The eco-evolutionary dynamics of microbial populations
By
David V anlnsberghe
B.Sc. Microbiology and Immunology University of British Columbia, 2012 Submitted to the Microbiology Graduate Program in partial fulfillment of the requirements for the degree ofDoctor of Philosophy at the
Massachusetts Institute of Technology April 2019
[JIJl\e
'2.o\4\'\
© 2019 Massachusetts Institute of Technology. All rights reserved.
,,,/
Signature redacted
Signature of Author ...
/ 7
iMicrobiology Graduate Program January 22, 2019
Signature redacted
Certified by ... .Martin F. Polz Professor of Civil and Environmental Engineering
_____,
"
Thesis SupervisorSignature redacted
Accepted by .... ~JUN 2 1
2019
LIBRARIES
ARCHIVES
1 Jacquin C. Niles Associate Professor of Biological Engineering Chair of Microbiology ProgramThe eco-evolutionary dynamics of microbial populations
David Vanlnsberghe
Submitted to the Microbiology Graduate Program in partial fulfillment of the requirements for the degree of Doctor of Philosophy
ABSTRACT
Microbes have adapted to life in complex microbial communities in a large variety of ways, and they are continually evolving to better compete in their changing environments. But identifying the conditions that a particular microbe thrives under, and how they have become adapted to those condition can be exceedingly difficult. For instance, Clostridium diffici/e became widely known for being the world's leading cause of hospital associated diarrhea, but people can also have C. difficile in their gut without developing diarrhea. Although these asymptomatic carriers are now thought to be the largest source of infection, we know very little about how these people become colonized. In the first chapter of my thesis I use publicly available microbiome survey data and a mouse model of colonization to show that C. difficile colonizes people immediately after diarrheal illnesses, suggesting C. difficile is a disturbance adapted opportunist. However, the differences between very recently diverged microbial populations that are adapted for growth in different conditions can be very difficult to detect. To address this limitation, I developed a method of identifying regions that have undergone recent selective sweeps in these populations as a means of distinguishing them, and specifically quantifying their abundance in complex environments. But part of what makes microbial evolution so difficult to interpret is the vast diversity of genes that are only shared by a fraction of all the members in a population. To better understand how these flexible regions are structured, I systematically extracted all contiguous flexible regions in nine marine Vibrio populations and compared their organization and
evolutionary histories. I found that horizontal gene transfer and social interactions have led to the evolution of modular gene clusters that mediate forms of social cooperation, metabolic tradeoffs, and make up a substantial portion of these flexible genomic regions. The observations made in these studies help us understand how microbes are organized into socially and ecologically cohesive groups, and how they have evolved to interact with complex and changing
ACKNOWLEDGEMENTS
I am extremely appreciative of all of the amazing people who have supported me in getting to this point in my life, and I can't begin to express the gratitude I feel for your friendship, guidance, and encouragement. To my advisor, Martin Polz, your enthusiastic and unwavering support made my time in your lab a profoundly enjoyable experience. Your thoughtful feedback and advice was invaluable in developing my research and helped me become a better scientist. To all of the members of the Polz lab, you have all been such spectacular friends and colleagues. From the moment I started my rotation, you all welcomed me with open arms and made me feel at home. Each of you have taught me so much, and I sincerely appreciate everything you've done to help me. I am also grateful to the past members of the Polz lab who laid the substantial
foundations that made all of my thesis work possible. To everyone else who has been kind enough to share their time, advice, and support, I am extremely thankful. Otto Cordero, Eric Alm, Jeff Gore, Libusha Kelly, and Penny Chisholm, you all provided invaluable feedback, advice, and opportunities that shaped the course of my degree. To the members of the BE Communication Lab who taught me so much about scientific communication and how to help others with their own work, I am very grateful for the opportunities you gave me.
I would also like to thank all of the people from my time at the University of British Columbia who supported and encouraged me. William Mohn, the support and freedom you gave me during
my time in your lab to try new things and make mistakes was an incredible experience, and I would not be the person I am today without it. Steven Hallam, although I never formally worked
in your lab, you always treated me like a valued member of your team and strongly advocated for my personal and professional development. Cameron Strachan, I certainly would not be where I am today without your friendship and selfless support.
To my amazing wife, Ariana, none of this would have been possible without your love and support. To my family, you taught me to see the beauty in nature and set me on the path that led me here, and I am extremely grateful for everything you have done to encourage my curiosity passion for learning along the way.
TABLE OF CONTENTS
Introduction
6
References 9
Chapter 1: Diarrheal events can trigger long-term Clostridium difficile colonization
with recurrent blooms
11
Introduction 12 Results 13 Discussion 20 Methods 23 References 26 Supplementary Figure 31
Chapter 2: A biological definition of microbial populations
32
Introduction 32 Results 35 Discussion 42 Methods 58 References 68 Supplementary Figures 73 Supplementary Tables 80
Chapter 3: Gene transfer and social interactions promote the evolution of modular
gene clusters
100
Introduction 101 Results 103 Discussion 114 Methods 116 References 119Conclusion
123
References 125INTRODUCTION
Microbiology changed forever with the introduction of cheap sequencing technologies. It suddenly became feasible to sequence dozens, then hundreds, and now thousands of microbial genomes. Microbial ecologists also leveraged new sequencing methods to study the structure of microbial communities with unprecedented resolution using 16S rRNA PCR amplicon
sequencing. But we are still trying to account for the vast diversity we observe, not only in the complexity of species representation in microbial communities that came into focus, but also the diversity within each species.
In particular, sequencing more microbial genomes only made the boundaries between groups with fundamentally different ecology harder to define because it highlighted the frequency of genetic exchange between distantly related groups of microbes (Gogarten, Doolittle and Lawrence, 2002; Tettelin et al., 2005; Cohan and Perry, 2007; Doolittle and Zhaxybayeva, 2009). For macroscopic plants and animals, these boundaries are much easier to define, because those organisms can be physically mapped and their interactions with each other and their environment can be much more easily observed. But microbes exist in much more abstract and intangible niche spaces, and have much more promiscuous genetic exchange with more distantly related organisms than macroscopic eukaryotes. Consequently, the boundaries between
fundamental units of bacterial diversity are difficult to identify, which limits our ability to understand how microbes evolve in the environment.
Although the problem of delineating the boundaries between ecologically and genetically cohesive groups of bacteria is difficult and often contentious, progress on this problem is building. A growing body of literature argues that these boundaries ought to be drawn between groups of bacterial strains with a history of co-existence and elevated rates of gene-transfer (Cohan and Perry, 2007; Cadillo-Quiroz et al., 2012; Shapiro et al., 2012). Reasoning that alleles can flow through microbial populations in a way that is consistent with macroscopic eukaryotes. However, identifying discontinuities in gene flow as a means to predicting populations remains difficult, as recent attempts have failed to differentiate very recently diverged Vibrio
cyclotrophicus and Prochlorococcus populations (Bobay and Ochman, 2017). Nevertheless, these advancements place us closer to a unified microbial species than ever before.
Another major issue that has arisen from the rapid accumulation of sequenced bacterial genomes, is that as we sequence more genomes from the same species, the amount of accessory genomic content that is only shared by a subset of strains has increased, seemingly without end (Welch et al., 2002; Medini et al., 2005; Kettler et al., 2007). Indeed, even strains that otherwise appear identical can differ substantially in gene content. Although we have been aware of this sequence diversity for quite some time, there is currently a contentious debate over whether it is generally beneficial, neutral, or deleterious to its host cells (Shapiro, 2017; Rocha, 2018). Certainly
individual examples provide adaptive functions to cells, from genomic islands with efflux pumps that confer antibiotic resistance (Van Bambeke, Balzi and Tulkens, 2000; Davies and Davies, 2010), to pathogenicity factors that enable Escherichia coli to compete in the human gut by causing diarrhea (Nougayrede et al., 2006). But these are only examples, and the enormous size and complexity of the flexible genome resists systematic categorization.
The works presented in this thesis attempt to improve our understanding of how microbial diversity is organized, and how those units evolve and interact with their environment. First, in chapter 1 I leverage the availability of publically available human microbiome sequencing data to show that the human pathogen C. difficile colonizes people following diarrheal illnesses. Following the observation that nine different people became colonized by C. difficile after Salmonella and Vibrio cholerae infections, we searched microbiome surveys of healthy people to
show C. difficile carriage is rare in people with no history of gastrointestinal disturbances. Then, by developing a mouse model of asymptomatic colonization that relied on feeding mice avirulent
C. difficile spores along with increasing doses of laxatives, we confirmed our observations in people. The findings we present in this chapter disrupt the widely held belief that C. difficile is a hospital and antibiotic associated pathogen. Our study reveals that short periods of increased intestinal flow are sufficient to trigger susceptibility to colonization, and characterizes the day-to-day dynamics of C. dif icile carriage over year-long time scales. Further, our study provides insight into why C. difficile is often detected as a co-infecting pathogen with other sources of infectious diarrhea (Murphy et al., 2017). Our results suggest that many of these co-infected patients could be asymptomatic carriers, and the secondary infection responsible for their symptoms triggered C. difficile shedding. If true, this could imply that the actual prevalence of disease caused by C. difficile is lower than current estimates.
Through a collaboration with Phil Arevalo, in the second chapter we develop a method for identifying the boundaries between microbial populations, then implement a reverse-ecology framework for identifying what differentiates closely related populations and measuring their abundance in the environment. In doing so, we reproduce predicted population structure in
Vibrio cYclotrophicus (Shapiro et al., 2012), Prochlorococcus (Kashtan et al., 2014), and Sulpholobus (Cadillo-Quiroz et al., 2012) model systems, and make novel insights into the
structure of Ruminococcus gnavus populations. Specifically, we identified regions of the genome that have recently undergone population specific sweeps in each of the three predicted
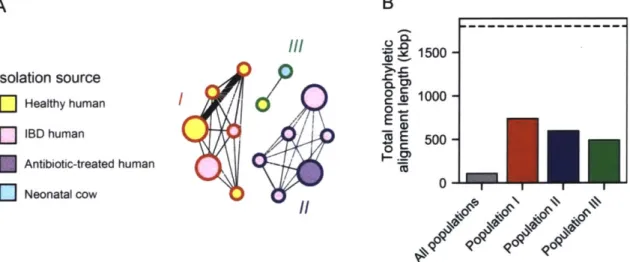
populations, and show these regions are enriched for specific functional categories of enzymes. By mapping metagenomic reads from human fecal samples of healthy and inflammatory bowel
disease patients, we show that two R. gnavus populations are differentially associated with healthy and Crohn's disease patient groups, despite less than 7% of the genome supporting a monophyletic division between all groups. In fact, less than 40% of the genome supported a monophyletic grouping for any one of the three populations. In that way, our reverse ecology
approach provided an unbiased method for delineating microbial populations and identifying the variables that may be driving their diversification without any prior knowledge of their ecology.
In the final chapter I present work that highlights the potential roles of social interactions and gene transfer in structuring the flexible genome. Using a collection of 641 genomes from nine marine Vibrio populations, we systematically identified all flexible regions and compare their
distribution and evolutionary histories. Our observations suggest most flexible regions are gained and lost as entire modules, suggesting accretionary gene gain or loss is rare. At least one quarter of the flexible loci we identified were made up of multiple modular gene clusters, where each module evolved independently and transfer allowed strains to rapidly transition between
different genotypes. We present examples of these multi-module loci that mediate social
interactions, metabolic tradeoffs, and maintain exceptionally high levels of allelic heterogeneity in populations. This modular organization and pattern of transfer enables strains to rapidly transition between different fitness peaks without having to incur the evolutionary cost of
REFERENCES
Van Bambeke, F., Balzi, E. and Tulkens, P.M. (2000) Antibiotic efflux pumps. Biochemical
Pharmacology, 60, 457-470.
Bobay, L.-M. and Ochman, H. (2017) Biological Species Are Universal across Life's Domains.
Genome Biology and Evolution, 9, 491-50 1.
Cadillo-Quiroz, H., Didelot, X., Held, N.L., Herrera, A., Darling, A., Reno, M.L., et al. (2012) Patterns of Gene Flow Define Species of Thermophilic Archaea (ed NH Barton). PLoS Biology,
10,e1001265.
Cohan, F.M. and Perry, E.B. (2007) A Systematics for Discovering the Fundamental Units of
Bacterial Diversity. Current Biology, 17, R373-R386.
Davies, J. and Davies, D. (2010) Origins and evolution of antibiotic resistance. Microbiology and
molecular biology reviews: MMBR, 74, 417-3 3.
Doolittle, W.F. and Zhaxybayeva, 0. (2009) On the origin of prokaryotic species. Genome research, 19, 744-56.
Gogarten, J.P., Doolittle, W.F. and Lawrence, J.G. (2002) Prokaryotic Evolution in Light of
Gene Transfer. Molecular Biology and Evolution, 19, 2226-223 8.
Kashtan, N., Roggensack, S.E., Rodrigue, S., Thompson, J.W., Biller, S.J., Coe, A., et al. (2014) Single-Cell Genomics Reveals Hundreds of Coexisting Subpopulations in Wild Prochlorococcus.
Science, 344, 416-420.
Kettler, G.C., Martiny, A.C., Huang, K., Zucker, J., Coleman, M.L., Rodrigue, S., et al. (2007) Patterns and Implications of Gene Gain and Loss in the Evolution of Prochlorococcus. PLoS
Genetics, 3, e231.
Medini, D., Donati, C., Tettelin, H., Masignani, V. and Rappuoli, R. (2005) The microbial
pan-genome. Current Opinion in Genetics & Development, 15, 589-594.
Murphy, C.N., Fowler, R.C., Iwen, P.C. and Fey, P.D. (2017) Evaluation of the BioFire FilmArray@ GastrointestinalPanel in a Midwestern Academic Hospital. European Journal of
Clinical Microbiology & Infectious Diseases, 36, 747-754.
Nougayrede, J.-P., Homburg, S., Taieb, F., Boury, M., Brzuszkiewicz, E., Gottschalk, G., et al. (2006) Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science (New
York, N.Y), 313, 848-5 1.
Rocha, E.P.C. (2018) Neutral Theory, Microbial Practice: Challenges in Bacterial Population
Genetics (ed S Kumar). Molecular Biology and Evolution, 35, 1338-1347.
Shapiro, B.J. (2017) The population genetics of pangenomes. Nature Microbiology, 2,
1574-1574.
Shapiro, B.J., Friedman, J., Cordero, O.X., Preheim, S.P., Timberlake, S.C., Szab6, G., et al. (2012) Population genomics of early events in the ecological differentiation of bacteria. Science
(New York, N.Y.), 336, 48-5 1.
Tettelin, H., Masignani, V., Cieslewicz, M.J., Donati, C., Medini, D., Ward, N.L., et al. (2005) Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial "pan-genome". Proceedings of the National Academy of Sciences of the United States ofAmerica, 102, 13950-5.
Welch, R.A., Burland, V., Plunkett, G., Redford, P., Roesch, P., Rasko, D., et al. (2002) Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America, 99, 17020-4.
Chapter 1:
Diarrheal events
can
trigger long-term
Clostridium difficile colonization with recurrent blooms
David VanInsberghe1, Joseph A. Elsherbini', Bernard Varian2, Susan Erdman2, Martin Polzi
'The Department of Civil and Environmental Engineering, Massachusetts Institute of Technology, 02139, USA
2The Division of Comparative Medicine, Massachusetts Institute of Technology, 02139, USA
Abstract
Although it is widely held that Clostridium difficile is an antibiotic and hospital associated pathogen, recent evidence indicates that this is an insufficient depiction of the risks and reservoirs. In fact, the rate of infection continues to rise despite the implementation of patient
isolation, increased hospital sanitation, and antibiotic stewardship protocols. A common thread that links all known major risk factors of infection is that they are all associated with
gastrointestinal disturbances, but the relationship with C. difficile colonization has never been tested directly. Here we show that disturbances caused by diarrheal events trigger susceptibility to C. difficile colonization. We first detected C. difficile blooms in human gut microbiome surveys following food poisoning and Vibrio cholerae associated diarrheal disease that had remained untreated with antibiotics. Carriers remained colonized for year-long time scales with highly variable patterns of C. difficile abundance, characterized by short one-to-two day periods of increased shedding and weeks where C. difficile was undetectable. Since the short shedding events are often linked to subsequent GI disturbances, our results help explain why C. difficile is often detected as a co-infecting pathogen in diarrhea patients. To directly test the impact of diarrhea on susceptibility to colonization, we developed a mouse model that allowed us to monitor colonization without the influence of disease caused by C. dif/7cile toxins. As mice ingested increasing quantities of laxatives while being exposed to avirulent C. dfficile spores, more mice experienced blooms of C. difficile in their feces, while control groups experience none. Our results suggest that the likelihood of colonization is highest in the days immediately following acute disturbances, where antibiotics and hospitals are common examples of large disturbances and locations with a high burden of exposure. The recovery period after diarrheal
disturbances could therefore be an important window for preventing transmission and lowering the incidence of infection.
INTRODUCTION
As the world's leading cause of diarrhea in hospitalized patients, Clostridioides (Clostridium) difficile imposes a heavy burden on healthcare systems (Loo et al., 2011; Dubberke and Olsen, 2012; Desai et al., 2016). Although C. difficile infections (CDI) have become increasingly treatable with more widespread use of fecal microbiota transplants, the incidence of infection of
C. difficile continues to rise (Reveles et al., 2014; Kelly et al., 2016). Indeed, the prevalence of C. difficile infections continues to rise despite extensive interventions that limit patient-to-patient transmission and ration the use of specific antibiotics strongly linked to developing CDI
(Bruminhent et al., 2014; Patton et al., 2017; Ray et al., 2017). These observations suggest meaningful interventions will require targeting stages along the path from colonization to disease. However, the mechanisms of colonization and factors that govern the progression to disease are not well understood.
C. difficile is often referred to as hospital and antibiotic associated (Leffler and Lamont, 2015), but there is mounting evidence to suggest these are overgeneralizations of the reservoirs and risks of transmission for this pathogen (Tschudin-Sutter et al., 2015; Crobach et al., 2018; Durovic, Widmer and Tschudin-Sutter, 2018). Fine-scaled epidemiological studies have shown that at most one third of infections in hospitalized patients can be attributed to within hospital transmission, suggesting the largest reservoirs of infection are elsewhere (Eyre et al., 2013a; Kociolek et al., 2018). Furthermore, only a few antibiotics carry a substantially elevated risk for developing CDI - particularly clindamycin, fluoroquinolones, and cephalosporins (Tedesco, Barton and Alpers, 1974; McFarland, Surawicz and Stamm, 1990; Buffie et al., 2012; Brown et
al., 2013; Dubberke et al., 2015; Guh et al., 2017). Beyond these classes of antibiotics, several non-antibiotic risk factors are important - particularly receiving an enema and using stool
softeners or gastrointestinal (GI) stimulants (McFarland et al., 1990; Leffler and Lamont, 2015). Sequencing 16S rRNA PCR amplicons to survey the composition of fecal microbial
et al., 2013; Weingarden et al., 2014; Buffie et al., 2015; Schubert, Sinani and Schloss, 2015; Zackular et al., 2016; Davis et al., 2016; Seekatz et al., 2016; Daquigan et al., 2017). The ubiquity of publicly available microbiome datasets provides a useful opportunity to rapidly determine the prevalence of C. difficile in different demographics. However, previous analyses of microbiome surveys have focused on identifying species that are positively or negatively correlated with the presence of C. difficile (Buffie et al., 2015; Daquigan et al., 2017). But determining the timing of events that precede colonization, and the dynamics of C. difficile abundance in carriers over short and long term time scales are important components of comprehensively understanding the transmission cycle of C. difficile.
Since many of the major non-antibiotic risk factors for developing CDI cause GI disturbances to varying degrees, we hypothesized that C. difficile colonization is triggered by disturbances caused by diarrheal disturbances, regardless of their etiology. To test our hypothesis, we first analyzed human gut microbiome time series studies conducted on people who had major diarrheal illnesses without being treated with antibiotics. Because we observed C. difficile colonization soon after these illnesses, we tested our hypothesis experimentally by feeding mice increasing quantities of laxatives while being exposed to non-pathogenic C. difficile spores. Together, our results show that GI disturbances create a window of susceptibility to colonization by C. difficile during the recovery period. Since carriers shed highly variable quantities of C. difficile in their feces over year-long time scales, our results further suggest that carriers are important drivers of transmission. The observation that subsequent GI disturbances trigger periods of increased shedding in C. difficile carriers also help explain why C. difficile is often detected as a co-infecting pathogen. These results challenge the commonly held assumption that C. difficile is an antibiotic and hospital associated pathogen and instead suggest antibiotics and hospitals are simply important sources of GI disturbances and locations with a high burden of C. difficile exposure.
RESULTS
C. difficile blooms in the human gut during recovery from GI infections
To determine if GI disturbances trigger colonization by C. difficile, we searched for gut
microbiome surveys with high temporal resolution and incidence of diarrheal illnesses that were not treated with antibiotics. We focused on DNA sequencing based microbial community
profiling rather than culture based analyses since DNA extraction protocols are gentle enough to target vegetative cells over spores (Wunderlin et al., 2014). As a consequence, detecting C. difficile in these data suggests the presence of an actively growing population of cells, and not
spores that are only transiently present.
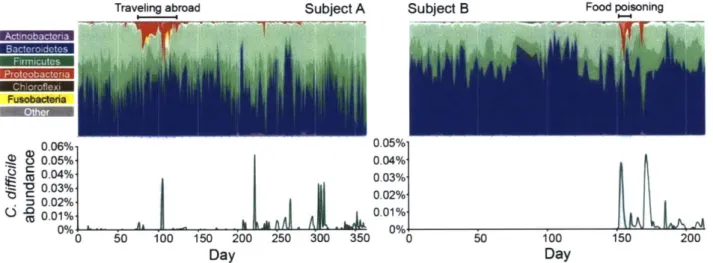
Re-analysis of a time-series study by David et al (2014), which sampled the microbiome of two adults for up to a year (291 and 159 samples total, respectively), revealed how colonization and increased shedding are linked to diarrheal disturbances (Fig.1). In subject B, C. difficile was not detectable for the first 151 days, but rose sharply in abundance two days after food poisoning
induced diarrhea, which remained untreated, suggesting colonization occurred during this period. Eighteen days after the illness began, subject B had another GI disturbance, and the abundance of C. difficile bloomed in their feces. Similarly, C. difficile bloomed immediately after subject A contracted a diarrheal illness that was not treated with antibiotics while traveling abroad.
Notably, neither subject was aware that they were colonized by C. difficile during this time.
Traveling abroad Subject A Subject B Food poisoning
Fusabeind.__ 0.06% 0.05% 0.05% 0.04% 5 0.04% 0.03%, 0.03% 0.02% . :C 0.02% S0.02%-
LA.AL
4
~ .0.01% 0.01% 0 50 100 150 200 250 300 350 0 50 100 150 200 Day DayFigure 1. C. difficile colonization and blooms are associated with recovery from diarrheal disturbances. The fecal microbial communities of two adult males were surveyed on daily intervals for up to one year using 16S rRNA PCR amplicon sequencing (David et al., 2014). Top panel summarizes the family level phylogenetic structure of the fecal microbial community over time. Different shades of the same color represent different families in the same Phyla. Bottom panel shows the relative abundance of C. difficile throughout the study.
Our re-analysis of the study by David et al (2014) also revealed the short and long tenn
dynamics of C. difficile carriage. After subject B became colonized, C. difficile persisted in their feces through the remaining 64 days of sampling. In subject A, C. difficile was present in the earliest days of sampling, suggesting the initial colonization event occurred beforehand, but similarly persisted through the entire 365 day study. During these periods, the abundance of C. difficile was highly variable from day-to-day, often fluctuating below the limit of detection to days of substantially increased shedding. Although the abundance of C. difficile was between 0.02%-0.05% during blooms, these abundances are within the range of abundances observed in actively symptomatic patients (Daquigan et al., 2017). In the context of the dense microbial community in the colon of >IO" cells/g of stool (Sender, Fuchs and Milo, 2016), even an abundance of 0.01% is substantial - implying each gram of feces has 107 C. difficile cells. Together, these observations demonstrate that people can remain colonized by C. difficile for year-long time scales, but the abundance of C. difficile during this time is highly variable and often associated with secondary diarrheal events.
Next, we analyzed a time series by Hsaio et al (2014) that sampled the feces of seven people who sought treatment for Vibrio cholerae infection in Bangladesh. Every bowel movement was collected while they cleared the infection, and after they had their first solid formed stool they were discharged from the hospital. Subjects where then sampled daily for two weeks, then weekly for several weeks after. All seven subjects showed similar patterns of community transformation and assembly - shifting from a Proteobacteria dominated community during infection, back to a healthy state dominated by Firmicutes and Bacteroidetes during recovery (Fig. 2). As well, all seven subjects had blooms of C. difficile either
just
prior to discharge, or in the days immediately after leaving the hospital. Even months after discharge, all seven subjects had detectible C. difficile in their feces. These observations provide further evidence that the critical window of susceptibility to colonization is during the early stages of recovery from an acute disturbance.BFW
1.0% 0.5%-AfJUIN C on C(0.
i
%o -Samples taken after discharged I1
iu.uui e fo hspItaJIO 3.0% bC 0% L Fusobsiwiae M o 0' 1b0 20 s Sample numberFigure 2. C. difficile colonizes patients recovering from Vibrio cholerae infections. The feces
of seven subjects who sought treatment for Vibrio cholerae infections but had not recently used
antibiotics were surveyed using 16S rRNA PCR amplicon sequencing (Hsiao et al., 2014). Every
bowel movement was collected during their hospital stay (1.5 samples taken per hour on
average), then patients were sampled daily for two weeks after discharge, then weekly for several
weeks, so sample number does not correspond linearly with time. The top panel summarizes the
family level phylogenetic structure of the fecal microbial community over time. Different shades
of the same color represent different families in the same phyla. The bottom panel shows the relative abundance of C. difficile throughout the study.
A
1.0 0.5 0CC
10 2b 30 40D
% 00.8% O 0.4% 10 20 30 Y% 0.60% % 0.3%A 110 Y lbI * 1'5 20 10 20 30 5 10 15 20 10 20 30 40 Sample number 4.0 2.0 0E
0.2 0.1 0' CU M 8 C .0 C C-) C., ciG
W
C. difficile is rare in people with no recent history of diarrhea
To determine if healthy people can become colonized by C. difficile if they are exposed without a GI disturbance, we next searched for studies that surveyed the microbiome of people who did not have recent diarrhea. These criteria first identified a study by Caporaso et al (2011) that
sampled the feces, tongue, and both palms of two adults on 332 and 130 occasions over the course of one year, and neither subject had a diarrheal event. Since C. difficile was detected on the palms of both subjects on two separate occasions each, spread throughout the year, we can
infer that these subjects were at least occasionally exposed to C. difficile in their daily activities. However, we did not detect C. difficile in the feces of either individual, suggesting that neither was infected in spite of repeated exposure.
The National Institute of Health Human Microbiome Project (NIH HMP) healthy cohort offered an additional opportunity to test whether healthy people are less likely to be colonized with C. difficile using a much larger number of subjects (NIH HMP Working Group et al., 2009). The extensive list of over 30 exclusion criteria ensures all subjects can be readily recognized as "healthy" (Aagaard et al., 2013). Importantly, these criteria excluded subjects that used antibiotics in the six months prior to sampling, and that have a history of gastrointestinal
disorders or acute illnesses. The 286 subjects were sampled up to three times each, with a total of 603 samples. We only detected C. difficile in one sample (relative abundance 0.001%) from a subject who was sampled three times, supporting that C. difficile colonization is rare in healthy people.
Laxatives facilitate C. difficile colonization in mice
To directly test the impact of diarrhea in facilitating C. difficile colonization, we developed a mouse model of tunable GI disturbance intensity that is not affected by C. difficile pathogenesis. We fed four groups of ten mice increasing quantities of osmotic laxatives while they were exposed to C. difficile spores, and monitored their feces for C. difficile blooms using 16S rRNA amplicon sequencing. The osmotic laxative polyethylene glycol 3350 (PEG3350) was chosen because it is an effective way of inducing diarrhea while minimizing impacts on the mouse, due to its low toxicity and negligible absorption, and minimal impacts on the microbiome because it is not readily metabolized (Pashankar and Bishop, 2001; Kashyap et al., 2013). PEG3350, a.k.a. MiraLAX, is available over-the-counter and often given to children due to its perceived safety.
To eliminate the influence of C. difficile toxins from exacerbating diarrheal symptoms, we used spores from a non-toxinogenic C. difficile strain (ATCC BA1801). Mice were single housed in cages with grated bottoms to minimize coprophagy and their feces was sampled daily, starting three days prior to treatment and extending for ten additional days after laxative treatment. At the end of treatment, the control group (group 1) had hard, well-formed stool pellets; group 2
(15 mg/mL PEG3350) had soft but well-formed pellets; group 3 (30 mg/mL PEG3350) had soft, but not watery pellets; and group 4 (150 mg/mL PEG3350) had loose watery stools. Groups 1-3 had no observable changes in behavior or measurable weight loss, but mice in group 4 became
inactive and lost on average 6% of their initial body weight. Accordingly, the treatment groups approximate mild laxative use (group 2), strong laxative use (group 3), and acute diarrhea (group 4).
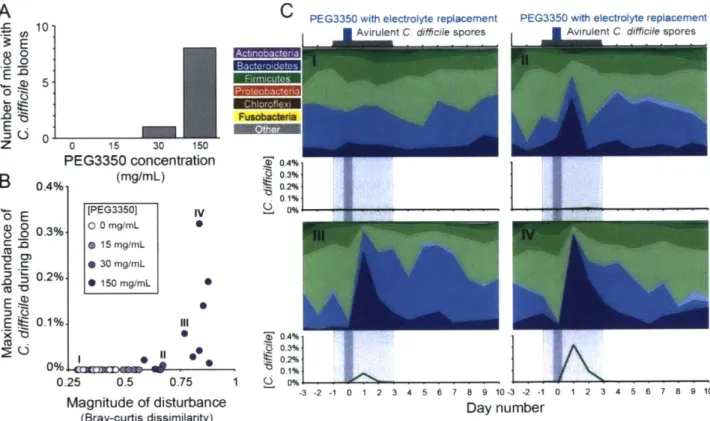
By increasing the concentration of laxatives in their drinking water, the number of mice with blooms of C. difficile in their feces, and the magnitude of those blooms, increased (Fig. 3). None
of the mice from either the control group (group 1 -0 mg/mL PEG3350) or lowest treatment group (group 2 - 15 mg/mL PEG3350) had C. difficile detectible in their feces. Whereas one mouse from group 3 (30 mg/mL PEG3350) and eight mice from group 4 (150 mg/mL PEG3350) had blooms of C. difficile in their feces. The amount of C. difficile detected also increased with the intensity of the disturbance caused by the laxative, measured by the overall change in the microbial community (Fig. 3 and S 1). Interestingly, B. thetaiotaomicron rapidly expanded to abundances of 50-90% of the microbial community the day after treatment in all mice that experienced C. difficile blooms. Although B. thetaiotaomicron did not consistently increase in abundance in the human subjects who had C. difficile blooms (Figs. I and 2). Overall, these results show that GI disturbances caused by laxatives make mice more susceptible to C. difficile colonization, corroborating our observations from humans with diarrheal disturbances.
0 .0
E n
0
C PEG3350 with electrolyte replacement 1 Avirulent C difficile spores
CO E 0 0 0 .1% I 0%L M60 -A - 0 0.25 0.5 0.75 1 Magnitude of disturbance (Bray-curtis dissimilarity) 0~ 0. 0. 0. 0
PEG3350 with electrolyte replacement
AvirulentC difficile spores
10. 0- 0 15 30 I50 PEG3350 concentration 0.4%. (mg/mL) [PEG3350] 0.3% 0 0 mg/mL 0 15 mg/mL 0 30 mg/mL 0.2%1 15 mg/mL S
Figure 3. Increasing disturbance intensity increases the likelihood and magnitude C. difficile blooms in mice. Four groups of ten C57BL/6J Mice were single housed in cages with grated bottoms. Non-toxinogenic C. difficile spores were suspended in drinking water at 104 spores/mL for one day prior, during, and two days following PEG3350 laxative treatment. Groups 1, 2, 3, and 4 had 0, 15, 30, and 150 mg/mL PEG3350 in their drinking water with electrolyte replacement for 8-10 hours only. Daily fecal samples were processed using 16S rRNA gene sequencing. (A) The number of mice in each group that had blooms of C. difficile in their feces (B) The magnitude of the disturbance is measured as the average Bray-Curtis
dissimilarity between the fecal microbial communities of each mouse on day 1 relative to the three days prior to treatment. (C) The top panels summarize the family level phylogenetic
structure of the fecal microbial community over time, where different shades of the same color represent different families in the same phyla. The lower panels show the relative abundance of
C. difficile in each mouse over time.
OA% 0.3% 02% .0.1%
V
B 0 E =C3 EU 2% -3 -2 -1 0 1 2 3 4 5 6 7 8 9 10 -3 -2 -1 0 1 2 3 Day number 4 5 6 7 8 9 10DISCUSSION
Antibiotics like clindamycin are thought to make people susceptible to CDI by disrupting barrier functions of the microbiota and altering the physical and chemical properties of the gut (Buffie et al., 2012). Here we show that this relationship is more general than previously thought by
demonstrating that diarrheal disturbances can trigger susceptibility to colonization, regardless of its etiology. By monitoring colonization in high-resolution time series we were able to resolve the timing of the critical window of susceptibility and the dynamics of C. difficile shedding in carriers over year-long time scales. These observations reconcile disparate risk factors and contribute to mounting evidence that the popular view of C. difficile as an antibiotic and hospital associated pathogen is an incomplete description of its ecology. Instead, our results suggest that
C. difficile is a disturbance associated opportunist, and colonization is tied to a history of GI disturbances caused by diarrheal events.
Our analysis highlights that the critical window of susceptibility occurs in the days immediately following the disturbance, during the recovery period. Four of the seven subjects recovering from V. cholerae infections had blooms of C. difficile while they were in the hospital just prior to having their first solid formed stool (Fig. 2). The remaining three subjects had blooms of C. difficile in their feces within days of being discharged. Likewise, all mice had their first bloom of
C. difficile within the first two days of the laxative treatment. Accordingly, the days following a diarrheal disturbance are an important target for interrupting the C. difficile transmission cycle. Our analysis of long-term time series studies reveals how carriage can persist for year-long time scales, and the burden of C. difficile shedding fluctuates substantially over that time (Fig. 1). In particular, secondary disturbances appear to consistently trigger short periods of substantially
elevated C. difficile shedding in carriers. This link to diarrheal events, regardless of their
etiology, further highlights the importance of targeting the window following GI disturbances for interrupting C. difficile transmission. Not all of the secondary blooms of C. difficile we detected were obviously associated with diarrheal illnesses, however, suggesting that once someone is a
carrier even mild disturbances could trigger increased shedding. The highly variable abundance of C. difficile within carriers over time also has major implications for diagnostics (van Prehn et al., 2015). Since C. difficile would often fluctuate below the limit of detection for several days at
a time, these data show how single-time point diagnostics can be unreliable for identifying carriers.
The observation that GI disturbances trigger transient periods of increased C. difficile shedding in carriers also helps explain why C. difficile is commonly identified as a co-infecting pathogen. The recent transition of medical laboratories to use culture-independent tests that detect dozens of different pathogens at once has revealed the large prevalence of co-infection in diarrhea patients (Halligan et al., 2014; Murphy et al., 2017). In particular, approximately 10% of patients with diarrhea will test positive for multiple GI pathogens, and 40% of those cases identify C. difficile as a source of co-infection (Murphy et al., 2017). However, it isn't clear if C. difficile is a driver of disease symptoms in these patients. Our results suggest that many of these co-infected patients could be asymptomatic carriers, and the secondary infection responsible for their
symptoms triggered C. difficile shedding. If true, this could imply that the actual prevalence of disease caused by C. difficile is lower than current estimates.
Diarrhea may be sufficient to stimulate susceptibility to C. difficile colonization, but none of the nine subjects identified here as long-term carriers developed symptoms. This could indicate that the factors responsible for triggering the progression to disease are separate from those that facilitate colonization. Most antibiotics are associated with similar increased relative risk for developing CDI as receiving an enema or taking stool softeners and GI stimulants (McFarland et al., 1990). But clindamycin, fluoroquinolones, and cephalosporin antibiotics have a substantially elevated risk for developing CDI (Guh et al., 2017). Since diarrhea is a common side effect of antibiotics (Bartlett, 1992), it is possible that many antibiotics primarily promote susceptibility to colonization by causing intestinal disturbances. While only a few antibiotics also affect the necessary changes to the physical, chemical, and microbial properties of gut to permit the progression to disease, such as altering the bile acid composition in the colon (Allegretti et al., 2016).
Although all of the people highlighted in this study were asymptomatic carriers, we do not know if any became symptomatic after these studies ended, or whether they transmitted C. difficile to others. Consequently, it will be important to determine how often these long-term carriers develop symptoms or act as sources of infection for others. There is evidence that C. difficile colonization reduces the likelihood of developing CDI, particularly if the strain is
non-toxinogenic (Shim et al., 1998; Nagaro et al., 2013). So if these carriers became colonized with a non-pathogenic strain of C. difficile, they could become resistant to infection when challenged by a different, virulent strain. But if the colonizing strain is from a hyper-virulent clade of C.
difficile, these carriers could feasibly transition to a disease state if conditions in the bowel change. Indeed, most asymptomatic C. difficile carriers harbor toxinogenic strains (Eyre et al., 2013b; Alasmari et al., 2014). Even if people who are asymptomatically colonized are less likely to develop CDI, regardless of the colonizing genotype, the pattern of recurrent blooms
accompanied by increased shedding suggests they are important drivers of transmission. A recent study by Tropini et al. (2018) showed that adding PEG3350 to the drinking water of mice at the same concentration as our highest treatment group (150 mg/mL) for six full days had persistent and long term effects on the microbiota. After only ten hours of the same treatment, we also observed changes to the microbiota that persisted for at least 10 days after treatment (Figs. 3 and Si). This observation highlights how even mild diarrheal events can cause long term
changes to the microbiota, and that the majority of changes occur very quickly. However, our lowest treatment group (15 mg/mL) was often indistinguishable from control samples in terms of the scale of the changes observed in the microbiota composition after treatment. Since our lowest treatment group was an approximation of mild laxative use, and none of those mice experienced C. difficile blooms, our study suggests there is little danger in limited prescribed use of laxatives but underscore the dangers of abusive use.
The perspective that C. difjicile is hospital and antibiotic associated has substantially shaped responses to the escalating rate of CDI. Considering mounting evidence that this view
inaccurately describes the ecology of C. difficile, our results suggest that future interventions will need to be more conscientious of the role that non-antibiotic GI disturbances have in the C. difficile transmission cycle. In particular, our analyses emphasize the importance of targeting the critical window of susceptibility in the days immediately following a diarrheal events. Each year in the United States roughly 48 million people contract a foodbome illness (Scallan et al., 2011), highlighting the days immediately following illnesses like these as a substantial target for
reducing the prevalence of C. dfficile. The development of new pharmacological or biological therapeutics that aim to increase colonization resistance during this window could be an important step towards this goal.
METHODS
Microbial community 16S rRNA gene sequence analysis
To determine how diarrheal disturbances affect C. difficile colonization in microbial community surveys, we first download the processed sequences that each study used in their analyses. The downloaded reads were trimmed with Vxtractor (Hartmann et al., 2010) (a HMM scan based method of isolating variable regions from 16S rRNA sequences) to ensure the amplicon
sequences could be aligned across consistent fractions of the 16S rRNA variable regions. These trimmed sequences were classified up to the family level using MOTHUR(Schloss et al., 2009) and Silva(Pruesse et al., 2007) reference sequences.
Detecting C. difficile 16S rRNA sequence types specifically
Methods for phylogenetically classifying 16S rRNA amplicon sequences from all bacterial phyla, such as MOTHUR (Schloss et al., 2009) and QIIME (Caporaso et al., 2010), are not consistently reliable below the level of family. To counter this and detect C. difficile specifically, we built a database of all known C. difficile 16S rRNA sequence types using all 754 available genomes on NCBI in October 2015. First, we extracted the full-length 16S rRNA gene sequences from these genomes, then used Vxtractor(Hartmann et al., 2010) to extract commonly amplified variable regions V4, VI-V3, V4-V5, and V7-V9 sequences. We identified 1, 11, 4, and 12
unique sequence types within from these regions, respectively. All reads with >99.5% sequence
identity to these unique sequence-types were classified as C. difficile reads. Mouse model of varied disturbance intensity
Mice (C57BL/6J; Jackson Laboratories) were single housed in cages with grated bottoms to reduce coprophagy. Sampling started three days prior to treatment, and extended for ten days after. C. difficile spores were suspended in the drinking water supplied to the mice at 104 spores/mL for one day prior, and two days following treatment. On the treatment day, all four groups were given water with strawberry flavored electrolyte replacement (BioServ, Flemington, NJ), but only groups 2, 3, and 4 had polyethylene glycol 3350 (PEG3350, aka Miralax; Sigma-Aldrich, St. Louis, MO) added to their water at 15, 30, and 150 mg/mL, respectively. Treatment lasted 8 hours for groups 1, 2, and 3, but 10 hours for group 4. Experiments were performed in
compliance with MIT institutional guidelines and approved by the Institutional Animal Care and Use Committee.
Non-toxinogenic spore preparation
Spores of the non-toxinogenic C. difficile strain ATCC BA 1801 were prepared as previously described (Lawley et al., 2009). Briefly, a 20 mL overnight inoculate of C. dfficile in Columbia broth was added to 500 mL sterile Clospore media and incubated for 10 days at 370C
anaerobically. Spores were collected by centrifugation and purified by incubating with 15 mg/g trypsin type 1 (Sigma-Aldrich, St. Louis, MO), washing with sterile deionized water a total of 6 times, and heating at 70'C for 15 min to kill all remaining vegetative cells. Spores were counted microscopically prior to dilution with sterile deionized water and inclusion in drinking water. Mouse fecal DNA extraction
Fecal DNA was extracted using DNeasy PowerSoil HTP 96 Kit (Quiagen, Hilden, Germany) as previously described (David et al., 2014). Briefly, approximately 25 mg feces was added to the lysis matrix and the manufacturer's instructions were followed with limited exceptions to improve extraction efficiency. Prior to lysis by bead-beating, 0.4 mg Proteinase K (Quiagen, Hilden, Germany) was added to each well before incubating at 65'C for 10 min, mixing every 2 minutes. After bead beating, the samples were incubated at 95'C for 10 minutes prior to
continuing with the manufacturer's instructions for the remaining steps. 16S rRNA PCR amplicon sequencing of mouse fecal DNA
Amplicon libraries were prepared using methods described in Preheim et al.(Preheim et al., 2013) and sequenced using Illumina MiSeq 2x250 paired-end at the BioMicroCenter
(Massachusetts Institute of Technology, Cambridge, MA). Primers were removed from forward and reverse reads using a custom python script, allowing one mismatch and discarding reads without the primer. The reads were then quality filtered and truncated to a common length, allowing up to two expected errors and truncating to 225 bases for the ward reads and 220 for the reverse. Dada2 (Callahan et al., 2016) was used to infer the Amplicon Sequence Variants (ASVs) from the raw reads. Pseudopooling was performed by running Dada2 twice and using all
identified ASVs from the first run as prior sequences in the second, as described on the pooling documentation on the dada2 website. This reduces the risk of false negatives with dada2 where
sequences with only one or two reads that are close to a more abundant sequence are erroneously corrected. After denoising and merging, the ASVs were classified up to the family level using the MOTHUR (Schloss et al., 2009) classifier and Silva (Pruesse et al., 2007) reference
sequences. The ASV abundance table and all necessary metadata to reproduce our analysis are available in table SI and the complete pipeline used is available on GitiHub.
DATA AVAILABILITY
The publicly available data that we re-analyzed here were generated by David et al (David et al., 2014) accessible on the European Nucleotide Archive (ENA) under the accession number ERP006059, Hsaio et al (Hsiao et al., 2014) on the NCBI Short Read Archive (SRA) under the accession number PRJEB6358, by Caporaso et a! (Caporaso et al., 2011) on MG-RAST under the accession number MG-RAST:4457768.3-4459735.3, and The NIH Human Microbiome Project (NIH HMP Working Group et al., 2009) on NCBI under the accession number PRJNA43017. The amplicon sequencing data from the mouse model of varied disturbance intensity are available on the NCBI SRA under the accession number PRJNA507320.
REFERENCES
Aagaard, K., Petrosino, J., Keitel, W., Watson, M., Katancik, J., Garcia, N., et al. (2013) The Human Microbiome Project strategy for comprehensive sampling of the human microbiome and why it matters. FASEB journal: official publication of the Federation ofAmerican Societies for Experimental Biology, 27, 1012-22.
Alasmari, F., Seiler, S.M., Hink, T., Burnham, C.A.D. and Dubberke, E.R. (2014) Prevalence and risk factors for asymptomatic clostridium difficile carriage. Clinical Infectious Diseases, 59, 216-222.
Allegretti, J.R., Kearney, S., Li, N., Bogart, E., Bullock, K., Gerber, G.K., et al. (2016) Recurrent Clostridium difficile infection associates with distinct bile acid and microbiome profiles.
Alimentary Pharmacology and Therapeutics, 43, 1142-1153.
Bartlett, J.G. (1992) Antibiotic-associated diarrhea. Clinical Infectious Diseases, 15, 573--581. Brown, K.A., Khanafer, N., Daneman, N. and Fisman, D.N. (2013) Meta-analysis of antibiotics and the risk of community-associated Clostridium difficile infection. Antimicrobial agents and chemotherapy, 57, 2326-32.
Bruminhent, J., Wang, Z.-X., Hu, C., Wagner, J., Sunday, R., Bobik, B., et al. (2014)
Clostridium Difficile Colonization and Disease in Patients Undergoing Hematopoietic Stem Cell Transplantation. Biology ofBlood and Marrow Transplantation, 20, 1329-1334.
Buffie, C.G., Bucci, V., Stein, R.R., McKenney, P.T., Ling, L., Gobourne, A., et al. (2015) Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature, 517, 205-208.
Buffie, C.G., Jarchum, I., Equinda, M., Lipuma, L., Gobourne, A., Viale, A., et al. (2012) Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infection and Immunity, 80, 62-73.
Callahan, B.J., McMurdie, P.J., Rosen, M.J., Han, A.W., Johnson, A.J.A. and Holmes, S.P. (2016) DADA2: High-resolution sample inference from Illumina amplicon data. Nature Methods, 13, 58 1-5 83.
Caporaso, J.G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F.D., Costello, E.K., et al. (2010) QIIME allows analysis of high- throughput community sequencing data Intensity
normalization improves color calling in SOLiD sequencing. Nature Publishing Group, 7, 335-336.
Caporaso, J.G., Lauber, C.L., Costello, E.K., Berg-Lyons, D., Gonzalez, A., Stombaugh, J., et al. (2011) Moving pictures of the human microbiome. Genome biology, 12, R50.
Crobach, M.J.T., Vernon, J.J., Loo, V.G., Kong, L.Y., Pechind, S., Wilcox, M.H., et al. (2018) Understanding Clostridium difficile colonization. Clinical Microbiology Reviews, 31, e00021-17. Daquigan, N., Seekatz, A.M., Greathouse, K.L., Young, V.B. and White, J.R. (2017) High-resolution profiling of the gut microbiome reveals the extent of Clostridium difficile burden. npj Biofilms and Microbiomes, 3, 3 5.
David, L.A., Materna, A.C., Friedman, J., Campos-Baptista, M.I., Blackburn, M.C., Perrotta, A., et al. (2014) Host lifestyle affects human microbiota on daily timescales. Genome Biology, 15, R89.
Davis, M.Y., Zhang, H., Brannan, L.E., Carman, R.J. and Boone, J.H. (2016) Rapid change of fecal microbiome and disappearance of Clostridium difficile in a colonized infant after transition from breast milk to cow milk. Microbiome, 4, 53.
Desai, K., Gupta, S.B., Dubberke, E.R., Prabhu, V.S., Browne, C. and Mast, T.C. (2016) Epidemiological and economic burden of Clostridium difficile in the United States: estimates from a modeling approach. BMC Infectious Diseases, 16, 303.
Dubberke, E.R. and Olsen, M.A. (2012) Burden of Clostridium difficile on the Healthcare System. Clinical Infectious Diseases, 55, S88-S92.
Dubberke, E.R., Reske, K.A., Seiler, S., Hink, T., Kwon, J.H. and Burnham, C.A.D. (2015) Risk factors for acquisition and loss of Clostridium difficile colonization in hospitalized patients. Antimicrobial Agents and Chemotherapy, 59, 4533-4543.
Durovic, A., Widmer, A.F. and Tschudin-Sutter, S. (2018) New insights into transmission of Clostridium difficile infection-narrative review. Clinical Microbiology and Infection, 24, 483-492.
Eyre, D.W., Cule, M.L., Wilson, D.J., Griffiths, D., Vaughan, A., O'Connor, L., et al. (2013a) Diverse sources of C. difficile infection identified on whole-genome sequencing. URL
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3868928&tool=pmcentrez&renderty pe=abstract [accessed 15 October 2015]
Eyre, D.W., Griffiths, D., Vaughan, A., Golubchik, T., Acharya, M., O'Connor, L., et al. (2013b) Asymptomatic Clostridium difficile Colonisation and Onward Transmission (ed Y-F Chang). PLoS ONE, 8, e78445.
Guh, A.Y., Adkins, S.H., Li,
Q.,
Bulens, S.N., Farley, M.M., Smith, Z., et al. (2017) Risk Factors for Community-Associated Clostridium difficile Infection in Adults: A Case-Control Study. Open Forum Infectious Diseases, 4.Halligan, E., Edgeworth, J., Bisnauthsing, K., Bible, J., Cliff, P., Aarons, E., et al. (2014) Multiplex molecular testing for management of infectious gastroenteritis in a hospital setting: a comparative diagnostic and clinical utility study. Clinical Microbiology and Infection, 20, 0460-0467.
Hartmann, M., Howes, C.G., Abarenkov, K., Mohn, W.W. and Nilsson, R.H. (2010) V-Xtractor: An open-source, high-throughput software tool to identify and extract hypervariable regions of small subunit (16S/1 8S) ribosomal RNA gene sequences. Journal of Microbiological Methods, 83, 250-253.
Hsiao, A., Ahmed, A.M.S., Subramanian, S., Griffin, N.W., Drewry, L.L., Petri, W.A., et al. (2014) Members of the human gut microbiota involved in recovery from Vibrio cholerae infection. Nature, 515, 423-426.
Kashyap, P.C., Marcobal, A., Ursell, L.K., Larauche, M., Duboc, H., Earle, K.A., et al. (2013) Complex interactions among diet, gastrointestinal transit, and gut microbiota in humanized mice. Gastroenterology, 144, 967-977.
Kelly, C.R., Khoruts, A., Staley, C., Sadowsky, M.J., Abd, M., Alani, M., et al. (2016) Effect of fecal microbiota transplantation on recurrence in multiply recurrent clostridium difficile infection a randomized trial. Annals ofInternal Medicine, 165, 609-616.
Kociolek, L.K., Gerding, D.N., Espinosa, R.O., Patel, S.J., Shulman, S.T. and Ozer, E.A. (2018) Clostridium difficile Whole Genome Sequencing Reveals Limited Transmission Among
Symptomatic Children: A Single-Center Analysis. Clinical Infectious Diseases.
Lawley, T.D., Croucher, N.J., Yu, L., Clare, S., Sebaihia, M., Goulding, D., et al. (2009) Proteomic and genomic characterization of highly infectious Clostridium difficile 630 spores. Journal of Bacteriology, 191, 5377-5386.
Leffler, D.A. and Lamont, J.T. (2015) Clostridium difficile infection. (ed DL Longo). The New England journal of medicine, 372, 1539-48.
Loo, V.G., Bourgault, A.-M., Poirier, L., Lamothe, F., Michaud, S., Turgeon, N., et al. (2011) Host and pathogen factors for Clostridium difficile infection and colonization. The New England journal of medicine, 365, 1693-703.
McFarland, L. V., Surawicz, C.M. and Stamm, W.E. (1990) Risk Factors for Clostridium difficile Carriage and C. difficile-Associated Diarrhea in a Cohort of Hospitalized Patients. Journal ofInfectious Diseases, 162, 678-684.
Murphy, C.N., Fowler, R.C., Iwen, P.C. and Fey, P.D. (2017) Evaluation of the BioFire FilmArray@ GastrointestinalPanel in a Midwestern Academic Hospital. European Journal of Clinical Microbiology & Infectious Diseases, 36, 747-754.
Nagaro, K.J., Phillips, S.T., Cheknis, A.K., Sambol, S.P., Zukowski, W.E., Johnson, S., et al. (2013) Nontoxigenic Clostridium difficile protects hamsters against challenge with historic and epidemic strains of toxigenic BI/NAP 1/027 C. difficile. Antimicrobial Agents and
Chemotherapy, 57, 5266-5270.
NIH HMP Working Group, Peterson, J., Garges, S., Giovanni, M., McInnes, P., Wang, L., et al. (2009) The NIH Human Microbiome Project. Genome Research, 19, 2317-2323.
Pashankar, D.S. and Bishop, W.P. (2001) Efficacy and optimal dose of daily polyethylene glycol 3350 for treatment of constipation and encopresis in children. The Journal ofpediatrics, 139, 428-32.
Patton, A., Davey, P., Harbarth, S., Nathwani, D., Sneddon, J. and Marwick, C.A. (2017) Impact of antimicrobial stewardship interventions on Clostridium difficile infection and clinical
outcomes: segmented regression analyses. Journal ofAntimicrobial Chemotherapy, 73, 517-526.
Preheim, S.P., Perrotta, A.R., Martin-Platero, A.M., Gupta, A. and Alm, E.J. (2013) Distribution-based clustering: using ecology to refine the operational taxonomic unit. Applied and
environmental microbiology, 79, 6593-603.
van Prehn, J., Vandenbroucke-Grauls, C.M.J.E., van Beurden, Y.H., van Houdt, R., Vainio, S. and Ang, C.W. (2015) Diagnostic yield of repeat sampling with immunoassay, real-time PCR, and toxigenic culture for the detection of toxigenic Clostridium difficile in an epidemic and a
non-epidemic setting. European Journal of Clinical Microbiology and Infectious Diseases, 34,
2325-2330.
Pruesse, E., Quast, C., Knittel, K., Fuchs, B.M., Ludwig, W., Peplies, J., et al. (2007) SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data
compatible with ARB. Nucleic Acids Research, 35, 7188-7196.
Ray, A.J., Deshpande, A., Fertelli, D., Sitzlar, B.M., Thota, P., Sankar, T.C., et al. (2017) A multicenter randomized trial to determine the effect of an environmental disinfection
intervention on the incidence of healthcare-associated clostridium difficile infection. Infection
Control and Hospital Epidemiology, 38, 777-783.
Reveles, K.R., Lee, G.C., Boyd, N.K. and Frei, C.R. (2014) The rise in Clostridium difficile infection incidence among hospitalized adults in the United States: 2001-2010. American journal
of infection control, 42, 1028-32.
Scallan, E., Hoekstra, R.M., Angulo, F.J., Tauxe, R. V., Widdowson, M.A., Roy, S.L., et al. (2011) Foodbome illness acquired in the United States-Major pathogens. Emerging Infectious
Diseases, 17, 7-15.
Schloss, P.D., Westcott, S.L., Ryabin, T., Hall, J.R., Hartmann, M., Hollister, E.B., et al. (2009) Introducing mothur: Open-source, platform-independent, community-supported software for
describing and comparing microbial communities. Applied and Environmental Microbiologv, 75,
7537-7541.
Schubert, A.M., Sinani, H. and Schloss, P.D. (2015) Antibiotic-induced alterations of the murine gut microbiota and subsequent effects on colonization resistance against Clostridium difficile.
mBio, 6, e00974.
Seekatz, A.M., Rao, K., Santhosh, K. and Young, V.B. (2016) Dynamics of the fecal
microbiome in patients with recurrent and nonrecurrent Clostridium difficile infection. Genome