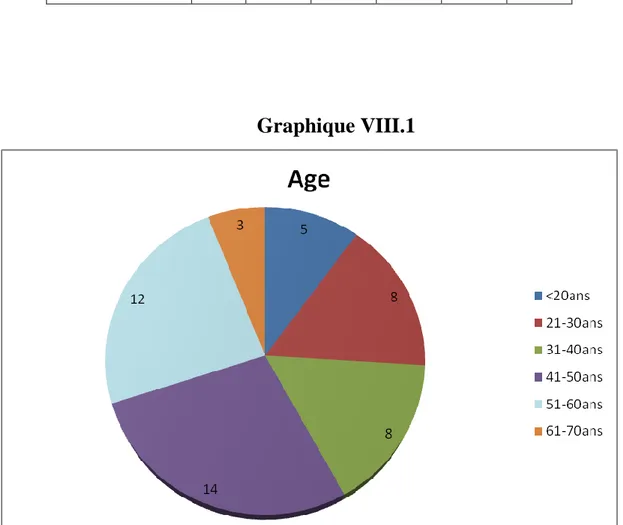
3.1. Résultats selon le sexe et l’âge………...91
3.2. Résultats selon le type de tumeur, le siège et la fréquence………..97
4. Présentation des résultats selon la méthode diagnostique………..102
5. Cas particuliers………..104
5.1. Les récidives………104
5.2. Les tumeurs multiples………105
5.3. Les éclatements lors de la chirurgie……….105
5.4. Les relectures anatomopathologiques………..106
5.5. Les lipomes remaniés………106
IV. Discussion……….107
1. Le critère « sexe »………...107
2. Le critère « âge »………107
3. Les antécédents des patients………108
4. La fréquence des tumeurs………..109
5. Le siège des tumeurs………110
6. L’aspect clinique………..111
7. Le bilan complémentaire………112
8. La prise en charge thérapeutique………...113
10. Les biais de notre étude………..115
a) L’état des dossiers……….115
b) La taille de notre échantillon……….115
c) L’absence de thèse ancienne traitant du sujet et de données nationales épidémiologiques sur ces tumeurs……….116
CONCLUSION……….117
RESUME……….119
REFERENCES BIBLIOGRAPHIQUES………...121
REMERCIEMENTS
A notre maître et président de thèse
Monsieur le professeur MAHFOUD Mustapha
Professeur de traumatologie-ORTHOPEDIE
avicenne
En acceptant de présider notre jury de thèse, vous nous comblez d’un immense plaisir et d’un grand honneur.
Vos qualités humaines et professionnelles sont pour nous, exemplaires Veuillez trouver ici, l’expression de notre considération et notre profond respect.
A notre maître et rapporteur de thèse
Monsieur le professeur B. CHAGAR
Professeur de traumatologie orthopédie
HMIMV
Nous vous devons beaucoup, cher maître, et ces quelques lignes exprimeront mal notre gratitude envers vous. Vous avez été attentif à notre orientation scientifique et à la réalisation de notre thèse dans une atmosphère de respect mutuel, de bienveillance et d’efficacité.
Votre rigueur scientifique, votre disponibilité et votre constante gentillesse sont source d’inspiration pour nous.
A notre maitre et juge de thèse,
Monsieur le professeur al bouzidi abderrahmane Professeur d’anatomopathologie
avicenne
C’est pour moi un grand honneur que vous acceptiez de siéger au sein de mon jury de thèse. J’admire vos qualités humaines, professionnelles et scientifiques. En vous demandant de juger mon travail, je cherche à ce que vous y apportiez votre expérience et la rigueur de votre technicité.
Je voudrais vous dire avec respect, combien je vous suis reconnaissante pour tout ce que vous m’apporterez
A notre maître et juge de thèse,
Madame le professeur amil touria
Professeur de radiologie
HMIMV
Je suis très sensible à l’honneur que vous me faites en acceptant de siéger au sein de mon jury de thèse.
J’ai toujours respecté en vous, cher maître, le médecin, la scientifique dévouée et l’enseignante dynamique.
C’est à la lumière de votre expérience très formatrice que j’ai souhaité que vous puissiez juger ma thèse. Vous l’avez accepté avec gentillesse et intérêt, je vous suis respectueusement très reconnaissante.
A notre maître et juge de thèse,
Monsieur le Professeur achemlal LAHCEN
Professeur agrégé de rhumatologie
HMIMV
Vous avoir comme membre de jury de ma thèse, est pour moi, un grand honneur. Vous avez accepté avec toute la modestie des grands maîtres,
d’apporter vos idées à mon travail. Je suis profondément sensible à la confiance que vous me portez, et je m’efforcerai, cher maître, de ne pas démériter.
Acceptez donc, monsieur, mes sincères sentiments de reconnaissance et de respect.
Au docteur KADI Saïd, professeur assistant au service de traumatologie orthopédie I à l’HMIMV
Je vous remercie vivement de l’intérêt que vous avez bien voulu porter à ce travail en acceptant de sacrifier de votre temps afin d’apporter vos
connaissances à l’élaboration de cette thèse.
Votre simplicité, votre rigueur scientifique, votre persévérance dans le travail et surtout votre dévouement pour les malades, font de vous un modèle pour moi. Veuillez trouver ici, l’expression de mon profond respect et de ma
reconnaissance.
A tout le personnel du service de traumatologie-orthopédie I de l’HMIMV, grand merci pour votre patience et votre gentillesse à mon égard. Le dynamisme que j’ai vu dans le service à travers tout un chacun me pousse à vouloir aller de l’avant. Merci.
Index des tableaux
Tableau I : classification des tumeurs bénignes musculo-squelettiques Tableau II : classification des tumeurs cutanées
Tableau III.1, graphique III.1: Motif de consultation
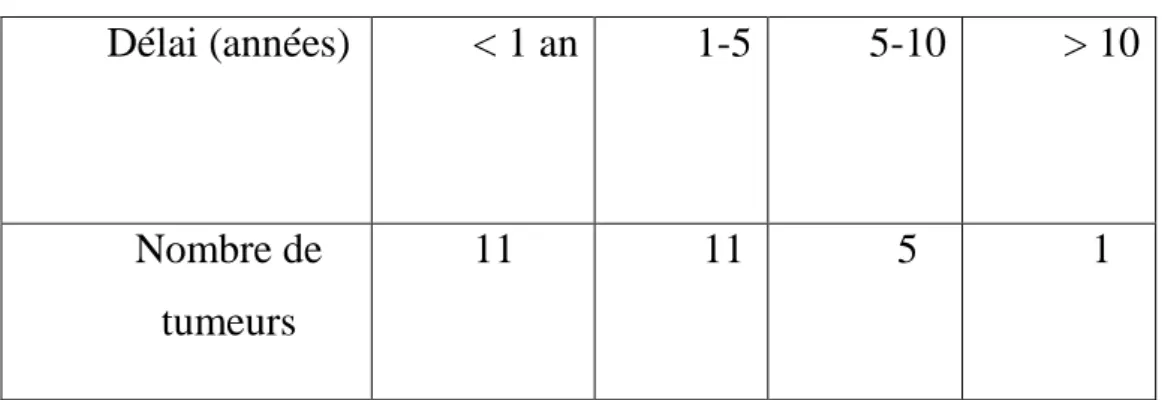
Tableau III.2, graphique III.2 : Motif de consultation selon le type de tumeur Tableau IV.1, graphique IV.1: délai d’évolution
Tableau IV.2, graphique IV.2: délai d’évolution selon le type de tumeur Graphique IV.3 : dimensions des tumeurs
Graphique IV.4 : consistance des tumeurs
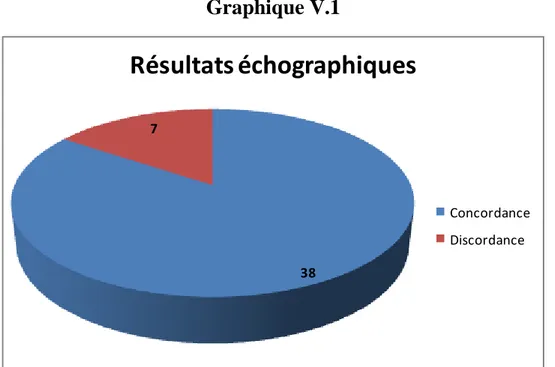
Tableau V, graphique V: type de bilan para clinique Graphique V.1 : résultats anatomopathologiques
Tableau VI, graphique VI : prise en charge chirurgicale Graphique VII.1, tableau VII.2 et graphique VII.2 : Sex-ratio Tableau VIII.1, graphique VIII.1 : tranches d’âge
Tableau VIII.2, graphique VIII.2 : tranches d’âge combinées au sexe Tableau VIII.3 : tranches d’âges des différents types de tumeur
Tableau IX : Effectif-Pourcentage-Siège en fonction du type de tumeur
Graphique IX.1 : effectif ; Graphique IX.2 : pourcentage ;Graphique IX.3 : siège Graphique IX.4 : siège en fonction du type de tumeur
Tableau X, graphique X : méthode diagnostique Graphique XI : récidives
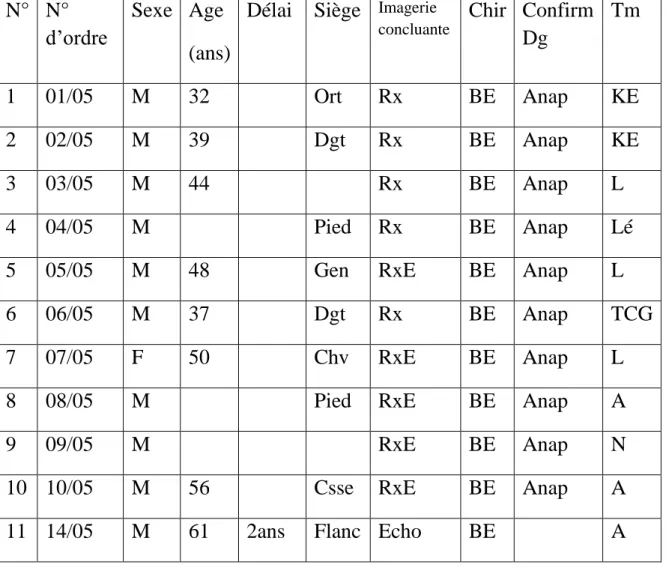
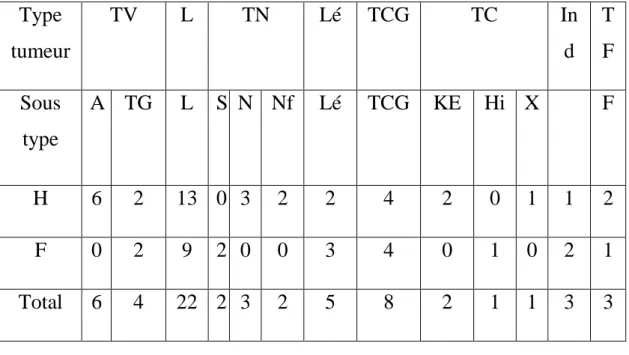
LISTE DES ABREVIATIONS Tumeurs : TV : tumeurs vasculaires A : angiome TG : tumeur glomique L : lipome Lé : Léiomyome TN : tumeurs nerveuses S: schwannome N: névrome Nf: neurofibrome TF: tumeurs fibreuses F: fibrome
TCG: tumeur à cellules géantes des gaines et des tendons TC : tumeurs cutanées
KE : kyste épidermoïde Hi : hidradénome X : xanthome Ind : indéterminé Bilan : Rx : radiographie standard Echo : échographie TDM : Tomodensitométrie
IRM : imagerie par résonnance magnétique RxE : radiographie + échographie
Clin : clinique Chir : chirurgie
Anap, Anapath : anatomopathologie
Termes courants : Dg : diagnostic MS : membre supérieur MI : membre inférieur H : homme F : femme SF : signe fonctionnel NF : neurofibromatose
Tableau de résumé Csse : cuisse Dgt : doigt Pgt : poignet Ort : orteil Gen : genou Chev : cheville Hche : hanche Jbe : jambe Diss : disséminé (r) : remanié (h) : hibernome BE : biopsie-exérèse B-E : biopsie puis exérèse B : biopsie
4.
Définition
Les tumeurs bénignes sont des proliférations non invasives de cellules. On entend par tumeurs bénignes des parties molles, les lésions bénignes développées à partir du tissu conjonctif et de ses variétés différenciées que sont le tissu adipeux, le tissu musculaire, le tissu vasculaire, les tissus synovial et aponévrotique, ainsi que celui des enveloppes des nerfs périphériques [1].
Nous nous intéresserons donc aux tumeurs qui ont pour origine un de ces tissus. Ceci exclura de l’étude les tumeurs d’autres origines comme les myosites calcifiantes et autres qui peuvent aussi se localiser au niveau des parties molles.
L’OMS définit les tissus mous comme étant << les tissus extrasquelettiques, non épithéliaux, à l’exclusion de la glie, du tissu lymphoïde, des séreuses et des tissus de soutien des organes et des viscères>> [2].
Les tissus mous relient, soutiennent et entourent les organes du corps humain. Ils se trouvent entre la peau et les organes internes et comprennent différents tissus tels que les muscles, les tendons, le tissu adipeux et fibreux ainsi que le tissu nerveux. Les parties molles représentent plus de la moitié du poids du corps humain.
Notons que ces définitions des tissus mous excluent le tissu cutané qui fera pourtant partie de notre travail.
Les tumeurs cutanées touchent le tissu épithélial, conjonctif ou annexiel. Elles feront partie de cette étude car bien que prises en charge habituellement dans les services de dermatologie ou de chirurgie plastique, ces tumeurs nécessitent parfois une exérèse chirurgicale large qui peut être l’apanage d’un service de traumatologie.
De plus, ces tumeurs posent souvent un problème de diagnostic différentiel avec les tumeurs sous cutanées.
5.
Intérêt de l’étude
La crainte majeure devant la découverte d’une anomalie quelconque est de le voir dégénérer ou de se retrouver porteur d’une tumeur maligne;
L’intérêt de notre travail sera:
D’abord diagnostic: nous détaillerons la conduite pratique aboutissant au diagnostic final de tumeur bénigne des parties molles en nous aidant de la clinique, la paraclinique, la macroscopie et enfin l’anatomopathologie. Ensuite thérapeutique: nous exposerons les différentes méthodes
thérapeutiques proposées à ces tumeurs.
Enfin pronostique et évolutif : nous présenterons les risques liés à ces tumeurs et leur évolution avant et après traitement.
Evaluer l’épidémiologie de ces tumeurs : fréquence, sexe ratio, âge d’apparition, siège
Evaluer les méthodes thérapeutiques et diagnostiques de ces tumeurs
Donner les éléments de diagnostic différentiel entre tumeurs bénignes et malignes et dénombrer les récidives.
GENERALITES
VI. Epidémiologie
4. Fréquence
Les tumeurs bénignes au niveau des parties molles des membres sont plus fréquentes que les tumeurs malignes [2, 1, 3]. Ce sont des tumeurs très fréquemment rencontrées de la deuxième à la cinquième décennie de vie. L’incidence des tumeurs bénignes dans la population générale est inconnue, parce qu’un certain nombre de patients présentant une grosseur qui ne les gêne pas, ne consultent pas. Dans la population hospitalière, Enzinger et Weiss [4] avancent la proportion approximative de 100 lésions bénignes pour une lésion maligne avec une incidence annuelle d’environ 3000 tumeurs bénignes par million d’habitants [1].
Il est difficile de donner un sex-ratio pour l’ensemble de ces tumeurs; en effet, il y a des variations selon le type histologique de la tumeur (par exemple, les lipomes se développeraient beaucoup plus fréquemment chez la femme [5]) et les facteurs favorisants (génétiques, traumatiques…) de chaque tumeur.
6. Répartition géographique
A nos jours, aucune étude n’a clairement établi une prédominance raciale ou ethnique pour ces tumeurs ; il en existe bien sûr comme la tumeur à cellules granuleuses (tumeur bénigne primitive des enveloppes périphériques) qui se développerait plus chez les sujets de race noire [6, 7, 8]. Mais il est bien évident que ces tumeurs seront plus diagnostiquées dans les pays où le système sanitaire et les moyens diagnostiques sont plus évolués.
VII. Histogenèse [9, 4]
Les cellules néoplasiques bénignes ont une forme semblable à celles des tissus normaux et des lésions hyperplasiques. Elles ne présentent aucune monstruosité. Elles sont aussi typiques que possible.
Elles se multiplient par mitoses bipolaires régulières et par division directe. Tel mode domine dans certains cas, tel autre dans d’autres cas. Ainsi la mitose est
peu fréquente dans les néoplasmes bénins du tissu conjonctif et exceptionnelle dans ceux des muscles lisses [9].
Le rythme de ces divisions est manifestement plus rapide que dans les tissus normaux, mais les figures caractéristiques ne sont jamais très nombreuses. Il semble que leur abondance est moindre que dans les régénérations et que dans les périodes actives des hyperplasies [4].
S.R. BABIN et Al affirment que 10 à 15% de ces tumeurs gardent une histologie incertaine ou demeurent inclassées [1].
VIII. Caractéristiques des tumeurs bénignes [9]
Les néoplasmes bénins sont caractérisés par leur accroissement exclusivement local, expansif et non extensif. Ils refoulent les tissus normaux environnants mais ne les envahissent pas [9, 4, 10].
Deux catégories tissulaires de signification bien différente les constituent: un seul tissu y est doué de l’autonomie prolifératrice et celle-ci induit une prolifération adaptative conjonctivo-vasculaire. Mais comme la première est indéfiniment persistante, la seconde l’est également. De là résulte un complexe organoïde indéfiniment accru [9].
Formes
Lorsque le tissu néoplasique provient d’un revêtement superficiel, la tumeur forme une saillie sessile, souvent villeuse, qui en s’accroissant, tend à se pédiculiser.
Lorsqu’il se développe à partir d’un tissu profond, il forme une masse globuleuse qui refoule les tissus environnants à mesure que son volume augmente [4].
Capsule
Les néoplasmes bénins comportent toujours du tissu conjonctivo-vasculaire auquel sont associés suivant les régions, d’autres éléments différenciés: glandes, muscles…. Ces derniers, comprimés par la tumeur en expansion, s’atrophient et disparaissent ; de sorte que seul le tissu conjonctif persiste à son contact. Du fait même du refoulement qu’il subit, ces faisceaux, tassés, s’orientent parallèlement à la surface de la tumeur et forment autour d’elle une sorte d’enveloppe, la capsule [4, 9].
La capsule est constituée par une lame fibreuse qui se moule exactement, sous tension, sur le néoplasme bénin, mais n’en fait pas partie [9]. Elle est reliée à la surface par de délicats tractus conjonctifs et par des vaisseaux (nés du réseau vasculaire régional et irriguant la tumeur). La tumeur n’adhère donc que très faiblement à la capsule; il existe ainsi un plan de glissement perceptible à la palpation. Après incision de la capsule, la tumeur fait hernie par l’ouverture et se laisse aisément énucléer grâce à ce plan de glissement qui est aussi un plan de clivage [4]. Du côté de la tumeur, la limite de la capsule est donc parfaitement nette; ce n’est pas le cas du côté des tissus sains: si l’on étudie de dedans en dehors, on y reconnaît d’abord une série de couches collagènes compactes et pauvres en cellules, comparable à celles d’une aponévrose. Peu à peu, le parallélisme de ces couches perd de sa régularité. Des éléments différenciés en
voie d’atrophie, d’abord clairsemés, puis plus nombreux, apparaissent entre ces strates et, peu à peu, le refoulement s’étant moins fait sentir, les tissus reprennent leur morphologie régionale. Ainsi, la capsule fait extérieurement corps avec les tissus environnants [4, 9].
Conséquence du refoulement exercé par la tumeur sur les tissus environnants, la capsule est d’autant plus nette que la tumeur est volumineuse. Il ne faut donc pas s’étonner si elle est peu évidente ou même manque autour des néoplasmes bénins de faibles dimensions, au début de leur évolution [9].
Accroissement
Il est indéfini dans les néoplasmes bénins du fait de la prolifération du tissu initiateur, mais il est soumis en outre à l’apport nutritif que fournit le tissu conjonctivo-vasculaire induit [9,10].
Qu’ils soient superficiels, pédiculés ou profonds, encapsulés; c’est par leur périphérie que leurs vaisseaux nourriciers les pénètrent. A mesure que leur diamètre augmente, la circulation devient de plus en plus précaire dans leur profondeur et, de plus, tend à se repartir inégalement.
L’insuffisance circulatoire locale a pour effet, un ralentissement voire un arrêt de l’accroissement. Son inégalité se traduit par l’apparition de foyers en active prolifération contrastant avec d’autres, plus ou moins stabilisés. Les premiers poursuivent leur croissance tandis que les seconds la ralentissent. Ainsi se constituent, dans une tumeur primitivement sphérique, des noyaux qui, lui donnant un aspect lobé, festonnent ses bords, et même tendent à s’en séparer. La
capsule épouse encore la surface du néoplasme, mais devient irrégulière comme elle [4, 9]. Dans ces conditions, l’exérèse par clivage peut être incomplète. Or tout fragment tumoral laissé en place peut continuer sa prolifération et donner lieu à une récidive locale. Une récidive n’est donc pas forcément un signe de malignité. Le mieux, pour éviter un tel accident est d’éviter le clivage et de réséquer la capsule, avec sa tumeur.
IX. Classification
Le tableau I donne la classification OMS des tumeurs des parties molles (proposée par Enzinger et Weiss) [2].
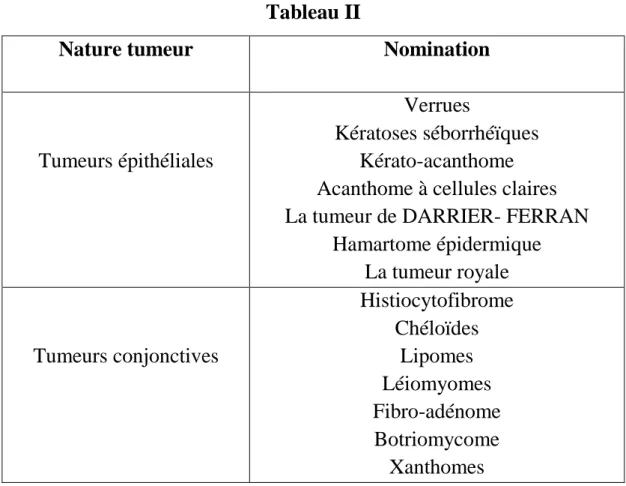
Notons que cette classification donne les tumeurs bénignes des parties molles musculosquelettiques excluant ainsi les tumeurs cutanées bénignes qui feront partie de notre étude et dont voici une classification sommaire dans le tableau II: Classification des tumeurs bénignes cutanées [11].
Tableau I
Type de tissu Tumeurs bénignes
Tissu conjonctif Structures fibreuses
Structures histiocyto-fibreuses
o Fibrome: Fasciite nodulaire Fasciite proliférative
Myosite proliférative Chéloïdes
o Fibromatose superficielle Fibromatose radiogénique de l’enfant
o Histiocytome fibreux
o Xanthogranulome, réticulohistiocytome, xanthome, fibroxanthome
Tissu graisseux
Lipomes: Angiolipome, lipome polymorphe, lipoblastome, angéiomyélolipome, myélolipome,
lipome intramusculaire, lipomatose, hibernome Rhabdomyome
Tissu musculaire Léiomyome : angiomyome, léiomyomatose, léiomyoblastome
Vaisseaux sanguins
Hémangiome : capillaire, artérioveineux, caverneux, veineux, épithélial, pyogénique,
hémangiomatose, tumeur glomique, hémangiopéricytome, hyperplasie papillaire
endothéliale
Hémangioendothéliome (tumeur semi maligne) Vaisseaux
lymphatiques
Lymphangiome, lymphangiomatose, lymphangiomyomatose
Tissu synovial Tumeur à cellules géantes
Tissu mésothélial Mésothéliome : localisé, épithélial, fibreux, mixte Mésothéliome multikystique et péritonéal Tissu mésenchymateux Mésenchymome Ganglions périphériques Ganglioneurone Tissu paraganglionnaire Paragangliome Nerfs périphériques
Neurilemmome ( schwannome bénin) Neurofibrome,
Neurofibromatose
Tableau II
Nature tumeur Nomination
Tumeurs épithéliales
Verrues
Kératoses séborrhéïques Kérato-acanthome Acanthome à cellules claires La tumeur de DARRIER- FERRAN
Hamartome épidermique La tumeur royale Tumeurs conjonctives Histiocytofibrome Chéloïdes Lipomes Léiomyomes Fibro-adénome Botriomycome Xanthomes
Tumeurs annexielles
Kystes épidermiques et sébacées Hidradénomes ou syringomes Trichoépithéliomes Cylindromes Tumeurs vasculaires Angiomes Malformations vasculaires Tumeur glomique
Tumeurs nerveuses Schwannome, neurofibrome, nevrome
Naevus
Naevi communs Formes particulières
X. Etude anatomopathologique
1. Les tumeurs cutanées 2.1. D’origine épithéliale
2.1.1. Les verrues [11]
Ce sont des proliférations épidermiques donnant une saillie circonscrite à surface verruqueuse [3,11].
On distingue les verrues:
- D’origine virale dues au Papilloma virus humain (HPV). On a: Les
verrues vulgaires (HPV 2, 4), les verrues plantaires (HPV1), les verrues planes (HPV3, 10),
- La verrue séborrhéique ou kératoses séborrhéiques ou verrue sénile
2.1.2. Le kérato-acanthome [3, 11]
Formation tumorale en bulbe d’oignon, de la taille d’un pois chiche à celle d’une noisette siégeant surtout au visage mais aussi sur le dos des mains des sujets ayant dépassé la quarantaine.
2.1.3. L’acanthome à cellules claires [3, 11]
Petit nodule de teinte rosée, assez souvent recouvert d’une croûte, qui s’observe chez l’adulte âgé et se localise sur la jambe. L’évolution se fait sur plusieurs années sans tendance à la disparition.
2.1.4. L’hamartome épidermique [3,11]
Lésion congénitale le plus souvent unilatérale, formée d’élevures ou de plaques mamelonnées grises, d’aspect crasseux, hyperkératosique.
2.2. D’origine conjonctive 2.2.1. Histiocytofibrome [3, 11]
Petite formation intradermique, dure, en pastille de 0.5 à 1cm, légèrement en relief, de couleur brun foncée, parfois pigmentée. Se voit surtout chez la femme et au niveau de la jambe.
2.2.2. Chéloïdes [3, 11]
Formations intradermiques, saillantes, de consistance très ferme, à surface lisse, parfois bosselée, blanche ou rosée ou télangiectasique, de forme allongée ou ovalaire, assez souvent spontanément sensibles ou prurigineuses. Elles sont surtout localisées au niveau des régions pré sternales, scapulaires et brachiales.
On distingue les chéloïdes:
o Provoquées, secondaires à des traumatismes divers : vaccination, brûlures, lésions inflammatoires.
o Spontanées: très fréquentes dans la race noire et pouvant prendre des dimensions importantes et devenir invalidantes: ‘‘ maladie chéloïdienne’’.
2.2.3. Lipomes [5, 12]
Les lipomes cutanés se développent à partir des cellules graisseuses de l’hypoderme. Ils peuvent être solitaires ou multiples réalisant alors des lipomatoses. Une prolifération adipocytaire est probable dans les lipomes encapsulés, une hypertrophie adipocytaire est prédominante dans les lipomatoses non encapsulées.
d’adipocytes matures, le plus souvent encapsulée et de croissance lente. Il survient surtout à l’âge adulte sans prédilection de race avec une légère prédominance féminine dans certaines études. Cliniquement, il s’agit d’une masse siégeant profondément dans la peau, mobile par rapport aux plans profonds, sans adhérence aponévrotique ou musculaire. La peau de recouvrement n’est pas inflammatoire et se mobilise par rapport à la tumeur. La taille du lipome solitaire est relativement variable en fonction de l’ancienneté ou du potentiel de croissance ; leur grand axe est le plus souvent de 2 à 4 cm. Certains lipomes peuvent devenir volumineux allant jusqu’à 20 cm de grand axe. Le siège est ubiquitaire avec une prédilection pour la région cervicale postérieure, les régions scapulaires ou dorsales hautes, antébrachiales, fessières et enfin la racine des cuisses.
Les lipomatoses: on a:
La lipomatose multiple familiale ou lipomatose mésosomatique ou
maladie de Roch-léri [5, 13, 16]. Le mode de transmission le plus fréquent est autosomique dominant avec pénétrance variable.
Les lipomes apparaissent généralement à partir de l’âge de 20 à 30 ans ; on note une prédominance masculine (sexe ratio= 2). Sa prévalence est d’environ 2 pour 100000 habitants [13, 16]. Le nombre de lipomes est très variable allant de quelques éléments à une centaine. La répartition est grossièrement symétrique avec atteinte du tiers inférieur des bras, de la région anté-brachiale sans dépasser le poignet. Au niveau du membre inférieur, la racine des cuisses est la localisation de prédilection. Les lipomes siègent aussi à la base du thorax, sur
l’abdomen et la région lombaire. Le visage, les extrémités, le cou et la région dorsale sont habituellement respectés [13].
La LMF est une affection bénigne avec apparition progressive de lipomes au cours de l’âge adulte. L’évolution vers l’ulcération, la nécrose et la calcification est très rare. D’exceptionnelles dégénérescences liposarcomateuses sont signalées, de même que des régressions spontanées [16].
La lipomatose multiple symétrique (LMS) ou maladie de Launois Bensaude [17, 18].
La lipomatose douloureuse de Dercum [17, 20].
Il existe également des lipomatoses associées dans les syndromes de Gardner-Richards, Protée [21]….
1.2.4. Léiomyomes [4, 9]
Ils se développent aux dépens des muscles lisses pilomoteurs, des parois artériolaires. Ce sont des tumeurs arrondies ou allongées, de petite taille, douloureuses à la pression et au froid. On y distingue le piloléiomyome et l’angioléiomyome [3].
1.2.5. Les tumeurs vasculaires
Hémangiomes [3]
C’est une prolifération tumorale bénigne de cellules endothéliales vasculaires. Tumeur glomique: prolifération de cellules ou hamartome aux
dépens du glomus neuromyoartériel [23]. C’est une petite tumeur douloureuse des extrémités surtout digitales [3, 23, 24].elle survient entre 30 et 50ans avec une prédominance féminine (sex-ratio=1/3) [23, 25, 26]. L’IRM en fait le diagnostic au moindre doute [27, 28, 29, 30].
1.2.6. Xanthomes [11]
Ce sont des infiltrations cutanées de couleur jaunâtre faites de cellules histiocytaires de plus ou moins grande taille à cytoplasme spumeux ou de très grande taille avec nombreux noyaux disposés en couronne centrale. On a les xanthomes:
Plans: dont le xanthélesma des paupières est le plus fréquent
Tubéreux: intéressent surtout les convexités (coude, genou), les faces d’extension des bras. Ils représentent une des manifestations de l’hypercholestérolémie familiale (type II de FREDRICKSON).
Eruptifs: petite taille, groupées sur une surface (fesse, membres…), ils apparaissent brutalement souvent à la suite d’un repas riche en graisses. Ils font partie du tableau de l’hypertriglycéridémie exogène (type I de FREDRICKSON).
1.2.7. Le naevus
C’est une hyperplasie circonscrite bénigne d’origine hamartomateuse (dysembryoplasique) [3] ou acquise des mélanocytes dans la peau [11].
On a:
Les formes particulières : les naevi congénitaux, les naevi bleus, le naevus de Sutton, le naevus de SPITZ et les naevi atypiques.
1.2.8. La tumeur à cellules géantes des gaines et des tendons [3]
C’est une tumeur bénigne de la gaine tendineuse et de la synoviale. Elle se présente comme une masse dermique ou sous cutanée, ferme de croissance lente, siégeant en regard des articulations surtout digitales [31]. Elle survient chez l’adulte jeune entre 20 et 40ans avec une faible prépondérance féminine
[32]. L’IRM en est l’examen de choix [33].
Histologiquement, c’est une masse lobulée jaunâtre, renfermant une population cellulaire riche, constituée surtout de cellules de type macrophagique et de cellules géantes multinuclées. Quelques mitoses et des irrégularités nucléaires peuvent se voir [31, 34, 35].
Les récidives après le traitement ne sont pas rares, liées en général à une synovectomie incomplète [31].
2. Les tumeurs conjonctives d’origine musculo- squelettiques
2.6. Les tumeurs bénignes du tissu graisseux: les lipomes
Le lipome est une tumeur mésenchymateuse bénigne composée de cellules graisseuses matures sans atypie cellulaire. Il est fréquent entre 30 et 50 ans
2.6.1. Caractéristiques du tissu adipeux [4, 9]
Le tissu adipeux est un dérivé du mésenchyme primitif mais ne doit plus être considéré comme formé de fibrocytes chargés de graisses. En effet, le fibrocyte banal contient le plus souvent de minuscules enclaves graisseuses mais non cette goutte unique, enclose dans une large vacuole creusée dans un cytoplasme aminci et qui refoule latéralement un noyau aplati comme c’est le cas dans le lipocyte. Cette bulle cytoplasmique est elle-même entourée par une délicate résille de réticuline dont on ne retrouve l’analogue qu’autour de ces éléments mésenchymateux hautement spécialisés que sont les fibres musculaires.
Aussi, le tissu adipeux a les caractères d’une glande endocrine mésenchymateuse ; elle entrepose la graisse reçue du sang et peut l’éliminer et le remettre en circulation [4, 9].
2.6.2. Siège des lipomes
Leur siège est un point quelconque du mésenchyme [5, 9]. La plupart naissent dans les régions les plus riches en tissu adipeux à l’état normal.
Ils sont souvent multiples (parfois symétriques), parfois uniques. Leur taille varie dans les plus larges proportions. On en a décrit qui pesaient 30 kilogrammes et davantage.
Chose remarquable, les lipomes semblent en dehors du métabolisme général. L’amaigrissement de leurs porteurs n’entraîne pas, sauf exceptions, leur diminution de volume (inversement dans les lipomes pararénaux, leur croissance rapide coïncide avec une régression générale du tissu adipeux normal) [9].
2.6.3. Constitution [9]
Ce sont des masses globuleuses, souvent multilobées, plus ou moins nettement limitées. Leur structure est ordinairement simple et reproduit assez bien celle du tissu adipeux banal : amas de vésicules adipeuses ou lobules, séparées les uns des autres par des cloisons fibreuses plus ou moins complètes. Entre les vésicules, on trouve un réseau capillaire d’une grande richesse [4, 9].
La taille des lobules est variable. On peut trouver ça et là, des éléments petits, dont le cytoplasme contient plusieurs gouttelettes adipeuses, et qui sont vraisemblablement les éléments jeunes et fertiles de la tumeur, ceux qui assurent son accroissement. Les vésicules adipeuses ne se divisent pas. Elles ne contribuent à l’accroissement de la masse tumorale qu’en se surchargeant de graisse.
Entre les cellules lipomateuses et surtout dans les septas conjonctifs, les mastzallen sont toujours nombreuses.
2.6.4. Variétés particulières de lipomes cutanés et sous cutanés
L’angiolipome [22, 37]: c’est une variété clinique fréquente et particulière qui survient chez l’adulte jeune avec une prédilection pour les membres et sans facteur déclenchant particulier. La taille est presque toujours inférieure à 2cm de grand axe. Parfois les lésions sont multiples avec un regroupement régional, la pression est souvent douloureuse, la coloration bleutée si le contingent vasculaire est important.
L’exérèse montre une tumeur encapsulée avec un contingent adipocytaire mature et un contingent vasculaire constitué de capillaires dilatés parfois thrombosés et de fibres musculaires lisses provenant des vaisseaux. Lorsque la composante vasculaire est importante, on utilise la dénomination d’angiolipome cellulaire ; lorsque le contingent de fibres musculaires lisses est important, on peut poser la question d’un angioléiomyome avec une petite composante adipocytaire( angio-lipo-léio-myome) [4].
L’hibernome ou lipome du tissu adipeux immature ou lipome fœtal
ou lipome de la graisse embryonnaire ou lipome à cellules granuleuses [9, 20,
38, 39, 40]: il s’agit d’une variété rare de lipome bénin développé à partir de la
graisse fœtale de couleur brune. Le terme d’hibernome a été proposé en raison de la ressemblance avec la graisse des animaux hibernants.
Il se manifeste chez l’adulte jeune, peut-être un peu plus précocement que le lipome solitaire classique. Les 2 sexes sont touchés avec une légère prédominance féminine. L’allure clinique est celle d’une tuméfaction ferme, de taille variable pouvant aller jusqu’à 10cm de grand axe. La lésion siège de préférence dans les zones où existe de la graisse fœtale à l’état normal (cou, région interscapulaire). D’autres sièges sont possibles, mais encore plus rares : cuisses, fesses, aisselles…….
L’allure anatomopathologique montre sur le plan :
Macroscopique : une couleur allant du jaune-brun au rouge-foncé.
Microscopique : une prolifération adipocytaire organisé en lobules, séparés par du tissu conjonctif. La population adipocytaire est un mélange d’adipocytes matures de grande taille à cytoplasme vacuolaire, contenant des graisses
birefringentes, de cellules multivacuolées à contenu adipeux, de cellules rondes à noyau central et à cytoplasme éosinophile. Ces cellules ont des cytoplasmes riches en mitochondries. Il existe d’exceptionnelles formes malignes.
Le lipome périsudoral ou adénolipome cutané [41, 42, 43]
C’est un lipome solitaire d’individualisation récente caractérisé par la présence au sein d’un lipome encapsulé, de glandes sudorales eccrines. L’aspect est celui d’une formation nodulaire siégeant le plus souvent à la racine des cuisses ou sur la fesse. Il touche l’adulte avec une légère prédominance féminine.
Le lipoblastome bénin et la lipoblastomatose [45, 46, 47]
Il s’agit d’une entité rare, caractérisée par son siège et son âge de survenue (avant l’âge de 3 ans) ; il est plus fréquent chez le petit garçon (sex-ratio=3) ; pas de terrain génétique. Le siège de prédilection se situe au niveau des extrémités des membres. La forme circonscrite se présente sous la forme d’une tuméfaction souple, indolore, bien circonscrite. Il existe une forme infiltrative, mal limitée, pouvant toucher les muscles et qui est désignée sous le terme de lipoblastomatose. La biopsie montre des lobules adipocytaires avec des adipocytes à des stades de maturation différents, sans anomalies cytologiques. Les cellules adipeuses vont subir une maturation progressive au cours de l’évolution. La tumeur bien circonscrite récidive rarement après exérèse tandis que celle infiltrative est plus difficile à enlever et peut récidiver.
Le fibrolipome perinerveux ou macrodystrophie lipomateuse digitale
[38, 47].
C’est une malformation rare, siégeant avec prédilection aux poignets ou aux doigts avec développement progressif chez l’adulte jeune pouvant s’accompagner de douleurs et de paresthésies. Le siège de prédilection est sur le trajet du nerf médian. La lésion peut s’accompagner d’une macrodactylie. La biopsie montre la présence de nerfs avec un périnèvre épaissi entouré par des cellules adipocytaires matures.
C’est une entité autonome, sans parenté avec les formes localisées de neurofibromatose ou avec le syndrome de Protée [21].
Le lipome infiltrant ou lipome profond [5]
Il peut être vu dans le cadre des consultations dermatologiques en raison de la déformation cutanée qu’il entraîne. Il s’agit d’un lipome sous aponévrotique développé aux dépens de la graisse inter ou intramusculaire et dont la croissance peut entraîner une voussure cutanée ou une compression vasculaire ou nerveuse. Il est rare (2% des lipomes), survient chez l’adulte et en cas de localisation superficielle, touche le tronc et les racines des membres.
2.7. Les tumeurs bénignes du tissu musculaire 2.7.1. Les léiomyomes
a. Caractéristiques des muscles lisses [9]
Ils sont formés par des fibres lisses, cellules fusiformes, parcourues d’une extrémité à l’autre par des fibrilles contractiles parallèles, réfringentes et acidophiles [4, 9].
Les noyaux occupent la région moyenne de chaque cellule. Ils sont allongés, étroits, en forme de bâtonnet, fortement colorables.
Ces cellules sont incluses dans une mince gaine de réticuline (membrane pellucide) et isolées les unes des autres par une substance fondamentale délicatement spongieuse que parcourent de fines fibres élastiques. Elles s’ordonnent en faisceaux anastomosés les uns avec les autres en plexus [9]. On admet en outre que certains éléments contractiles sont d’origine épithéliale : ce sont les cellules myo-épithéliales des glandes. A l’état normal, ces cellules sont rameuses et situées entre les cellules glandulaires et la basale. Mais il arrive que, subissant une hyperplasie, elles se libèrent plus ou moins de l’épithélium et acquièrent une forme semblable à celles des fibres lisses mésenchymateuses. Les léiomyomes sont constitués par des fibres lisses associées à des vaisseaux [9, 48]. Ces tumeurs sont ubiquitaires [49, 50, 51, 52, 53]. La pathogénie des léiomyomes est obscure et sans doute, variable avec leur siège.
b. Aspect macroscopique [9, 50]
Les léiomyomes sont des tumeurs de dimensions très variables (microscopiques ou énormes).
D’ordinaire, ce sont des masses sphériques, à contours géographiques, ou bosselées. Quand ils sont très petits, les léiomyomes font corps avec le tissu où ils ont pris naissance ; plus gros, ils en sont séparés par une capsule. Après une
phase d’accroissement interstitiel, ils tendent à s’extérioriser, à faire saillie hors de l’organe où ils se sont développés et restent unis à lui par un pédicule plus ou moins grêle.
Leur consistance est ferme, souvent extrêmement dure. Leur surface de coupe est d’aspect variable, tantôt rose pâle, tantôt blanc pure, sèche ou humide, homogène ou formée par un mélange de bandes fibreuses compactes séparées par des régions molles, œdémateuses, parfois tachées de suffusions hémorragiques ou creusées de cavités de désintégration. En somme, leur aspect extérieur est celui de fibromes (c’est d’ailleurs ce nom que les chirurgiens utilisent habituellement) et la nature musculaire ne s’affirme qu’après examen microscopique.
Leur nombre est variable, il en est de solitaire [48] mais souvent ils sont multiples, particulièrement au niveau de l’utérus.
c. Histogénèse [9]
L’examen microscopique montre des anomalies localisées dans la disposition des muscles ; quelques faisceaux tranchent sur leurs voisins. Ils sont plus serrés, moins acidophiles, plus riches en cellules et par conséquent en noyaux. Ils forment un nodule mal limité, dont les faisceaux périphériques se continuent avec les faisceaux normaux, et dont les faisceaux profonds, épaissis, ont une orientation particulière et contournée.
A un grossissement plus fort, on voit que les cellules du nodule sont relativement pauvres en myofibrilles. Leurs noyaux sont moins allongés, plus larges que ceux des cellules normales.
D’autres nodules, plus volumineux, manifestent plus clairement leur autonomie. Ce sont des masses arrondies, formées par des faisceaux musculaires semblables à ceux précédemment décrits. Leurs fibres conservent une forme allongée. Ces faisceaux sont ramifiés, épais, anastomosés, tourbillonnants. Dans leurs intervalles, se ramifient des capillaires engainés d’un collagène où l’on reconnaît des cellules fixes plus ou moins nombreuses. Les cellules tumorales sont séparées par des fibres conjonctives plus ou moins abondantes suivant les cas.
d. Constitution [4, 9, 50]
L’architecture de ces tumeurs est semblable au premier abord à celle des sarcomes fibroblastiques. Leurs constituants sont différents ; la plupart sont des cellules musculaires lisses imparfaites mais assez riche en myofibrilles pour être franchement acidophiles. Elles ne présentent ni monstruosités nucléaires, ni mitoses. Leur prolifération par amitose est accompagnée d’une production de fibrilles collagènes intercellulaires et de membranes pellucides. Des capillaires parcourent les interstices interfasciculaires, accompagnés de rares cellules adventielles. Autour d’eux les faisceaux musculaires ont une orientation très variée. Ça et là, ils les entourent de couches concentriques qui simulent une sorte de média.
Le léiomyome grandit sur place et refoule autour de lui les tissus voisins. Il s’en isole plus ou moins complètement par une capsule. Souvent des centres de proliférations y apparaissent et sont l’origine d’une lobulation incomplète, traduite extérieurement par un bossèlement de sa surface.
e. Accidents évolutifs [9]
Parfois, des ramollissements oedémateux et dégénératifs accompagnés d’ectasies vasculaires et d’hémorragies, modifient profondément les caractères macroscopiques des léiomyomes et les font considérer souvent comme « en dégénérescence sarcomateuse ». L’histologie reforme presque toujours ce diagnostic.
2.7.2. Le rhabdomyome [4, 9]
Le rhabdomyome est la tumeur bénigne qui se développe à partir des fibres musculaires striées. Il est plus rare que le léiomyome. Les tumeurs du muscle strié sont plus souvent malignes que bénignes.
La genèse des tumeurs bénignes se calquent grossièrement sur celui des tumeurs des muscles lisses.
Siège de prédilection [9]: cœur, langue
2.8. Les tumeurs bénignes du tissu vasculaire sanguin: les hémangiomes
2.8.1. Structure du tissu vasculaire [9]
L’élément essentiel d’un vaisseau est un tube formé de cellules aplaties et minces, associées en un revêtement ou endothélium, qui repose sur une gaine de collagène réticulé. Celle-ci le sépare incomplètement des tissus mésenchymateux environnants.
Les propriétés du tissu vasculaire: o La polarisation
o Secondairement la structure primitive se complique ou se spécialise
o La plasticité et la facilité d’adaptation aux circonstances ; un capillaire peut devenir une veine ou une artère, une veine peut s’artérialiser.
o L’endothélium peut proliférer sans que ses éléments entrent en contact avec le sang circulant. Alors ses bourgeonnements ne se canalisent pas et restent massifs, comme ceux que l’on obtient dans les cultures de plasma.
2.8.2. Types de tumeurs vasculaires [4, 9]
Toutes les tumeurs contiennent des vaisseaux sanguins ; ceux-ci y figurent comme éléments associés ou comme constituants du stroma. Ici, nous n’envisagerons que les tumeurs résultant d’une prolifération des endothéliums vasculaires et aboutissant à l’édification de vaisseaux plus ou moins complexes, et non celles qui peuvent provenir de leurs seules tuniques musculaires (léiomyomes) ou adventitielles.
On distingue trois catégories d’hémangiomes: dysgénétiques, hyperplasiques et néoplasiques.
Les hémangiomes dysgénétiques: hémangiomes capillaires et artériels.
Les hémangiomes capillaires: aucune région n’est exempte mais la peau est le siège de prédilection (On les appelle hémangiomes cutanés ou naevi vasculaires).
Aspect macroscopique: on distingue les hémangiomes plans et les hémangiomes tubéreux. Les premiers, pouvant rétrocéder, sont constitués par de simples
taches sans relief, rouges ou violacées de forme et de dimensions variables. Les seconds ont une surface irrégulière, mamelonnée, verruqueuse et peuvent pénétrer les muscles, comprimer les nerfs et corroder les os.
Histogenèse et constitution [9]: au début de son accroissement, on distingue dans le derme (et plus particulièrement au voisinage des glandes sébacées et sudoripares) de larges vaisseaux bordés par deux ou trois couches de cellules. Ce revêtement stratifié est propre aux angiomes dysgénétiques et s’observe dans la plupart de leurs vaisseaux dont la paroi bourgeonne des ramifications dont chacune produit une sorte de lacis pelotonné. Chacun de ces pelotons s’accroît par lui-même, sans se relier, sauf exceptions avec les capillaires du voisinage. Il ne possède guère qu’un gros vaisseau afférent et un gros vaisseau efférent. De ce fait, le peloton angiomateux forme dès le début, une petite masse arrondie et relativement autonome. Sa régularité initiale est pourtant troublée par la constitution dans sa masse de centres de prolifération secondaire qui lui donne un contour festonné et s’individualisent plus ou moins par la suite. Du peloton initial naissent de multiples autres pelotons conglomérés.
L’endothélium de ces hémangiomes présente de nombreuses figures mitotiques d’où résultent des cellules qui perdent tout contact avec la lumière vasculaire, se divisent à leur tour, et font saillie en marge du tube vasculaire. Ainsi se constituent localement de petits amas puis des cordons cellulaires au sein desquels se produisent des fissures. Celles-ci se mettront en rapport avec la cavité sanguine et deviendront de nouveaux capillaires. Tout en progressant, les vaisseaux s’entourent de délicates gaines collagènes argyrophiles. Ces jeunes vaisseaux se réunissent en anses et forment un lacis d’abord lâche puis serré
parce que de nouveaux bourgeonnements et de nouvelles anastomoses viennent réunir les anses préexistantes et combler leurs interstices.
Les hémangiomes artériels et veineux
Les hémangiomes hyperplasiques : c’est un granulome hyperplasique et plus
précisément une hyperplasie angiomateuse exubérante. Il est formé essentiellement de glomérules et de néovaisseaux baignant dans un infiltrat inflammatoire qui se localise au fur et à mesure et laissant la place à un infiltrat collagène.
D’autres auteurs tels que M. WASSER et AL [76] ont proposé une classification des hémangiomes tenant compte de plusieurs facteurs combinés.
S. CHAGNON et Al [54] classent les hémangiomes en hémangiomes caverneux, capillaires et veineux.
2.9. Les tumeurs bénignes du tissu lymphatique: les lymphangiomes
Ils sont plus rares que les hémangiomes. On les trouve, eux aussi, un peu partout mais la peau et le cou sont leurs sièges de prédilection. Tous sont vraisemblablement dysgénétiques (cf hémangiomes dysgénétiques) [4, 9].
Les uns sont diffus, les autres encapsulés et localisés. Tous résultent de la prolifération de vaisseaux lymphatiques qui, progressivement, se dilatent irrégulièrement et se fusionnent ou forment des kystes. On distingue [9]:
Les lymphangiomes diffus: habituellement cutanés, ils donnent un aspect verruqueux.
Constitution [4]: au microscope, le derme papillaire et le derme fibreux sont creusés de cavités plus ou moins larges, toujours inégales, remplies par une sérosité où nagent quelques lymphocytes. La paroi de ces cavités est formée par un endothélium très mince qui repose sur une lame collagène. Leurs contours ont le dessin le plus capricieux : arrondis, anguleux ou étoilés. Dans les plus volumineux, on voit souvent des vestiges de cloisons qui forment un éperon dans leurs cavités et indiquent une fusion latérale de deux vaisseaux d’abord séparés.
Les lymphangiomes encapsulés: ils forment des tumeurs
sphériques ou bosselées, fluctuantes. Leur siège habituel est le cou mais on en a décrit dans la région axillaire, le mésentère….
Les lymphangiomes kystiques : c’est une agglomération de kystes inégaux séparés les uns des autres par des cloisons plus ou moins épaisses. Ces kystes ont une paroi revêtue par un endothélium très mince. Ils sont remplis de lymphe sous pression. Leurs cloisons contiennent des vaisseaux lymphatiques de toutes tailles et sont formées par du tissu conjonctif, graisseux et des vaisseaux sanguins.
Les tumeurs primitives des nerfs périphériques sont des tumeurs assez rares [55]. Elles représentent 2% des tumeurs des parties molles (Alnot et al [56], Le Viet et al [57]), et 90% des cas sont bénins [56]. Elles se développent aux dépens des éléments constitutifs du nerf et pose des problèmes diagnostiques malgré l’apport de l’IRM, thérapeutiques notamment en cas de tumeur inextirpable et anatomopathologiques lorsque la bénignité de la tumeur est difficile à affirmer [55]. Il existe plusieurs classifications de ces tumeurs: anatomopathologique (qui tient compte de l’origine des cellules tumorales), sémiologique qui les classe en tumeurs nerveuses primitives solitaires et en tumeurs nerveuses primitives multiples (neurofibromatoses de type I, II, schwannomatoses…). Elles sont généralement bénignes mais la transformation en tumeurs malignes dans le cadre de la neurofibromatose est une éventualité peu fréquente mais gravissime. En dehors des neurofibromatoses, la survenue d’une tumeur primitive des nerfs périphériques maligne est exceptionnelle et l’origine nerveuse de ces tumeurs malignes est souvent difficile à affirmer du point de vue anatomopathologique. Ces tumeurs primitives, lorsqu’elles sont inextirpables, sans zone de clivage par rapport aux éléments nerveux sains posent des problèmes thérapeutiques. [6]
2.10.1. Les tumeurs primitives solitaires des nerfs périphériques
L’incidence des tumeurs primitives solitaires des nerfs périphériques est faible. Ces tumeurs sont surtout rencontrées à l’âge adulte rarement chez l’enfant. Même si globalement l’incidence des tumeurs primitives solitaires des
nerfs périphériques est un peu importante aux membres supérieurs, ces tumeurs représentent moins de 5% des tumeurs de l’avant-bras et de la main [59, 60]. Le schwannome est la tumeur nerveuse périphérique solitaire la plus fréquente [6,
61]. On y trouve aussi le lipome intra-nerveux, le neurofibrome, l’hémangiome
de la gaine de Schwann, les hamartomes lipofibromateux ou neurofibrolipomes qui sont plus rares.
Certains auteurs G. CHICK, J-Y ALNOT et al [55], C. CHANTELOT
[62] classent les tumeurs bénignes isolées des nerfs périphériques en:
c. Les tumeurs extirpables
Elles refoulent les groupes fasciculaires sans les pénétrer et peuvent donc être énucléées sans interrompre la continuité nerveuse. Leur aspect macroscopique est très évocateur : ce sont des tumeurs encapsulées, en général arrondies, de moins de 5cms de diamètre. Il existe dons un plan de clivage constant entre la capsule tumorale et les fibres nerveuses permettant de conserver la continuité nerveuse. Souvent, la tumeur est appendue à un fascicule que l’on ne peut séparer et qu’il faudra donc sacrifier (ce fascicule n’est déjà plus fonctionnel dans certains cas ; ce qui n’entraînera donc pas de déficit neurologique supplémentaire en post opératoire) [55]. On a donc:
a.1. Le schwannome
Il représente 80 % de ces tumeurs bénignes [62]. Les premières descriptions de cette tumeur remonte au début du XIXème siècle ; le schwannome s’observe à tous les âges mais surtout entre 30 et 60 ans (études de
Kehoe = 63 %) avec un sex ratio de 1 [55]. Il est connu sous les termes de neurilemmome ou de neurinomme et est constitué par une population de cellules de Schwann [61, 62].
- Localisation : Das Gupta [63] trouve une incidence plus importante aux membres supérieurs (sur une série de 303 avec un rapport de ½). Aux nerfs périphériques, les schwannomes sont localisés aux nerfs médian, radial et ulnaire dans 20% des cas. [6]
- Aspects macroscopiques et histologiques: les schwannomes se
développent habituellement de manière excentrique dans le nerf, refoulant les éléments nerveux ; ceci explique leur caractère clivable du reste du nerf. Leur aspect macroscopique est celui d’une tumeur arrondie ou ovalaire, grisâtre ou jaunâtre, brillante, bien encapsulée par l’épinèvre. [6]
Du point de vue anatomopathologique: [6]
Macroscopiquement : la tumeur est excentrée par rapport au trajet du nerf, bien limitée, de forme ovoïde ou sphérique et de couleur brunâtre. Il s’agit d’un tissu mou facilement fragmentable. Les tissus voisins, et notamment le nerf atteint, ne sont que refoulés.
En microscopie optique: il existe 2 types A et B d’Antoni:
Le type A d’antoni, hypercellulaire, se caractérise par une disposition compacte des cellules de Schwann qui sont entrecroisées. L’accolement des noyaux cellulaires donne un aspect en « palissade ». Arrangés circulairement, ils forment les nodules de Verocay.
Le type B d’Antoni, hypocellulaire, à structure lâche, se caractérise par une dégénérescence myxoïde avec des cellules étoilées.
Les deux types sont le plus souvent associés sous une forme mixte. Les mitoses sont rares ; les vaisseaux sont dilatés à paroi épaissies et parfois thrombosées. Des plages hémorragiques ou nécrotiques avec dégénérescence cellulaire peuvent exister. Il n’y a pas de structures axonales dans le schwannome. Il existe d’autres variétés telles que les schwannomes mélanocytiques, notamment en fonction des caractéristiques cellulaires. Le diagnostic est souvent évident en microscopie optique. Toutefois une confirmation par immunohistochimie peut être utile.
En microscopie électronique, les schwannomes sont uniquement constitués par une population de cellules de Schwann.
Pronostic: les schwannomes bénins peuvent entraîner un déficit neurologique si la tumeur se développe au niveau d’un défilé anatomique. Après exérèse chirurgicale, il n’y a pas de récidive. La dégénérescence maligne d’un schwannome est controversée ; une méconnaissance du diagnostic de malignité initial ne peut être écartée [4, 64, 65]. En ce qui concerne les schwannomes isolés ou les schwannomes multiples de la schwannomatose, aucun cas de dégénérescence maligne n’a été décrit actuellement [4, 66].
a.2. Le lipome intra-nerveux [55]
C’est une tumeur encapsulée à développement intra-neural ; elle est exceptionnelle et n’a pas fait l’objet de nombreuses publications. Elle est à distinguer du lipome extrinsèque comprimant le nerf. Sa bonne tolérance explique le délai entre l’apparition de signes cliniques (précoce) et le traitement (tardif).
d. Les tumeurs inextirpables
Elles infiltrent l’ensemble des éléments constitutifs du nerf et leur exérèse complète est impossible sans altérer les fibres nerveuses. Ce sont des tumeurs non encapsulées qui ont des particularités anatomopathologiques propres à chaque type [55]. On distingue:
b.1. Les neurofibromes solitaires
Ils touchent habituellement l’adulte jeune et concernent environ 10% des tumeurs bénignes isolées (G. Chick et al). Ils apparaissent plus tôt que les schwannomes mais le délai pour en faire le diagnostic est plus long. Le reste de l’examen clinique est normal, c'est-à-dire qu’il n’y a aucun signe clinique de neurofibromatose (toutes les investigations doivent d’abord être faites dans le sens d’éliminer ce diagnostic) [55].
Pour Kehoe, 65% des neurofibromes sporadiques sont observés dans la tranche d’âge de 30-59 ans, rarement chez l’enfant sauf en cas de NF.
La distribution des neurofibromes entre membres supérieurs et inférieurs est assez homogène et ne montre pas de prédominance particulière [6].
Aspect macroscopique et histologie [6, 55]
Le neurofibrome est caractérisé par une grande diversité cellulaire avec principalement des cellules de Schwann, des fibroblastes et des cellules périneurales. Il peut y avoir aussi des cellules endothéliales, des mastocytes et des cellules mélaniques. Le tissu tumoral se développe de façon infiltrante parmi
les éléments nerveux, ce qui explique son caractère non extirpable chirurgicalement sans dommage pour les éléments nerveux sains.
Classification [6]
Les neurofibromes sont classés en plusieurs variétés selon leur mode de développement : localisé, diffus, plexiforme:
Le neurofibrome localisé: c’est la forme la plus fréquente. Bien que des neurofibromes localisés multiples soient caractéristiques de la NF1, ces neurofibromes sont pour la plupart du temps isolés et sans rapport avec cette phacomatose. Il n’y a pas de prédisposition de sexe. Le neurofibrome localisé superficiel ou cutané est une tumeur assez bien limitée qui siège essentiellement sur la peau du tronc. Les mains sont rarement atteintes. Il se développe au centre d’une terminaison nerveuse cutanée et, compte tenu de la destruction de l’épinèvre, le neurofibrome n’est pas encapsulé. Sa taille est très variable et il peut être pédiculé de type molluscum pendulum, sessile, intradermique ou nodulaire sous-cutané. Sa consistance est molle et gélatineuse. Il n’est généralement pas douloureux et a parfois un aspect violacé. Un neurofibrome localisé d’un nerf profond entraîne un épaississement fusiforme du nerf de dimension variable. La tumeur est le plus souvent bien limitée par l’épinèvre, ici plus épais et résistant.
Le neurofibrome diffus: il est associé à la NF1 dans 10% des cas. Se développant à l’extrémité d’un nerf cutané, sa localisation est cutanée. Du point de vue anatomopathologique, comme son nom semble l’indiquer, la prolifération traverse le derme et le tissu sous cutané. L’aspect clinique est une accentuation des plis cutanés ou un épaississement en plaque de la peau.
Le neurofibrome plexiforme: il est pathognomonique de la NF1
[67] et peut intéresser les nerfs superficiels et les nerfs les plus profonds. La
prolifération s’étend parmi les fascicules nerveux sur parfois une longue distance avec possible envahissement des collatérales. Ceci est à l’origine d’un aspect macroscopique tortueux ou vermiculaire du nerf et d’une impression de « paquet de ficelles » à la palpation des nerfs superficiels. Ce sont les neurofibromes qui dégénèrent le plus dans la NF1.
Pronostic [6]
Le développement d’une tumeur maligne primitive des enveloppes nerveuses périphériques à partir d’un neurofibrome peut seulement être affirmé par l’étude histologique lorsqu’une continuité est retrouvée entre la tumeur maligne et le neurofibrome.
DUCATMAN et Al [64] ont mis en évidence cette association dans une série de 120 patients atteints de tumeur maligne primitive des enveloppes nerveuses périphériques traités à la Mayo clinique. L’association neurofibrome/tumeur maligne primitive des enveloppes nerveuses périphériques (TMPENP) a été observée chez 81% des tumeurs malignes primitives des enveloppes nerveuses périphériques notées dans la NF1, contre 41% des TMPENP observés chez des patients indemnes de NF1. Dans cette même étude, d’après l’analyse de 1124 patients atteints de NF1, il a été déterminé qu’un patient porteur de la NF1 avait 4600 fois plus de risques de développer ce type de sarcome que la population générale. Approximativement, 2 à 29% des patients atteints de NF1 pourront développer une TMPENP, habituellement après une période de latence de 10 à 20 ans.
b.2. L’hémangiome de la gaine de Schwann [6]
C’est une tumeur exceptionnelle, décrite presque toujours chez la femme et apparaissant le plus souvent au cours de la première décade de vie. Le nerf médian est la localisation la plus fréquente
Trois types ont été individualisés par Y. ALLIEU et Al selon l’extension locale de l’hémangiome:
Le type : intraneural extrafasciculaire, facilement extirpable car situé en périphérie de la gaine;
Le type 2: intrafasciculaire non extirpable sans dommage pour le nerf-hôte;
Le type 3 à développement intra et extraneural.
b.3. Le neurofibrolipome [55]
C’est une tumeur exceptionnelle probablement congénitale dont l’étiologie est inconnue. Le nerf médian et ses branches en sont le site de prédilection. Il n’a pas été constaté de dégénérescence maligne.
D’autres auteurs comme Y. ALLIEU et Al [6] proposent plutôt la classification suivante:
o Tumeurs bénignes primitives des enveloppes nerveuses périphériques:
Neurofibrome
Tumeur à cellules granuleuses Neurothécome
o Tumeurs bénignes primitives d’origine extraneurale : L’hamartome fibrolipomateux
Le pseudokyste mucoïde L’hémangiome
Le lipome intranerveux
Le choristome neuromusculaire
2.10.2. Les tumeurs bénignes primitives multiples ou neurofibromatoses [4]
Ce sont des maladies héréditaires caractérisées par la présence de neurofibromes multiples, de lésions cutanées, osseuses, ophtalmologiques et autres.
RICCARDI et Al [68] ont classé les neurofibromatoses comme suit: NF1: maladie de Von Recklinghausen
NF2: neurofibromatose acoustique
NF3: neurofibromatose mixte tenant à la fois de la NF1 et de la NF2 NF4: formes inclassables par ailleurs
NF5: neurofibromatose segmentaire NF6: taches café au lait isolées
NF7: neurofibromatose à début tardif: absence de neurofibrome avant la troisième décennie