HAL Id: hal-02785639
https://hal.inrae.fr/hal-02785639
Submitted on 4 Jun 2020HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Test de validation d’un kit de purification d’ADN
génomique de sols
Solene Perrin
To cite this version:
Solene Perrin. Test de validation d’un kit de purification d’ADN génomique de sols. Sciences du Vivant [q-bio]. 2017. �hal-02785639�
Plateforme Genosol UMR 1347 AgroEcologie INRA de Dijon 17 rue Sully-BP 86510 20 065 DIJON Cedex
Test de validation d’un kit de purification
d’ADN génomique de sols
PERRIN Solène
1ère année BTS Bio-Analyses et Contrôles Promotions 2016-2017
Lycée le Castel 22, rue Daubenton 21000 Dijon
Stage effectué du 29/05/2017 au 07/07/2017 Maître de stage : LELIEVRE Mélanie
SOMMAIRE
Présentation de la structure………. 1
I) Présentation générale de l’INRA……… 1
II) Présentation de l’INRA de Dijon………. 1
III) Présentation de l’UMR AgroEcologie……….. 2
IV) Présentation de la plateforme Genosol………. 2
Partie scientifique……….. 5
I) Contexte……… 5
II) Méthodes………. 6
1) Extraction standard d’ADN du sol………. 6
2) Purification de l’ADN……….. 8
a) Méthode standard (S)………. 8
b) Kit de purification : ZymoBIOMICS DNA Mini Kit (Z)……….. 10
c) Kit de purification : NucleoSpin Soil (N)……….. 11
d) Kit de purification : PoweLyzer PowerSoil DNA Isolation kit (P)…………. 11
3) Analyse de la qualité de l’ADN par électrophorèse sur gel d’agarose……….. 12
4) Dosage des ADN propre par fluorescence………. 13
5) Vérification de l’inhibition par la qPCR ou PCR quantitative………. 14
a) La PCR classique ou PCR en point final……….. 14
b) La PCR quantitative……….. 15
III) Résultats……… 19
1) Validation de la quantité de l’ADN par dosage fluorométrique……….. 19
2) Validation de la qualité de l’ADN……… 19
a) Analyse de la qualité de l’ADN par électrophorèse sur gel d’agarose…… 19
b) Analyse de la qualité de l’ADN par test d’inhibition par qPCR………. 20
3) Validation de l’outil diagnostic de routine par PCR quantitative………. 23
Conclusion……….. 24
I) Conclusion scientifique……….. 24
II) Conclusion personnelle……….. 25
Annexes……….. 26
Remerciements
Avant tout développement de cette expérience professionnelle au sein de l’INRA, je tiens à adresser mes remerciements à Madame Nathalie Munier-Jolain présidente du centre INRA de Dijon et à Monsieur Philippe Lemanceau directeur de l’UMR AgroEcologie pour m’avoir permis d’effectuer ce stage dans leurs locaux.
Je tiens à remercier Monsieur Samuel MONDY, responsable de la plateforme Genosol pour m’avoir accueilli au sein de son équipe.
Je souhaite particulièrement à adresser mes remerciements à Mélanie LELIEVRE ma tutrice de stage et à Julie TRIPIED pour avoir pris de leur temps et de leur patience afin de me former et de m’avoir apporté un certain nombre de connaissances. Je les remercie pour la confiance et l’autonomie qu’elles m’ont accordé tout au long de ce stage et de leur bonne humeur sans laquelle il aurait été moins plaisant d’effectuer ce stage.
Je remercie les autres personnels et stagiaires, Virginie, Samuel, Sebastien, Thomas, Almina, Marion, Cécile... qui ont pu m’aider ou avec qui nous avons pu passer des bons moments.
LEXIQUE
INRA : Institut National de Recherche en Agronomie UMR : Unité Mixte de Recherche
CRG : Conservatoire des Ressources Génétiques microbiennes LADM : Laboratoire d’analyses et de Développements Moléculaires SIE : Système d’Information Environnemental
AFNOR : Association Française de Normalisation ISO : Organisation Internationale de Normalisation LIMS : Laboratory Information Management System ADN : Acide DésoxyRibonucléique
EDTA : Ethylène Diamine Tétra Acétique TBE : Tris Borate EDTA
PVPP : PolyVynil PolyPirrolidone BET : Bromure d’Ethidium UV : Ultra-Violet
PRESENTATION DE LA STRUCTURE
I)
Présentation générale de l’INRA (Institut National de Recherche en Agronomie) :
L’INRA est un organisme français de recherche en agronomie qui a été fondé en 1946 après la seconde guerre mondiale pour répondre aux besoins de la France qui était en pénurie alimentaire et dont l’agriculture ne permettait pas de subvenir aux besoins alimentaires du pays.
Depuis, l’INRA est devenu le premier institut de recherche agronomique en Europe et le deuxième institut en sciences agricoles dans le monde. Il est placé sous la double tutelle du ministère chargé de la Recherche et du ministère chargé de l’agriculture.
Il a pour but de mener des recherches pour une agriculture durable, pour une alimentation saine et de qualité et pour un environnement préservé et valorisé. La mission de l’INRA est donc d’associer science et technologie afin d’améliorer et d’optimiser les techniques de l’agriculture et de l’élevage en France.
Avec 74 % de ses effectifs implantés en province sur plus de 150 sites, l'institut est présent dans la quasi-totalité des régions françaises, y compris l'outre-mer. Il est composé de plus de 8000 agents titulaires qui sont des chercheurs, des ingénieurs, des techniciens, des administratifs, des thésards, des étudiants et des chercheurs étrangers.
II)
Présentation de l’INRA de Dijon
:
Le centre INRA de Dijon est un institut pluridisciplinaire dont les recherches sont axées sur la sociologie et l’économie du développement territorial, l’Agroécologie et le goût de l’alimentaire. Ce sont donc trois thématiques essentielles qui sont les préoccupations de notre société et qui ont pour objectif de répondre aux grands enjeux de demain.
Le centre comprend différentes unités de recherche :
UMR AgroEcologie à Dijon
UMR Centre des Sciences du Goût et de l’Alimentation (CSGA) à Dijon
A ces unités s’ajoutent des unités qui dépendent administrativement du centre INRA de Dijon :
Unité ChronoEnvironnement à Besançon.
Unité de Recherche Technologique et Analyses Laitières (URTAL) à Poligny.
Unité Expérimentale « Domaine d’Epoisses » à Bretenières. , Mon stage s’est déroulé au sein de l’UMR AgroEcologie.
III)
Présentation de l’UMR AgroEcologie :
Crée en 2012, cette unité travaille en collaboration avec l’INRA, AgroSup Dijon, l’Université de Bourgogne-Franche-Comté et avec le CNRS (Centre National de la Recherche Scientifique).
Le but de cette Unité Mixte de Recherche est de développer une agriculture durable permettant une production qualitative et quantitative en phase avec les besoins alimentaire tout en respectant l’environnement. Face à l’augmentation de la population mondiale, la recherche doit faire se converger des objectifs agronomiques et environnementaux. Cette unité réunit donc des enjeux de tailles :
Assurer une production agricole de qualité et en quantité suffisante. Réduire l’utilisation d’intrants.
Proposer des systèmes agricoles innovants qui respectent et valorisent la biodiversité et l’environnement.
Cette unité comprend différents pôle de recherches : BioME (Biologie et fonctions éco systémiques des sols), GEAPSI (déterminismes Génétiques et Environnementaux de l’Adaptation des Plantes à des Systèmes de culture Innovants), GESTAD (Gestion Durable des Adventices) et IPM (Interactions Plantes Microorganismes). Elle comprend également différentes plateformes : Genosol, les serres, un ensemble de ressources biologiques et un ensemble de microscopie
La répartition des services et du personnel de l’UMR se trouve dans un organigramme (annexe1).
En période de stage, l’effectif du personnel est multiplié par deux.
IV)
Présentation de la plateforme Genosol :
Genosol est une plateforme de l’Unité Mixte de Recherche AgroEcologie dont le métier est la conservation et l’analyse des ressources génétiques des communautés microbiennes du sol.
Elle est constituée de trois centres d’activités principales, un Conservatoire des Ressources Génétiques microbiennes (CRG), un Laboratoire d’Analyses et Développements Moléculaires (LADM) et d’un Système d’Information Environnemental (SIE) qui s’articulent sur la diversité microbienne des sols et de l’environnement.
Le Conservatoire : c’est un Centre de Ressources Génétiques (CRG) sur les sols, unique en Europe qui a pour but de stocker et conserver les ressources génétiques. Les échantillons de sols conservés proviennent en majorité de réseaux de surveillance de sols, d’observatoire de recherche en environnement, de sites expérimentaux, de réseau d’exploitation agricole. Plus de 14000 sols sont stockés et environ 1000 nouveaux sols sont en prévision d’acquisitions par année. L’activité de ce service est certifié ISO 9001 depuis 2015.
Le Laboratoire d’Analyses et de Développements moléculaires (LADM) : Ce service s’articule autour de 300 m2 de laboratoires de biologie moléculaire dans lesquels sont analysés les échantillons de sols issus du CRG dans le cadre de projets scientifiques. Ce service occupe d’améliorer la standardisation des procédures et des outils moléculaires avec des objectifs de normalisation (AFNOR et ISO) par la mise en place de veille technologique. Ainsi, le LADM réalise du développement sur les méthodes qu’elle utilise en routine ou encore sur des outils de biologie moléculaire de nouvelle génération comme le séquençage haut débit. Elle développe notamment des outils comme l’extraction des acides nucléiques des sols, de PCR quantitative et les outils de caractérisation des ressources génétiques microbiennes. Le LADM permet d’aborder la diversité microbienne des sols.
Le LADM a la particularité de faire beaucoup de formations et de mettre ses équipements à disposition du personnel de l’UMR AgroEcologie.
Environnemental (SIE) : c’est un service qui regroupe les différents outils informatique, statistiques pour gérer les données produites par le LADM. Son fonctionnement est surtout basé sur :
- Une base de données, Microsol, qui renferme toutes les données produites par le LADM et permet d’une part de gérer le CRG et le LADM ainsi que la traçabilité des échantillons et d’autre part, de stocker et d’analyser les données de caractérisation génétique des sols. Cette base de données est le premier référentiel d’interprétation et de traduction de la biodiversité des sols.
- Un pipeline d’analyses nommé GnS-Pipe qui permet l’analyse des millions de données issues de séquençage haut débit.
- Des applications de gestion de la plateforme telles que la gestion documentaire, la gestion des ressources et des équipements.
La plateforme est en cours d’acquisition d’un LIMS (Laboratory Information Management System) qui gérera d’ici 2 ans l’activité complète de la plateforme depuis la demande d’analyse jusqu’à l’édition des résultats.
La plateforme est composée de 3 personnels permanents et un personnel CDD. Elle est dirigée par un ingénieur sous la tutelle d’un comité de gestion regroupant des chercheurs de l’UMR AgroEcologie.
Mon stage se situe dans le contexte d’une veille technologique au sein du Laboratoire d’Analyses et de Développements Moléculaires dans le cadre de mise en place d’un nouveau kit de purification d’ADN en remplacement d’une méthode, posant problème actuellement.
PARTIE SCIENTIFIQUE
I)
Le contexte :
Le sol est un ensemble complexe qui possède des rôles clef dans l’environnement autre qu’un rôle dans l’agriculture ou la construction :
La régulation des flux de matière
Il fournit des éléments nutritifs pour la production agricole Il lutte contre les inondations en tant que réservoir tampon
Le sol est également la principale réserve de la diversité microbienne. Dans 1 g de sol, plusieurs milliards de microorganismes comme les bactéries, les champignons microscopiques, les protozoaires ou encore les débris de végétaux y sont dénombrés.
Ces microorganismes jouent un rôle fondamental dans le fonctionnement du sol. Ils régulent le cycle du Carbone et de l’Azote ou encore dégradent des polluants organiques tels que les pesticides et les dérivés de pétrole. Ce sont des bons indicateurs de l’évolution de la qualité d’un sol car les communautés microbiennes intègrent rapidement les stress environnementaux touchant le sol du fait que ces derniers subissent beaucoup de perturbations (pollution, érosion, acidification, tassement des sols..) et l’usage intensif de pesticides conduit à une réduction importante de la biodiversité microbienne. Il est donc important de veiller à la qualité biologique et microbiologique des sols.
C’est pourquoi, dans la plateforme Genosol, les analyses sont axées sur l’étude de populations bactériennes et fongique présentes dans ces sols. Ainsi, la plateforme travaille sur deux gènes représentatifs de ces populations à savoir le gène 16s pour l’étude des bactéries et le 18s pour l’étude des champignons. Ces gènes ont la particularité d’être hautement conservés, présents chez tous les microorganismes puisqu’ils codent d’une part pour la sous-unité ribosomique 16s pour les bactéries et pour la sous-unité ribosomique 18s pour les champignons.
Pour exploiter ces gènes, la plateforme Gensosol extrait en routine l’ADN génomique de sols. Cette extraction comporte une double étape de purification pour éliminer les polluants (acides humiques) co-extraits avec l’ADN des sols. La purification d’ADN permet surtout d’obtenir une matrice d’ADN propre, de qualité et en quantité suffisante pour permettre d’effectuer des analyses moléculaires sur les gènes 16s et 18s. Depuis quelques mois, le LADM rencontre des problèmes avec le protocole standard de purification d’ADN. Depuis toujours, la purification d’ADN de sol est réalisée en deux étapes, un tamis moléculaire et une affinité exclusion mais depuis quelques temps les ADN extraits sont de moins bonne qualité et les pertes d’ADN après purification sont importantes.
Au mois de décembre 2016, le LADM a mis en évidence le problème qui causait cette forte perte. En effet, le produit utilisé pour réaliser le tamis moléculaire a changé de lieu de production. Les nouveaux lots ont été produits en Chine alors qu’ils étaient auparavant produits en Allemagne. Le fournisseur étant le seul sur le marché à fournir ce produit, Genosol souhaite alors valider une autre méthode de purification de ses ADN.
Le but de mon stage est de tester des kits de purification d’ADN et d’en valider au moins un qui permette à la plateforme d’obtenir des ADN purifiés de qualité, en quantité suffisante et dont la nouvelle purification n’ait pas d’impact sur les résultats habituellement obtenus en comparaison avec le protocole standard.
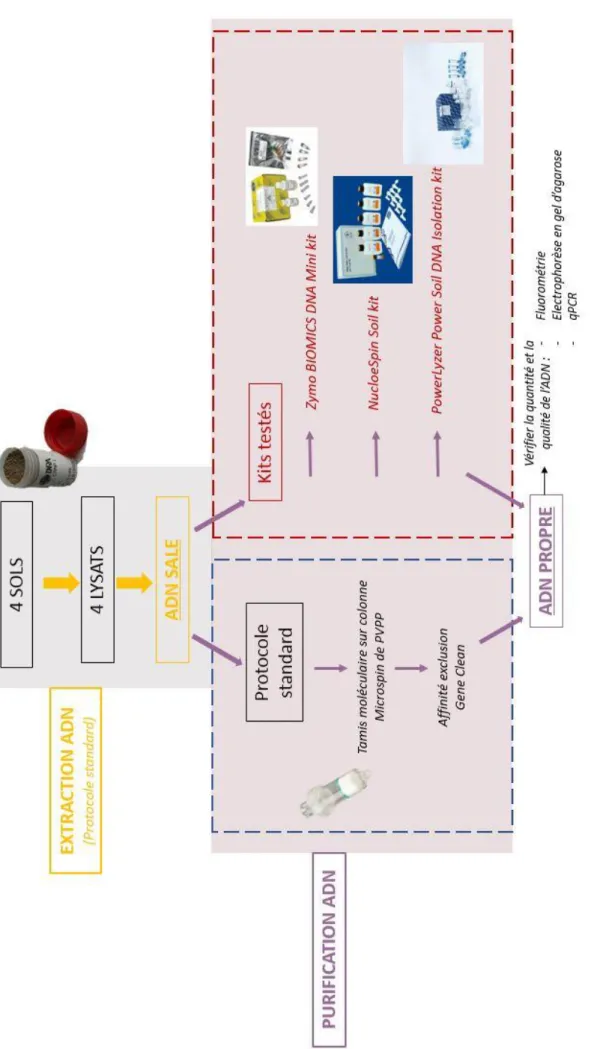
Fi gur e 1 : Sch ém a de c o nt ext e
Ainsi, le stage s’est déroulé en plusieurs parties comme le montre la figure 1 (Page de gauche).
Dans un premier temps, l’ADN de 4 échantillons de sol différents a été extrait en grande quantité (6 répétitions) pour obtenir un grand volume d’ADN à purifier. En effet, l’extraction d’ADN est composée de deux étapes qui mènent à l’obtention d’un lysat puis d’un ADN sale. C’est à partir de cet ADN que seront effectuées les purifications et donc les tests.
Dans un second temps, les ADN de ces 4 sols ont été purifiés en 3 répétitions sur quatre protocoles. Cela a représenté 48 échantillons à traiter. Le tableau ci-dessous énumère la liste des protocoles qui seront testés en incluant le protocole standard habituel de la plateforme. Chaque méthode a été codée afin de mieux gérer les échantillons.
Protocoles Standard (Genosol)
ZymoBIOMICS DNA Mini Prep kit
NucleoSpin Soil PowerLyzer DNA
Isolation
Marque Sigma et MP Biomedicals
MOBIO-QIAGEN Zymo Research Macherey-Nagel
Code S Z N P
Enfin, des outils moléculaires ont été appliqués sur ces ADN obtenus pour vérifier leur qualité et leur quantité. Pour évaluer la qualité, l’électrophorèse en gel d’agarose, la PCR quantitative ont été choisis et un dosage fluorométrique a été réalisé pour évaluer la quantité.
A l’issu de ces analyses, la plateforme a sélectionné un ou plusieurs protocoles sur lesquels seront appliqués les outils de diagnostics habituel. Si les résultats sont comparables, la plateforme changera le protocole par le kit qui conviendrait le mieux en tenant compte également des offres de prix et de disponibilité du SAV.
II)
Méthodes :
1) Extraction standard d’ADN du sol
L’extraction d’ADN des microorganismes est réalisée directement du sol lyophilisé issu du Conservatoire de Ressources Génétique (CRG). L’objectif est d’obtenir des ADN non dégradés et suffisamment purifiés pour qu’aucunes impuretés ne viennent interférer dans les analyses suivant l’extraction.
L’extraction a donc été réalisée sur 4 sols (194, 238, 189, 208) en 6 répétitions.
Etape 1 : Broyage et lyse des membranes :
L’extraction se fait à partir d’un gramme de sol par une lyse mécanique à l’aide de différentes billes couplées à une lyse chimique par action d’un tampon de lyse. Dans un même tube sont ajoutés avec le sol, 2g de billes de 0.01 mm en silice ; 2.5g de billes de 1.4 mm en céramique et 4 billes de verres et 5mL de tampon de lyse. Annexe 2 : point sécurité.
Composition du tampon de lyse (5 mL par échantillon) :
Composants Rôle Proportions en mL (pour 1 échantillon)
Tris-HCl Tampon à pH=8 0.5
EDTA Chélateur de cations (ions Mg2+ et Ca2+) 1
NaCl Précipitation des protéines 0.5
SDS Lyse membranaire 0.5
H2O Ultra-Pur (UP) Solvant 2.5
Remarque : Le tampon de lyse, après préparation, doit être filtré sur une membrane dont les pores font 0.2 µM pour éliminer les microorganismes potentiels dans les différents composants pour ne pas contaminer les sols et fausser les résultats. Annexe 2 : point sécurité
Chaque échantillon est ensuite mis sous agitation dans un appareil appelé FastPrep-24 à 4m/sec (figure 2) pendant 90 secondes pour broyer le sol et réaliser la lyse mécanique des cellules grâce aux billes contenues dans le tube. Le broyat est ensuite placé au bain marie à 70°C pendant 30 minutes pour que la lyse chimique soit réalisée. Après centrifugation à 7000g pendant 5 minutes à 20°C, les débris cellulaires, le reste de sol et les billes de verre sont éliminés dans le culot. Le surnageant est récupéré en totalité. Il contient l’ADN est appelé lysat.
Etape 2 : Déprotéinisation
Cette étape a pour but d’enlever les particules en suspension et les protéines dénaturées présentes dans le lysat. Pour cela, 100µL d’acétate de potassium est ajouté à 1mL de lysat pour permettre la précipitation des protéines. Une incubation de 10 minutes dans de la glace favorise la formation de cristaux qui vont piéger les protéines. Après centrifugation à 14000g pendant 5 minutes à 4°C, l’ADN est récupéré dans le surnageant, les protéines et les molécules en suspension sont éliminées dans le culot. Annexe 2 : point sécurité
Pour les 4 échantillons testés, la totalité du lysat a été utilisé pour permettre d’obtenir une grande quantité d’ADN à purifier.
Etape 3 : Précipitation de l’ADN
L’ADN est précipité en présence de 900µL d’isopropanol à -20°C, après 30 minutes à -20°C. Il est récupéré dans 200µL d’eau ultra-pure après centrifugation à 13000rpm pendant 30minutes. Annexe 2 : point sécurité
Figure 2 : FastPrep
L’ADN obtenu est dit « sale » car il contient quelques résidus de sol, des éléments polluants, tels que les sucres ou des macromolécules phénolique mais il contient essentiellement des acides humiques qui sont des polymères à haut poids moléculaires, chargés négativement, de couleur noire à brin résultant d’un processus de condensation oxydative des composés phénoliques et liés à des acides aminés ; peptides et polysaccharides. Ils sont riches en carbone mais moins riches en oxygène.
Cet ADN sale est habituellement dosé pour évaluer la biomasse microbienne d’un sol c’est-à-dire la quantité d’ADN présente dans un gramme de sol. Cette donnée est exploitée par la plateforme en regard avec les analyses physico-chimiques et l’utilisation du sol.
A ce stade, ces ADN dit « sale » ont été aliquotés puis conservés à -20°C pour être traités par les quatre protocoles de purification.
2) Purification de l’ADN :
A cette étape, les répétitions d’extraction ont été mélangées par sol pour former un échantillon unique qui sera purifié par le protocole standard d’une part et par les kits de purification à tester d’autre part.
Pour chaque protocole, 3 répétitions sur chaque sol ont été effectués. Pour rappel trois kits ont été testés en parallèle du protocole standard comme le montre le schéma ci-dessous
Cette étape permet donc de purifier les ADN sales extraits, c’est-à-dire d’éliminer des éléments polluants ; protéines ; sucres ; macromolécule phénoliques ou encore les acides humiques. Le but est d’obtenir des ADN limpides de qualité et en quantité suffisante pour réaliser les analyses moléculaires
a. Méthode standard (S)
La purification standard se fait pat deux méthodes successives différentes : un tamis moléculaire réalisé sur colonne Microspin de PVPP puis une colonne d’affinité exclusion.
Tamis moléculaire sur colonne Microspin de PVPP (PolyVynil PolyPirrolidone) :
Le PVPP est un polymère de hauts poids moléculaire qui réalise un complexe avec des composés phénoliques et alcaloïdes présents dans le sol. Il permet dans une colonne de retenir les polluants et de laisser passer l’ADN.
Préparation des colonnes : Il faut remplir une colonne de PVPP qui est sous forme de poudre. La pesée se fait en fonction de la couleur du culot, entre 80 et 120 mg de PVPP par colonne pour réaliser le meilleur tamis moléculaire possible en évitant un maximum les pertes d’ADN liées à la purification.
Il faut ensuite laver la colonne pour enlever les impuretés présentent dans le PVPP en faisant passer de l’eau Ultra-Pure puis celle-ci est séchée par centrifugation.
Pré-purification des ADN sales : 100µL d’ADN sale sont introduits au centre de la colonne et cet ADN est récupéré après centrifugation. Une grande partie des impuretés auront été retenues par la colonne. L’ADN doit ressortir limpide.
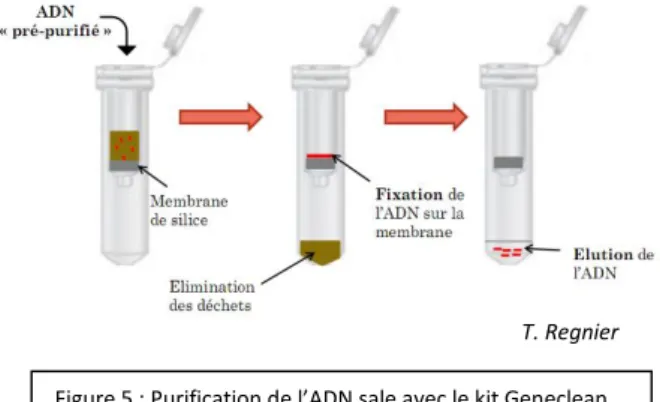
Purification sur colonne d’affinité-exclusion avec le kit Geneclean Turbo (MP Biomedicals) : Ce kit contient des colonnes de purification, des tubes de 2mL et différentes solutions nécessaires à la manipulation dont les compositions ne sont pas connues, le but est d’avoir un ADN limpide sans polluant.
L’ADN obtenu à l’étape précédente est mélangé avec la solution GTGNS (Geneclean Turbo GNomic Solution) puis est déposé dans une colonne de purification. L’ADN est alors retenu sur une membrane de silice lors d’une centrifugation à 10000g, c’est le principe d’affinité.
9
Figure 3 : Quantité de PVPP à peser en fonction de la couleur du culot
Figure 4: Pré-purification de l’ADN sale dans une colonne de PVPP.
L’ADN fixé sur la membrane de silice est alors lavé avec une solution à base d’éthanol nommée GTW (Geneclean Turbo Wash) avant d’être élué avec 60µL de tampon GTE (Geneclean Turbo Elution Solution), c’est l’exclusion.
L’ADN obtenu est dit ADN propre car il a été purifié de tout polluant. Il servira de matrice pour toutes les analyses moléculaires après avoir évalué la concentration par fluorométrie.
Quatre échantillons purifiés en 3 répétitions ont été obtenus avec cette méthode standard. Ils ont été codés S.
Les trois kits testés et présentés ensuite contiennent des solutions dont les compositions sont inconnues pour ne pas les fabriquer en interne. Ils contiennent également différentes sortes de colonnes et des tubes.
Les protocoles des kits détaillés ci-dessous sont initialement vendus pour extraire l’ADN à partir du sol. Puisque le protocole d’extraction d’ADN de la plateforme est certifié jusqu’à obtention de l’ADN dit « sale », il a été choisi d’utiliser ces kits à partir de l’étape de purification et à partir de 100µL d’ADN sale comme pour le protocole standard.
Les notices des différents kits testés sont fournies en anglais. Elles ont été traduites avant toute manipulation.
b. Kit de purification : ZymoBIOMICS DNA Mini Kit (Z)
Ce kit a été fourni par Zymo research gratuitement en échange de résultats. Il est basé sur le même principe que le kit Geneclean expliqué précédemment.
Annexe 2 : Protocole du kit en anglais
L’ADN sale extrait est déposé à raison de 100 µL dans une colonne de purification Zymo-Spin IV Filter qui permet de retenir les plus grosses impuretés contenues dans l’ADN après centrifugation. La colonne est ensuite jetée et un tampon ZymoBIOMICS DNA Binding Buffer est mélangé à l’ADN ce qui va permettre de retenir l’ADN dans la colonne suivante, c’est le principe d’affinité. Ce mélange est donc introduit dans une nouvelle colonne Zymo-Spin IIIC-Z.
Figure 5 : Purification de l’ADN sale avec le kit Geneclean T. Regnier
L’ADN fixé sur la membrane de la colonne est alors lavé avec différentes solutions. Un premier lavage avec le tampon ZymoBIOMICS DNA Wash Buffer 1 et un lavage avec un tampon à base d’éthanol ZymoBIOMICS DNA Wash Buffer 2. L’ADN est ensuite élué avec 100µL de ZymoBIOMICS DNase/RNase Free Water. L’ADN recueillit est pré-purifié.
En parallèle, il faut préparer la colonne ZymoBIOMICS IV - HRC Spin Filter en la mettant à centrifuger à vide puis en ajoutant 400µL de ZymoBIOMICS DNase/RNase Free Water.
L’ADN pré-purifié récupéré précédemment est donc transféré dans la colonne ZymoBIOMICS IV - HRC Spin Filter préalablement préparée. L’ADN est ensuite élué dans 30 µL. L’ADN obtenu est donc de l’ADN propre qui servira pour les mêmes analyses que l’ADN purifié obtenu avec le kit GeneClean.
c. Kit de purification : NucleoSpin Soil (N)
Annexe 3 : Protocole du kit en anglais
Ce kit vient du fournisseur et fabriquant Macherey-Nagel, il a été fourni gratuitement en échange de résultats. Il est également basé sur le principe d’affinité exclusion.
Pour ce kit, seul 3 échantillons de 3 sols ont pu être purifiés. Il n’y avait pas assez de colonnes, de tubes et de solution pour 12 analyses dans le kit gratuit. Il a donc été décidé d’enlever un sol puisque deux sols présentaient les mêmes caractéristiques, le sol 194 a été éliminé pour réaliser l’alnalyse avec 3 sols dont les couleurs étaient différentes.
100 µL d’ADN sale obtenu ont été introduits dans une colonne NucleoSpin Inhibitor Removal Column qui permet d’enlever les plus grosses impuretés contenues dans l’ADN. La colonne est jetée et l’ADN récupéré après centrifugation est mélangé à du tampon Buffer SB. Ce mélange est ensuite transféré dans une colonne NucleoSpin Soil Column et va permettre de retenir l’ADN sur la membrane de silice.
L’ADN fixé sur la membrane est ensuite lavé avec plusieurs solutions de lavage, avec un tampon Buffer SB, un tampon Buffer SW1 et avec une solution à base d’éthanol Buffer SW2. 60µL d’ADN sont ensuite élué avec la solution Buffer SE. L’ADN obtenu est donc de l’ADN dit « propre ».
d. Kit de purification : PowerLyzer PowerSoil DNA Isolation Kit (P)
Annexe 4 : Protocole du kit en anglais
Le vendeur de ce kit est la société Qiagen. Le fabriquant est MoBIO.
Le protocole de ce kit est légèrement différent des autres protocoles. Au départ, il faut mélanger 100 µL d’ADN sale obtenu précédemment à la solution C2 pour précipiter les protéines. Après centrifugation, Le surnageant est transvasé dans un nouveau tube où est ajouté 200µL de solution C3 qui sert à éliminer les inhibiteurs.
La même étape que précédemment est réalisée mais en ajoutant la solution C4 ce qui permettra à l’ADN de rester liée à la membrane. Le tout est transvasé dans la colonne Spin Filter puis l’ADN fixé à la membrane est lavé avec une solution à base d’éthanol appelé C5 et 60 µL d’ADN sont élués grâce à la solution C6. L’ADN obtenu est de l’ADN propre. Page de gauche, figure 6
3) Analyse de la qualité de l’ADN par électrophorèse sur gel d’agarose
L’électrophorèse en gel d’agarose est réalisée à partir de l’ADN purifié afin de vérifier la bonne qualité ou non des ADN.
Le principe d’une électrophorèse se trouve sous forme de schéma (figure 7) si dessous :
C’est une méthode qui permet de séparer des molécules à l’aide d’un champ électrique, la migration s’effectue selon la charge et la taille des molécules :
- Les molécules chargées négativement migrent vers l’anode (pôle +)
- Les petites molécules migrent plus loin car elles sont moins retardées par le réseau d’agar polymérisé obtenu avec le mélange d’agarose et de tampon TBE.
o Mode opératoire :
Un gel d’agarose (polyoside provenant d’une algue) à 1% est préparé à chaud dans du tampon TBE 1X (Tris Borate EDTA) avant d’être coulé sur un support à électrophorèse. Une fois polymérisé, le gel est plongé dans le tampon TBE dans une cuve à électrophorèse.
Les échantillons dilués au 1/5, sont ensuite déposés dans le gel à l’aide d’une micropipette. Ils sont préalablement additionnés de tampon de charge, une solution de bleu de bromophénol afin qu’ils soient assez denses afin de tomber au fond du puits. Il sert également de à visualiser le front de migration. Ces dépôts sont encadrés par des marqueurs de taille (solution de fragments d’ADN de tailles précises) qui permettent d’évaluer la taille de l’ADN déposé.
La migration sur le gel est laissée à courant constant de 70 mA et 100V pendant 2-3 heures.
o Coloration des gels et révélation de l’ADN au BET :
Afin de visualiser les acides nucléiques, le gel est soumis à un bain de bromure d’éthidium (BET) durant 15 à 30 minutes puis dans un bain d’eau pour désaturer le BET qui est une molécule qui s’intercale grâce à des liaisons faibles entre les paires de bases azotées de l’ADN ou de l’ARN.
Le BET émet une fluorescence sous ultra-violet (UV) quand il est lié à l’ADN. Cette propriété est utilisée pour les visualiser sous UV dans un appareil nommé imageur. Cet appareil est une chambre noire composée d’une table à UV reliée à une caméra qui transfère l’image sur un écran. Ainsi, il est possible de prendre en photo le gel contenant les ADN et l’image est récupérée directement via le réseau informatique.
Le BET étant est un produit hautement toxique, la manipulation de cette molécule doit donc se faire avec haute précaution. Annexe 2 : fiche sécurité
Si les ADN sont de bonnes qualités, une bande sera observée dans chaque puits. En revanche, si les ADN sont de moins bonne qualité, les pistes du gel seront « smireuses », signe que les ADN génomique ont été dégradés lors de la purification.
4) Dosage des ADN propre par fluorescence
Ce dosage est réalisé à partir de l’ADN propre obtenu grâce aux différents protocoles de purification et permet d’évaluer la quantité d’ADN extrait par un gramme de sol.
Le principe de cette méthode et de doser par fluorescence des ADN colorés avec un fluorochrome, le picogreen. Cette molécule est très sensible et s’intercale à l’ADN double brin. Il fluoresce à une émission de 538 nm après avoir été excité à une longueur d’onde de 485nm.
La fluorescence mesurée est proportionnelle à la quantité d’ADN ce qui permet le dosage en présence d’une gamme d’ADN connue.
Les ADN sont dosés dans une plaque de qualité optique dans un lecteur de plaque nommé Pro 200 de marque TECAN.
La plaque est alimentée par une gamme d’ADN de phage Lambda, de témoins et des ADN à doser :
- La gamme est réalisée à partir d’un ADN de phage lambda fournit dans le kit dans du tampon TE 1x (Tris-EDTA). Elle est constituée de 6 points entre 2 et 0.015 ng/µL.
- Les échantillons d’ADN à doser sont dilués au 1/5 après purification dans de l’eau.
- Les témoins servent à valider l’analyse. L’analyse comprend un témoin négatif composé uniquement de TE qui permet également d’identifier le bruit de fond. Un second témoin référencé est ajouté à l’analyse. Il s’agit d’un ADN de thymus de veau fournit dans le kit Fluorescent DNA Quantification préparé à 2ng/µL. L’appareil vérifiera que sa concentration est comprise entre 1 et 3 ng/µL pour valider l’analyse.
La préparation de la plaque suit la logique du tableau suivant :
Par puits : Gamme ADN Témoin négatif Témoin référence
V ADN (µL) 50 5 0 5
V Picogreen (µL) 50 50 50 50
V TE1X (µL) 0 45 50 45
Le lecteur de plaque lit directement la fluorescence dans chaque puits de la plaque préparée selon le tableau précédent. Le logiciel Magellan associé à l’appareil a été programmé pour fournir directement les concentrations en ng/µL dans les échantillons.
5) Vérification de l’inhibition par la qPCR ou PCR quantitative
Au sein de la plateforme Genosol, les analyses de PCR quantitative (qPCR) sont utilisées pour évaluer une quantité des gènes 16s et 18s dans les ADN du sol. Le dénombrement de ces gènes permet la densité microbienne dans les échantillons par évaluation du nombre de copies d’ADN ribosomal 16s des bactéries et 18s des champignons. Ainsi la plateforme diagnostique l’équilibre microbien dans les échantillons.
La PCR quantitative est une technique basée sur le principe de la PCR (Polymerase Chain Réaction) classique.
a. La PCR classique ou PCR en point final
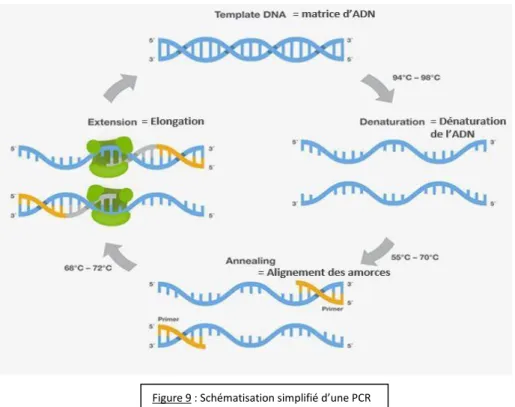
La PCR est une technique qui permet l’amplification rapide et en grande quantité d’une séquence spécifique d’un gène. Page de gauche figure 9 : principe simplifié de la PCR.
Pour réaliser une PCR, plusieurs composants sont indispensables :
Fi gur e 11 : P ri nc ipe d u Sy br G re en S. PE RR IN
Ensuite, la PCR s’effectue en plusieurs étapes dans un thermocycleur qui effectue des cycles de température pour multiplier l’ADN :
Etape 1, dénaturation initiale de l’ADN : L’ADN double brin est séparé en deux brins d’ADN constitutifs. Cette étape permet aussi l’activation de la Taq Polymerase qui change de conformation pour être active. Cette étape est réalisée à 95°C pendant 10 à 15 minutes.
Etape 2, dénaturation de l’ADN : Cette étape permet la séparation de l’ADN double brin, elle se fait à 94 °C.
Etape 3, hybridation des amorces : Les amorces se fixe de manière spécifique avec la séquence complémentaire de l’ADN grâce à la Tm qui est la température d’hybridation optimale de l’amorce, elle se situe entre 55 et 65 °C. La Tm se calcul par rapport aux nombres de nucléotides : Tm = 2°C*(nombre de bases AT de l’amorces) + 4°C*(nombre de bases CG de l’amorces)
Etape 4, élongation : C’est la synthèse du brin d’ADN complémentaire par la Taq Polymerase, la température optimale pour cette étape est de 72 °C.
Les étapes 2, 3 et 4 constituent un cycle PCR et sont répétées 35 à 40 fois. A la fin de chaque cycle, la quantité du gène souhaitée augmente de 2n (n est le nombre d’amplification).
b. La PCR quantitative
La PCR quantitative utilise donc le même principe que la PCR classique mais elle permet la lecture en temps réel du produit amplifié tout au long de la réaction grâce à la présence de fluorochrome. Cette technique est basée sur la détection de la fluorescence émise par les produits PCR en début de phase exponentielle (pour une plus grande précision).
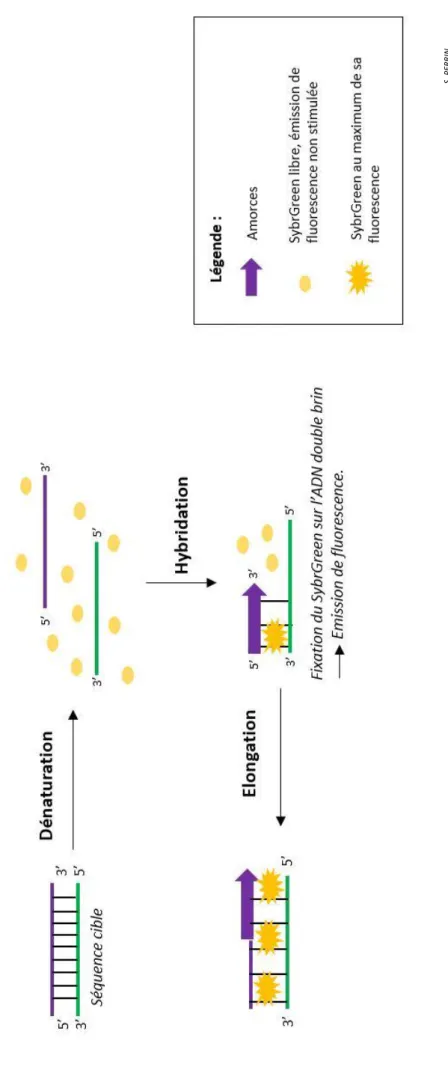
Le fluorochrome utilisé au laboratoire est le SybrGreen qui se lie directement à l’ADN double brin. Cette molécule est donc un intercalant de l’ADN double brin qui fluoresce à 535 nm sous une longueur d’onde d’excitation de 485 nm.
Cette mesure est effectuée à la fin de chaque cycle d’amplification puisque l’ADN amplifiée y est à l’état de double brin et que la fluorescence mesurée est proportionnelle à la quantité de produit PCR formé.
Page de gauche, figure 11 : Principe du SybrGreen
Figure 10 : Schématisation d’un cycle PCR
Pour effectuer cette qPCR, ce sont les mêmes éléments que la PCR classique mais avec ajout du SybrGreen. Les principaux composants de la PCR (Taq Polymerase, dNTPS, tampon) et le SybrGreen sont préalablement mélangés pour un gain de temps, ce mélange est appelé Mix qPCR. Dans un tube, ce mix, les amorces (515R et 341F pour une amplification du gène 16s ou FR1 et FF390 pour l’amplification du gène 18s), de l’eau et la T4 sont ajoutés. Le mélange est transféré dans une plaque optique 96 puits pour qPCR. Cette manipulation doit se faire sous enceinte PCR avec flux qui a été préalablement exposée aux UV pour dégrader l’ADN environnant afin d’éviter les éventuelles contaminations d’ADN et parce que le SybrGreen est un intercalant de l’ADN, donc une molécule mutagène.
Les ADN propres récupérés par purification ont été dilué en gradient de concentration à 0.5 ; 2 ; 5 et 10 ng/µL pour effectuer un test d’inhibition. Ces ADN dilués sont déposés dans les puits à raison de 2µL.
En parallèle, une gamme d’ADN plasmidique provenant d’Escherichia coli doit être préparée. 6 points de gammes sont préparés de 0.5*108 copies/µL à 0.5*103 copies/µL à partir d’une solution mère à 0.5*109 copies/µL pour une amplification des gènes 16s et 6 points de gammes sont préparés de 0.5*107 copies/µL à 0.5*102 copies/µL à partir d’une solution mère à 0.5*108 copies/µL pour une amplification du gène 18s.
La préparation de la plaque terminée, celle-ci est mise dans l’appareil pour la lecture des résultats que l’on peut suivre en direct sur le logiciel StepOne Plus, la lecture s’arrête au bout de 35 cycles.
Figure 12: Plan de plaque 96 puits pour la réalisation d’une qPCR
Le reste des puits : échantillons Légende :
Figure 14 : Courbe standard
Point de gamme Blancs
Echantillons
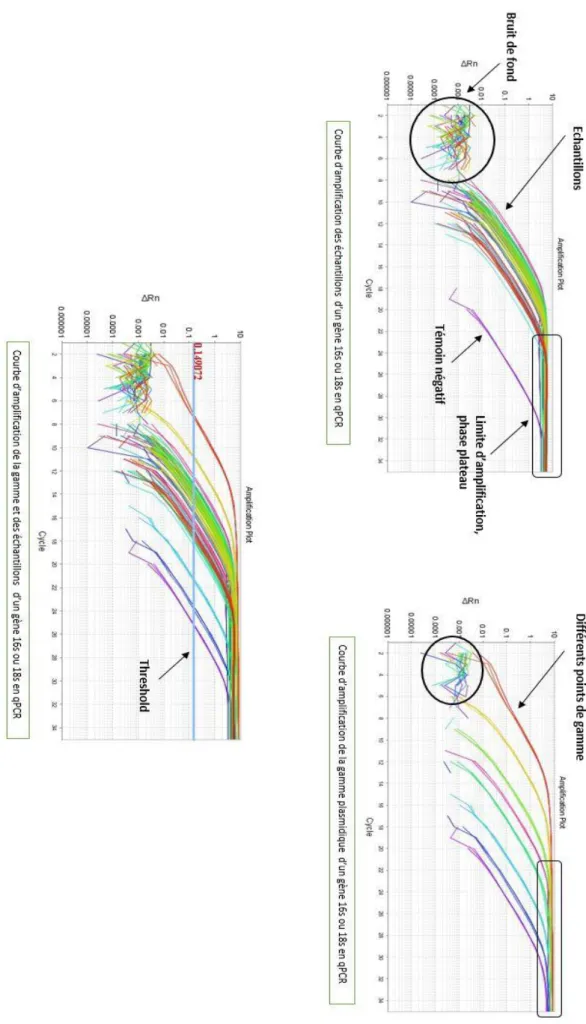
L’évolution de l’amplification du gène cible est représentée par la fluorescence détectée sous la forme de courbe. Sur cette courbe il y a trois phases (figure 13) :
Un bruit de fond dans lequel la fluorescence détectée est négligeable ou non détectable par la machine.
Une phase exponentielle, c’est là où il y a doublement du nombre de produits PCR à chaque cycle. C’est également à cette phase qu’est calculé le cycle de Threshold (Ct). Le threshold étant une valeur 10 fois supérieure au bruit de fond, c’est le début de la phase exponentielle, il est défini comme la limite de détection de l’appareil. Il correspond donc aux cycles ayant émis de la fluorescence car plusieurs cycles d’amplification du gène se suivent jusqu’à ce que la fluorescence des molécules amplifiées surpasse le bruit de fond, au point Ct.
Pour finir, il y a une phase plateau quand les composants de la PCR deviennent limitants.
Page de gauche, figure 13 : Courbe d’amplification de la gamme et des échantillons d’un gène.
Cette première courbe montre alors l’augmentation de la fluorescence en fonction du nombre de cycles. Il faut s’assurer qu’il y ait 1 log=3,3 entre chaque point de gamme c’est-à-dire 3.3 cycles entre chaque point pour la validation de la plaque. Cette courbe sert donc à valider la qPCR.
En parallèle, le logiciel calcule une courbe standard (figure 14) pour une autre validation de la qPCR. Les Ct des points de gammes et des échantillons de la courbe précédente ont été reportés sur la droite standard. Une droite des Ct en fonction du log de la quantité est alors obtenue. En traçant ces points sur cette courbe, la linéarité, l’efficacité, la sensibilité et la reproductibilité de nos essais sont ainsi déterminés.
Pour finir la validation des échantillons, l’appareil réalise une courbe de fusion appelé « Melt Curve » en fin de PCR pour évaluer la spécificité de la réaction. On évalue la spécificité car lors de de l’amplification, il peut y avoir des amplifications de contaminants, non spécifiques, anormales ou des productions de dimères d’amorces qui peuvent fausser la fluorescence et donc fausser les résultats.
La Melt Curve se fait en 3 étapes, une dénaturation des échantillons où la fluorescence est nulle du fait que le SybrGreen ne fluoresce qu’en double brin. Cette dénaturation est suivie d’une renaturation progressive de 0.5°C par minutes qui va montrer un pic de fluorescence. Pour finir, il y a une dénaturation qui va permettre à la fluorescence de diminuer, ce qui aboutira à la formation d’un pic.
La Melt Curve a donc pour principe une dénaturation progressive du matériel génétique présent dans les puits après amplification du gène suivi d’une lecture de l’absorbance après chaque augmentation de température.
III) Résultats
1) Validation de la quantité de l’ADN par dosage fluorométrique
Une analyse par dosage fluorométrique a été réalisée pour vérifier la quantité d’ADN après purification. A la fin du dosage, le logiciel fournit des données brutes qui sont exportées après avoir validé les résultats obtenus. Une courbe de la quantité d’ADN en ng/µL en fonction de la fluorescence est tracée et la validation se fait grâce au r2 et par la validation de la référence qui doit être comprise entre 1 et 2 ng/µL.
Pics des différents points de gammes
Pics des échantillons
Amplifications des amorces présentes dans la gamme Amplifications des amorces présentes dans les échantillons
Figure 15 : MeltCurve
0 20 40 60 80 100 194 238 189 208 Qu an tité d 'A D N e n n g/ µ L Sol
Comparaison de la quantité d'ADN récupéré
Protocole standard versus kits testés
S Z N P
Pour réaliser ce dosage, les 4 échantillons de sols purifiés avec les 4 protocoles ont été analysés en 3 répétions chacun, 1 plaque a été utilisé.
A partir des données brutes un graphique a été réalisé en faisant une moyenne des 3 répétitions de chaque échantillons et pour chaque protocole.
D’après le graphique, les quantités d’ADN observées entre les kits testés et le protocole standard sont différentes. Les ADN issus du kit N présentes une quantité nettement supérieure que les autres.
2) Validation de la qualité de l’ADN
a. Analyse de la qualité de l’ADN par électrophorèse sur gel d’agarose
Une analyse par électrophorèse a donc été réalisée pour vérifier la qualité des ADN après purification par les différents kits
Un gel d’agarose par électrophorèse a été réalisé pour chacun des quatre échantillons. Sur la figure ci-dessous, ont été déposés 3 répétitions de l’échantillon 238 de chaque protocole testé en parallèle à un marqueur de taille. Quelques soit l’échantillon, les résultats obtenus sont identiques.
Figure 16 : Résultat obtenus après une électrophorèse en gel d’agarose et une révélation au BET.
S Z N P ADN
ARN
Déchets et tâche de colorant Ladder
1Kb
Sur les gels les ADN ont bien été séparés, des ARN (encadrés en rouge) et des déchets (acides humiques encadrés en orange).
D’après cette photo, il n’y a pas d’impact sur la qualité des ADN concernant le mode de purification. En effet, seul la force du signal augmente dans les puits ce qui signifie qu’il y a plus d’ADN dans les échantillons purifiés dans certains kits.
Il est difficile de conclure sur la qualité des ADN avec cette méthode. La qualité sera évaluée par PCR quantitative.
b. Analyse de la qualité de l’ADN par test d’inhibition par PCR quantitative
Un test d’inhibition est réalisé pour vérifier la qualité de l’ADN en les diluant à 0.5 ; 2 ; 5 et 10 ng. Un ADN est de bonne qualité lorsqu’aucun inhibiteur de PCR n’empêche la réaction d’amplification de PCR. Une quantité d’ADN trop importante peut être un inhibiteur de PCR comme des polluants co-extraits avec l’ADN mal éliminés par la méthode de purification.
Pour ce test, les 4 échantillons ont été analysés en les diluant chacun à 0.5 ; 2 ; 5 et 10 ng en 1 répétition. 3 plaques qPCR ont donc été réalisées.
Avant d’exporter les résultats et de les analyser, il faut valider différents paramètre de l’analyse. o Validation de la PCR quantitative :
Validation de la gamme : Il faut analyser l’Amplification Plot et la courbe standard pour valider la gamme.
Pour valider la gamme, il doit y avoir un logarithme (1 log=1 carré sur le graphique) entre chaque point de gamme. La gamme doit être homogène c’est-à-dire des dilutions successives au 1/10e et les répétitions doivent être cohérente.
Le coefficient de corrélation est supérieur à 0.990, il est de 0.998 cela signifie qu’il y a une bonne linéarité. L’efficacité de la PCR est de 98,012% sachant qu’elle doit être comprise entre 85% et 110%. Le slope doit être compris entre -3,1 et -3,74, il est de 3,371.
Le coefficient de corrélation est supérieur à 0.990, il est de 0.998 cela signifie qu’il y a une bonne linéarité. L’efficacité de la PCR est de 98,012% sachant qu’elle doit être comprise entre 85% et 110%. Le slope doit être compris entre -3,1 et -3,74, il est de 3,371.
La gamme est donc validée. Les échantillons peuvent donc être analysés pour être validés à leur tour. La validation des échantillons se fait par :
- Le témoin négatif, tous les échantillons doivent se situer à au moins 1 log de celui-ci.
- La droite d’étalonnage, tous les échantillons doivent être compris dans l’intervalle défini par la gamme.
- La Melt-Curve, un seul pic doit être observable au niveau de la gamme et 1 pic décalé de la gamme doit être observable pour les échantillons.
Les échantillons sont donc validés, les analyses sur les tests d’inhibition peuvent être réalisées.
Témoins négatifs
VALIDE
VALIDE
10 ng
5 ng 2 ng
0,5
o Résltats du test d’inhibition :
Sur la figure ci-dessous sont représentées les courbes d’amplification (phases plateau) d’un échantillon apporté en ces 4 quantités différentes dans l’analyse. Quelques soit l’échantillon, les résultats obtenus sont identiques.
Ces courbes montrent un même niveau de phase plateau pour les dilutions à 0,5 ; 2 et 5 ng. En revanche, le plateau pour la dilution à 10ng est atteint plus vite. Il y a beaucoup de produits ADN apporté dans la réaction que cela gêne la bonne réalisation de l’amplification, la phase plateau arrive alors plus rapidement.
Quel que soit le protocole utilisé, une inhibition de PCR est constatée à 10ng quoiqu’elle ne soit pas systématique pour les kits N, S et Z.
Pour mieux visualiser les résultats, une courbe de la quantité d’ADN apporté dans le puits initialement en fonction du nombre de copies par puits est tracée pour vérifier qu’il y a bien une absence d’inhibition de 0,5 à 5 ng.
Le coefficient de corrélation est supérieur à 0,9, il vaut 0,9998 cela signifie qu’il y a une bonne linéarité donc qu’il y a une absence d’inhibition entre 0,5 et 5 ng. Cette inhibition signifie que l’ADN est de bonne qualité puisqu’aucun inhibiteurs de PCR n’empêche la réaction d’amplification de PCR avant 10ng. En effet les réactions de PCR quantitatives sont également effectuées sur des quantités d’ADN très faible et inférieures à 5ng.
R² = 0,9998 0 1 2 3 4 5 6
0,00E+00 1,00E+05 2,00E+05 3,00E+05 4,00E+05 5,00E+05
Q u an ti té d 'A DN ap p o rt é/p u its ( n g ) Nombre de copies/puits
Vérification de l'absence d'inhibition de l'ADN
par qPCR
Courbe d’amplification PCR pour l’échantillon 189 purifié avec le protocole N
0,00E+00 1,00E+05 2,00E+05 3,00E+05 4,00E+05 5,00E+05 6,00E+05 7,00E+05 8,00E+05 194 238 189 208 N o m b re d e cop ie s/n g Sol
Comparaison de la densité microbienne
Protocole standard versus kits testés
S Z N P
3) Validation de l’outil de diagnostic de routine par PCR quantitative
Dans le protocole standard de PCR quantitative, l’ADN est ajouté à raison de 2 ng/puits. Cette même analyse de routine a donc été effectuée sur les ADN propre obtenus avec les différents kits.
Le graphique ci-dessous montre la comparaison de densité microbienne obtenue avec le protocole standard par rapport aux kits testés.
Ce graphique montre que la densité microbienne des sols 189 et 208 est similaire. La densité microbienne ne varie pas du protocole standard par rapport aux kits de purification.
Les résultats de densités microbiennes du kit P pour le sol 194 et celle du S pour le sol 238 proviendraient d’une erreur de manipulation.
CONCLUSION
I) Conclusion scientifique
Ce stage rentrait dans le cadre d’une veille technologique dont le but était de tester différents kits de purification d’ADN afin de remplacer le protocole standard habituelle au PVPP et GeneClean qui pose problème actuellement au sein de la plateforme. La plateforme voudrait donc valider au moins un kit qui permette d’obtenir des ADN purifiés de qualité, en quantité suffisante et dont la nouvelle purification n’ait pas d’impact sur les résultats habituellement obtenus avec le protocole standard. Le temps de manipulation, le coût des kits et la disponibilité du fournisseur sont également des critères afin de trouver le meilleur kit.
Grâce aux résultats obtenus, on remarque que la quantité d’ADN varie entre les kits testés et le protocole standard. Le kit NucleoSpin soil et ZymoBIOMICS DNA Mini kit donne une quantité d’ADN plus importante alors que le kit PowerLyzer donne autant voire moins d’ADN que le protocole standard.
Pour la qualité de l’ADN, sur le gel comme avec la PCR quantitative, les kits donnent tous un ADN de la même qualité que le protocole standard.
Pour savoir quel kit de purification conviendrait le mieux afin de remplacer le protocole standard, un tableau récapitulatif est réalisé pour montrer les critères qui sont souhaités par la plateforme.
Protocole standard (PVPP + GeneClean)
ZymoBIOMICS DNA Mini kit
NucleoSpin soil PowerLyzer DNA Isolation Qualité qPCR + + + + Gel + + + + Quantité (fluorométrie) + +++ ++++ +/- Outils diagnostics de routine + + + + Temps - ++ ++ +/-
Prix (en €/échantillons) 3,443 6,38 3,447 4.04
Fournisseur
Nom MPbio
Sigma Ozyme
Macherey-Nagel Qiagen
SAV Etranger Etranger Local Etranger
Contact National National Local National
En conclusion des résultats ci-dessus, les kits ZymoBIOMICS DNA Mini kit et NucleoSpin soil seraient les kits les plus avantageux. En revanche, le kit NucleoSpin soil a un SAV qui est local qui peut donc intervenir rapidement en cas de problème et la purification peut se faire en colonne simple ou en plaque de 96 colonnes qui est un gain de temps encore plus avantageux.
Après ce stage et en vue des résultats obtenus, la plateforme souhaite approfondir les analyses sur le kit NucleoSpin soil. La plateforme envoie donc des banques de gènes à analyser, si les résultats sont les même qu’avec un ADN purifié avec le protocole standard, la plateforme utilisera ce kit pour effectuer la purification.
II) Conclusion personnelle
J’ai eu la chance d’effectuer mon stage au sein de l’INRA qui est un grand centre de recherche en France. Avoir effectué mon stage dans cette structure et avoir pu accéder à ces laboratoires étaient une bonne opportunité pour moi. Au départ je n’avais aucun rapport avec l’AgroEcologie mais grâce à ce stage ce domaine m’a beaucoup intéressé et j’ai pu apprendre de nombreuses choses que j’aimerais approfondir davantage.
Ce stage m’a permis d’avoir un regard réel sur le milieu du travail et plus particulièrement sur le domaine de la recherche en laboratoire, avec les inconvénients que l’on peut y rencontrer. J’ai été initié sur la sécurité au laboratoire et sur la gestion des déchets. J’ai également eu l’occasion de travailler au-delà de mon sujet de stage en étant intégrée dans la vie de la plateforme en participant aux réunions d’équipe, en travaillant sur la qualité puisque le Centre de Ressources Génétiques (CRG) de la plateforme est certifié ISO9001.
Au niveau du travail au laboratoire, j’ai réussi à prendre plus confiance en moi grâce à la confiance que l’on m’a très vite accordé. Ainsi, j’ai pris de l’assurance, de l’autonomie, j’ai réussi à gérer mon stress et à ne pas baisser les bras devant la difficulté. J’ai pu appliquer les enseignements techniques et théoriques étudiés en cours tout au long de cette première année de BTS ce qui m’a permis de ne pas être perdue. Travailler à la paillasse était très agréable du fait que les laboratoires de la plateforme sont neufs, avec beaucoup d’espace et avec assez de matériel pour tout le monde.
Grâce à ce sujet de stage, j’ai pu approfondir de l’anglais technique en analysant les protocoles des kits de purification à tester et j’ai également dû contacter des fournisseurs pour avoir certaines informations. Je me suis donc confrontée au métier de techniciens de recherche.
Humainement le stage m’a beaucoup apporté puisque j’ai rapidement été intégré au sein de l’équipe. C’est une ambiance conviviale et chaleureuse sans quoi le stage aurait été différent.
Travailler dans un tel environnement m’a conforté dans mes choix, depuis trois ans ce métier m’intéresse et ce stage était au-dessus de mes attentes et confirme réellement ce choix, les difficultés et les problèmes rencontrés ne m’ont pas fait peur ni ne m’ont fait changer d’avis, au contraire.
Pour finir, j’ai eu la chance de prolonger ce stage sur un CDD d’un mois. Cela m’a permis de continuer ma formation, d’acquérir d’autres connaissances et apprendre de mes erreurs. J’ai aussi eu l’opportunité de former l’équipe de la plateforme GENOSOL et certaines personnes de l’UMR AgroEcologie sur la manipulation du kit NucleoSpin soil dont j’étais la seule à maîtriser.
ANNEXES
Annexe 1: Organigramme de l’UMR, répartition des services et du personnel
Corrosif
Mortel
Inflammable
Annexe 2 : Points sécurité
Produit Etape Danger Protection (risque)
Billes de silice Extraction de l’ADN
(prépération du tube pour la lyse)
Blouse, gants (contact cutané) Masque à poussière (inhalation)
Lunettes (contact oculaire)
Tris
(Trisma Base, Tris [hydroxy méthyl] aminometane) Extraction de l’ADN (Préparation du tamon de lyse) Blouse, gants Acide chlorhydrique (NaCl) Extraction de l’ADN (Préparation du tampon de lyse)
Blouse, gants (contact cutané) Travailler sous sorbonne
EDTA (EthyleneDiamineTetracetic Acid) Extraction de l’ADN (Préparation du tampon de lyse) Blouse, gants Lunettes SDS
(Sodium Dodecyl Sulfate)
Extraction de l’ADN (Préparation du tampon de lyse)
Blouse, gants Lunettes
Acétate de potassium Extraction de l’ADN
(Déprotéinisation) Blouse, gants
Lunettes
Isopropanol Extraction de l’ADN
(Précipitation) Blouse, gants
Lunettes
Travailler sous sorbonne
BET
(Bromure d’Ethidium)
Coloration des gels,
révélation de l’ADN surchausses, surmanches Blouse, gants, lunettes
Travailler sous sorbonne
PVPP
(Polyvinylpolypyrrolidone)
Purification Blouse, gants
Masque à poussière
Ethanol Purification Blouse, gants
Travailler sous sorbonne
Picogreen Dosage de l’ADN par
fluorescence Blouse, gants
Travailler sous sorbonne adapté
CybrGreen qPCR Blouse, gants
Travailler sous sorbonne
Cancérogène, mutagène, toxique Irritant
Annexe 3 : Protocole du kit de purification ZymoBIOMICS DNA Mini Kit
Annexe 4 : Protocole du kit de purification NucleoSpin Soil
PowerLyzer PowerSoil DNA Isolation Kit