Design of S-Substituted Fluorinated Aryl
Sulfonamide-Tagged (S-FAST) Anions To Enable
New Solvate Ionic Liquids for Battery Applications
The MIT Faculty has made this article openly available.
Please share
how this access benefits you. Your story matters.
Citation
Huang, Mingjun et al. "Design of S-Substituted Fluorinated Aryl
Sulfonamide-Tagged (S-FAST) Anions To Enable New Solvate Ionic
Liquids for Battery Applications." Chemistry of Materials 31, 18
(August 2019): 7558-7564 © 2019 American Chemical Society
As Published
http://dx.doi.org/10.1021/acs.chemmater.9b02353
Publisher
American Chemical Society (ACS)
Version
Final published version
Citable link
https://hdl.handle.net/1721.1/122641
Terms of Use
Creative Commons Attribution-NonCommercial-NoDerivs License
Design of S
‑Substituted Fluorinated Aryl Sulfonamide-Tagged
(S-FAST) Anions To Enable New Solvate Ionic Liquids for Battery
Applications
Mingjun Huang,
†,⊥Shuting Feng,
‡,⊥Wenxu Zhang,
†Jeffrey Lopez,
§Bo Qiao,
†,§Ryoichi Tatara,
§Livia Giordano,
§,∥Yang Shao-Horn,
*
,§,∥and Jeremiah A. Johnson
*
,††Department of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139,
United States
‡Department of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge,
Massachusetts 02139, United States
§Research Laboratory of Electronics, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts
02139, United States
∥Department of Mechanical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge,
Massachusetts 02139, United States
*
S Supporting InformationABSTRACT: Electrolytes with improved thermal and oxidative stability must be developed to achieve greater power and energy densities without compromising safety in modern energy storage devices. Because of their much-reduced solvent vapor pressure and expanded electrochemical windows, solvate ionic liquids (SILs) of lithium salts have recently attracted significant attention in this regard. The current palette of SILs is, however, limited to only a few suitable anions with limited chemical functionality. Guided by fundamental physical organic chemistry principles, we designed a new family of S-substituted fluorinated aryl sulfonamide-tagged anions that feature variable numbers of electronically neutral or withdrawing sulfide, sulfoxide, and
sulfone substituents. Several salts of these electron deficient anions display very high electrochemical oxidative stability, good solubility, and a weakly coordinating nature that enables the synthesis of Li-based SILs with high thermal and electrochemical oxidative stability. This new family of functional, noncoordinating anions will potentially expand the scope of applications of SILs as safe electrolytes in battery devices.
L
ithium-ion (Li-ion) batteries have revolutionized the consumer market, including small-scale consumer elec-tronics, power tools, and large-scale power sources driving hybrid electric transportation systems.1,2 The increasing demand for clean energy, however, necessitates the develop-ment of energy storage devices that feature greater power and energy densities as well as a lower cost without compromising safety.3,4Although high energy densities have been achieved in conventional Li-ion batteries using high-voltage electrodes (for example, the 5 V class), the use of highly volatile and flammable carbonate electrolytes can compromise battery safety.5,6 Various strategies for preparing electrolytes with improved oxidative stability and maximum safety, including using inflammable solvents,6 highly concentrated electro-lytes,7−9 and solid state electrolytes, have been explored,4,9,10 yet many unresolved issues and challenges drive the search for new electrolyte materials.Due to their advantages, including a greatly reduced solvent vapor pressure,8,11,12 expanded electrochemical win-dows,7,8,13,14inhibited Al current collector corrosion,14,15and reversible Li metal deposition/stripping,16−18 highly concen-trated (>2.5 M) solutions of Li salts in non-aqueous solvents have recently attracted significant attention as potential next-generation liquid electrolytes.19,20For example, Watanabe et al. reported that, in the equimolar mixture of lithium bis-(trifluoromethane sulfonyl)imide (LiTFSI) and triglyme (or tetraglyme) at room temperature, each Li+coordinates to one
glyme molecule, forming a cationic complex.8,21−23 Because few free glyme molecules exist in these“solvate ionic liquids” (SILs), they display ionic liquid-like thermal stability, high
Received: June 16, 2019 Revised: August 8, 2019 Published: August 8, 2019
Article pubs.acs.org/cm Cite This:Chem. Mater. 2019, 31, 7558−7564
Derivative Works (CC-BY-NC-ND) Attribution License, which permits copying and redistribution of the article, and creation of adaptations, all for non-commercial purposes.
Downloaded via MASSACHUSETTS INST OF TECHNOLOGY on October 10, 2019 at 16:03:06 (UTC).
electrochemical oxidative stability (>4.5 V vs Li/Li+), low
flammability, and low rates of current collector corrosion, making them excellent candidates for reliable electrolytes in high-energy density devices.24 Further studies showed that “good” SILs require highly noncoordinating anions with some degree of structural flexibility.21−23,25−27 As a result, only b i s (fl u o r o s u l f o n y l ) i m i d e ( F S I ) , T F S I , b i s -(pentafluoroethanesulfonyl)imide (BETI), and bis-(nonafluorobutanesulfonyl)imide (NFSI)28 have been shown to be appropriate anions for SIL formation, while other tested anions such as perchlorate (ClO4−), tetrafluoroborate (BF4−), nitrate (NO3−), and trifluoroacetate (TFA−) fail to form
homogeneous SILs. Given the limited set of available SIL-forming anions, the development of new weakly coordinating anions with functional group modularity is needed to take advantage of the SIL concept and further optimize the physical and electrochemical properties of SILs for high-energy density Li-ion battery devices. Moreover, such SILs could potentially be flexibly tuned via anion modification to fulfill different requirements and serve as electrolyte candidates in other battery chemistries such as Li−air29 and lithium−sulfur batteries.18
Herein, we describe a new class of S-substitutedfluorinated aryl sulfonamide-tagged (S-FAST) salts (Scheme 1) that
features variable numbers of sulfide, sulfoxide, and sulfone substituents. The electronic properties of these substituents (see Table S1 for σpara Hammett parameters) afford more
electron deficient and weakly coordinating anions compared to previously reported alkoxy- and amine-functionalized FAST salts;30 these features imbue S-FAST salts with significantly improved oxidative stability and lower Lewis basicity. Notably, equimolar mixtures of the most electron deficient S-FAST salts and tetraglyme (G4) show high electrochemical oxidative stability (>4.5 V vs Li/Li+) and high thermal stability, both of
which are typical properties of SILs. Our FAST salts offer a synthetically tunable platform for the identification of optimal anion structures for next-generation electrolytes.
S-FAST sodium salts featuring isopropyl sulfide substituents (“SiPr”) were prepared via SNAr reactions between
penta-fluorophenyl anion A (Scheme 1a) and 2-propanethiol. Subsequent chemical oxidation using urea H2O2 provided
the corresponding sulfoxide (“SOiPr”) and sulfone (“SO2iPr”) S-FAST anions (Scheme 1b). The sodium salts of thesefive
S-FAST anions were characterized by1H, 13C, and19F nuclear
magnetic resonance (NMR) spectroscopy and high-resolution mass spectrometry (see the Supporting Information). O-Substituted anion A-SOiPr3F2, which performed the best in
terms of electrochemical oxidative stability and ionic conductivity of 26 previously reported O- and N-substituted FAST anions,30is provided for comparison (Scheme 1a). We note that the sodium salts of these anions are studied initially due to their ease of handling; for SIL studies, the corresponding lithium salts were prepared via ion exchange (vide inf ra).
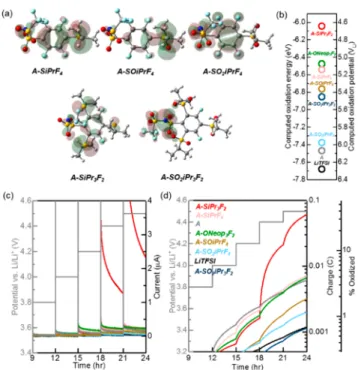
Density functional theory (DFT) calculations (see the
Supporting Information for details) were used to map the highest occupied molecular orbitals (HOMOs) of the S-FAST anions (Figure 1a). The HOMOs of the sulfide (A-SiPrF4and
A-SiPr3F2) and sulfoxide (A-SOiPrF4) anions were heavily localized on the sulfide and sulfoxide groups, respectively, while the HOMOs of the more electron deficient sulfone-based salts (A-SiPrF4and A-SiPr3F2) were shifted toward the
sulfonimide group. In contrast, the HOMO of A-ONeop3F2is uniformly distributed on the aromatic ring with little density on the oxygen atoms of the alkoxide substituents, highlighting the non-electron-donating nature of the S-based substituents of S-FAST anions compared to that of the ether substituents of A-ONeop3F2.30 As expected, the DFT-calculated
electro-chemical oxidation potential (VLi) for the S-FAST anions correlates with the oxidation state of the aryl substituents: sulfide < sulfoxide < sulfone for both mono- and trisubstituted anions (Figure 1b). S-FAST anions with three substituents (e.g., A-SO2iPr3F2) exhibited a calculated electrochemical oxidative stability lower than that of the monosubstituted Scheme 1. (a) Chemical Structures of Anions Studied
Previously30 and (b) Novel S-FAST Anions Described in This Work
Figure 1. (a) Computed HOMO maps of S-FAST anions. (b) Computed electrochemical oxidation energies of lithium S-FAST and traditional FAST salts. The electrochemical oxidation potentials on the experimentally measured scale vs Li/Li+, plotted on the right axis, were converted from the computed −Gox in electronvolts by the subtraction of 1.4 V.31,33 (c) Current response and (d) cumulative oxidative charge and estimated oxidized percentage of various sodium salts and LiTFSI in potentiostatic tests under oxygen.
Chemistry of Materials Article
DOI:10.1021/acs.chemmater.9b02353
Chem. Mater. 2019, 31, 7558−7564 7559
anions (e.g., A-SO2iPrF4). Notably, with the exception of A-SiPr3F2, all of the newly synthesized S-FAST anions are
predicted to be more stable than the comparatively electron-rich A-ONeop3F2.
Next, the electrochemical oxidative stabilities of dilute solutions (0.02 M) of S-FAST and A-ONeop3F2sodium salts and LiTFSI in propylene carbonate (PC), which is a comparatively stable solvent, were experimentally evaluated.30 We note that the anionic component has been shown to dominate the oxidative stability of dilute Li and Na salt solutions, thus making comparisons of oxidative stability between such solutions reasonable.30−32 Potentiostatic meas-urements were carried out in an electrochemical cell (glass fiber separator with a salt solution sandwiched between Li metal foil and a stainless steel mesh current collector) pressurized with oxygen. The measured currents (Figure 1c), cumulative charges (Figure 1d), and estimated percentages of oxidation [assuming one electron per anion (Figure 1d)] as a function of potential (from 3.8VLito 4.5VLi) are provided. In
agreement with our calculations, all of the S-FAST salts except for A-SiPr3F2exhibited currents and cumulative charges similar
to or lower than those of their parent salt A and A-ONeop3F2, highlighting the key role of the electron-withdrawing S-based substituents. Remarkably, S-FAST salts with sulfone groups (A-SO2iPrF4and A-SO2iPr3F2) showed comparable cumulative
oxidative charge to LiTFSI. In further agreement with our calculations (Figure 1b), the oxidation potential increases monotonically: sulfide < sulfoxide < sulfone. We note that contrary to the DFT prediction, trisubstituted A-SO2iPr3F2 exhibited an electrochemical oxidative stability higher than that of its monosubstituted counterpart, A-SO2iPrF4, possibly due to the improved kinetic stability of the bulkier anion. Altogether, most of these salts show excellent electrochemical oxidative stability up to 4.5VLi (<5% oxidation) and thus are promising electrolyte candidates for high-energy density battery chemistries.
Anions with decreased Lewis basicity provide more dissociated salts leading to a higher carrier concentration and conductivity. 23Na NMR was used to evaluate the extent of
dissociation of the sodium salts described above.34,35Figure 2a shows the23Na chemical shifts of each salt prepared as 0.1 M
solutions in acetonitrile (ACN) with 0.5 M NaClO4 in
deuterated water (D2O) as an internal reference. Previous studies have shown that more Lewis basic anions lead to more downfield 23Na NMR chemical shifts and considerable line
broadening.34Here, the monosubstituted S-FAST salts display relatively narrow23Na NMR signals at approximately−6 ppm. Notably, the23Na signal for the sulfone-based salt A-SO
2iPrF4
is similar to that of NaTFSI, which suggests that this salt is a comparably weak Lewis base. The signals for the trisubstituted salts are broader and downfield compared to those of the monosubstituted salts, suggesting that the former salts are more strongly coordinating. The 23Na NMR shifts and the
conductivity of 0.1 M S-FAST-Na solutions in 1,2-dimethoxy-ethane (DME) at 25°C were then correlated with the DFT-predicted free energy of association, ΔGasso (in an implicit
diethyl ether solvent with a dielectric constant set to 7.2 to mimic the solvation environment in DME), of each anion with Na+ (Figure 2b). In general, salts with less negative ΔGasso
values (lower Lewis basicity and less favorable anion−Na+
association) displayed more upfield-shifted signals (smaller relative shift compared to NaTFSI) and a higher conductivity in DME (Figure 2b). Most of the S-FAST salt solutions
exhibited ion conductivities that are greater than that of the A-OEt3F2 solution by a factor of at least 2.30 For example,
solutions of monosubstituted A series salts all had ion conductivities greater than 10−4S/cm at 25°C. Most notably, the A-SO2iPrF4 solution (1.8 × 10−4 S/cm) showed conductivity comparable to that of the NaTFSI solution (2.3 × 10−4 S/cm). The slightly lower conductivities for the
S-FAST salt solutions compared to that of the NaTFSI solution are likely due to the larger size of the S-FAST anion compared to TFSI; nevertheless, the identification of several S-FAST salts with solution ionic conductivities on par with that of NaTFSI, and comparable oxidative stabilities (vide supra), may represent a significant addition to the available salts for battery applications.
Considering the results described above, the combination of S substituents together with the electron-withdrawingfluorine substituents (Hammett parameters for fluorine: σmeta = 0.34 andσpara= 0.06) of our S-FAST anions led to their low Lewis
basicity (on par with that of TFSI). Thus, we hypothesized that these salts could be good candidates for the formation of SILs. Moreover, the modularity of the S-FAST platform could significantly expand upon the palette of available SIL-forming anions. To test this hypothesis, we prepared equimolar mixtures of G4 and representative S-FAST Li salts; the physical and electrochemical properties of these mixtures were compared with those of the most studied SIL, [Li(G4)]-[TFSI]. Here, we use the formula [Li(G4)][X] to represent the SILs formed by equimolar mixing of LiX salts (X = anion) and G4 solvent, including [Li(G4)][A], [Li(G4)][A-SiPrF4], and [Li(G4)][A-SO2iPrF4]. The Li S-FAST salts could be Figure 2.(a)23Na NMR spectra of S-FAST and control salts in 0.1 M ACN solutions (the 23Na signal from the inner reference, 0.5 M NaClO4in D2O, is set to 0 ppm). (b) Ion conductivities of 0.1 M salt solutions in DME at 25°C and23Na NMR chemical shifts (relative to NaTFSI) plotted vs the computed anion−Na+association free energy, ΔGasso, in implicit solvent with a dielectric constant set to 7.2. Chemistry of Materials
readily acquired by ion exchange of the corresponding Na salts. Notably, equimolar mixtures of G4 and these three salts were all liquids at room temperature. Unfortunately, Raman spectroscopy could not be used to characterize these salt mixtures due to strongfluorescence signals (between 700 and 900 cm−1) from the aromatic ring components of the S-FAST anions. Instead, attenuated total reflection (ATR) infrared spectroscopy was utilized to detect free G4 in these mixtures (Figures S1 and S2a). The largely suppressed peak for the G4 solvent (1109 cm−1) in [Li(G4)][A-SiPrF4] is consistent with
there being very little free G4 in this mixture (Figure S2a). Similar results were observed for [Li(G4)][A] and [Li(G4)]-[TFSI] (Figure S1). The upfield chemical shifts of G4 signals in the1H NMR spectrum (Figure S2c) and the downfield shift of the Li+ signal in the 7Li NMR spectrum (Figure S2b) of
[Li(G4)][A-SiPrF4] are indicative of coordination of Li+ to
G4. The diffusion coefficients (D) of G4, Li+, and each anion
were determined by diffusion NMR spectroscopy (Figures S4 and S5). The DG4/DLiratios are close to unity for [Li(G4)][A] [at 298 K, DLi= (7.1± 0.3) × 10−9cm2s−1and DG4= (7.2±
0.1)× 10−9cm2s−1] and [Li(G4)][A-SiPrF
4] [at 298 K, DLi=
(6.5± 0.3) × 10−9cm2s−1and DG4= (5.7± 0.1) × 10−9cm2
s−1], supporting the notion that Li+diffuses together with G4
in the form of a complex. The lithium transference numbers for [Li(G4)][A] and [Li(G4)][A-SiPrF4] are ∼0.58, slightly higher than that of [Li(G4)][TFSI] (0.52),25 presumably due to the larger size of the anion component of FAST-based SILs.
Figure 3a shows thermogravimetric analysis (TGA) curves for [Li(G4)][A], [Li(G4)][A-SiPrF4], [Li(G4)][A-SO2iPrF4],
and [Li(G4)][TFSI] as well as those of pure G4 and the pure salts. The onset of mass loss for G4 is greatly suppressed in these equimolar mixtures. The temperature corresponding to a 5% mass loss increased from 124°C for pure G4 to 172 °C for [Li(G4)][A-SO2iPrF4], 185 °C for [Li(G4)][A], 184 °C for [Li(G4)][A-SiPrF4], and 211 °C for [Li(G4)][TFSI]. We
further investigated the thermal stability of these mixtures using isothermal gravimetric measurements at 120°C (Figure 3b). Although the [Li(G4)][S-FAST] mixtures exhibited a mass loss (∼5%) that was greater than that of LiTFSI (∼2%) after 1 h at 120°C, these anions formed SILs as evidenced by comparison to neat G4 and the LiNO3/G4 mixture.22
To quantitatively analyze the kinetics of G4 evaporation and determine the fraction of G4 complexed with Li+ in these equimolar mixtures under the isothermal TGA conditions, we use the following reactions:
F
[Li(G4) anion][ ] G4 + [ ][Li anion] k k I,free 2 1 (1) → G4I,free k3 G4g (2)
where eq 1 describes a reversible equilibrium between the complexed G4 {[Li(G4)[anion]], i.e., the SIL} and free G4 in the liquid phase andeq 2describes irreversible evaporation of free G4. k1and k2are the rate constants of the forward and reverse reactions ineq 1, respectively, and k3is the evaporation
rate constant. Assumingfirst-order behavior for each species, a system of ordinary differential equations (ODEs) can be developed to describe the evolution of free, complexed, and evaporated G4 over time (see theSupporting Informationfor details). Moreover, assuming a constant density and that all mass loss is due to the evaporation of free G4, andfitting the
values of ki and initial concentrations so that the system of ODEs (eqs S1−S4) fits the recorded isothermal TGA mass loss data, we can obtain the mole fractions of free (xfree) and complexed (xcomplexed) G4, the equilibrium constant (Keq= k1/
k2), and the Gibbs free energy ofreaction 1[ΔG1=−RT ln Keq = −RT ln(k1/k2)] at 120 °C in each of the equimolar
complexes (Table S2 and Figure 3c). To demonstrate the influence of the degree of salt dissociation on SIL formation, ΔG1and xfreeare plotted against the computedΔGassofor each
mixture. As expected, a correlation between more dissociative salts (less negativeΔGasso) and more stable G4−Li+complexes
(less favorable ΔG1) as well as lower xfree values is observed.
The Keq values for [Li(G4)][TFSI], [Li(G4)][A], [Li(G4)]-[A-SiPrF4], and [Li(G4)][A-SO2iPrF4] were 0.008, 0.032, Figure 3.(a) TGA curves of equimolar mixtures of [Li(G4)][TFSI], [Li(G4)][A], [Li(G4)][A-SiPrF4], and [Li(G4)][A-SO2iPrF4] com-pared with those of the pure G4 and salts. (b) Isothermal mass loss plots for pure G4 and [Li(G4)][salt] at 120 °C. (c) Gibbs free energies of reaction 1 (circles) and mole fractions of free G4 (triangles) in equimolar mixtures of [Li(G4)][TFSI], [Li(G4)][A], [Li(G4)][A-SiPrF4], and [Li(G4)][A-SO2iPrF4], and [Li(G4)]NO3 at 120°C determined by numerical analyses of the isothermal TGA results in panel b vs the computed Li+−anion association free energy, ΔGasso. The dashed lines are added to guide the eye.
Chemistry of Materials Article
DOI:10.1021/acs.chemmater.9b02353
Chem. Mater. 2019, 31, 7558−7564 7561
0.217, and 0.046 (all corresponding toΔG1> 0 inFigure 3c), respectively, while the Keq value for [Li(G4)]NO3 was 95
(corresponding to ΔG1 = −3.6 kcal/mol in Figure 3c). In addition, xfreevalues for [Li(G4)][A], [Li(G4)][A-SiPrF4], and
[Li(G4)][A-SO2iPrF4] range from 0.03 to 0.05, slightly higher than that of [Li(G4)][TFSI] (0.01) while xfree for
[Li(G4)]-NO3under the same conditions was∼0.18. Altogether, these results demonstrate that our noncoordinating S-FAST salts form thermodynamically stable SILs with G4.
Next, linear sweep voltammograms (1 mV/s, 80 °C) were collected to investigate the electrochemical oxidative stability of these SILs (Figure 4a). In general, the electrochemical
oxidative stability is greater than 4.5VLi for all of these SILs, decreasing in the following order: [Li(G4)][A-SO2iPrF4] >
[Li(G4)][A] > [Li(G4)][TFSI] > [Li(G4)][A-SiPrF4]. The enhancement of oxidative stability for the SILs compared to that of 1.0 M G4 solutions is the combined result of the presence of very little free G4 in the former and the high oxidative stability of our S-FAST anions.
The ionic conductivities of these SILs as a function of temperature are provided in Figure 4b. The S-FAST-based SILs show ion conductivities decreased by a factor of 5−10 compared to that of [Li(G4)][TFSI], with room-temperature values of∼10−4S/cm, which is again likely due the larger size
of the FAST anions compared to the size of TFSI and the apparently increased viscosity of these SILs. In future work, it may be possible to enhance the ionic conductivity of these SILs by adding innocent, viscosity-decreasing molecular solvents as diluents (e.g., hydrofluoroethers).36,37Nevertheless, the results reported here represent a significant advance in the development of SILs, nearly doubling the number of available SILs for use in battery research, which is made possible by careful molecular design of noncoordinating S-FAST-based anions.
In summary, we have developed a family of S-FAST anions that feature three different S-containing substituents: sulfides, sulfoxides, and sulfones. These substituents combined with the electron-withdrawing fluorine substituents provide S-FAST salts with high electrochemical stability, low Lewis basicity, and high ion conductivity, confirmed by both DFT calculations and experimental measurements. The weakly coordinating nature of these salts enables their formation of novel SILs with G4 that feature high thermal and electrochemical oxidative stability, potentially expanding the scope of SILs for applications in battery devices. Lastly, this work highlights the synthetic modularity of the FAST platform, which enables facile tuning of properties for a wide range of potential applications guided by fundamental physical organic chemistry concepts (e.g., Hammett parameters).
■
EXPERIMENTAL SECTIONElectrochemical Stability Testing. The sulfonimide compounds were dried under vacuum at 75°C overnight before being transferred into a glovebox under an Ar atmosphere [<0.1 ppm H2O, <0.1 ppm O2 (MBraun)] without exposure to an ambient atmosphere. The oxidative stability of the sulfonimide compounds was studied using potentiostatic stability tests in electrochemical cells consisting of lithium foil [D = 15 mm (Chemetall)], 90μL of a 0.02 M sulfonimide sample in propylene carbonate [<20 ppm H2O by Karl Fischer titration (BASF)], one piece of glass fiber separator [D = 18 mm (Whatman, grade GF/A)], and 316 stainless steel mesh as the current collector (D = 12.7 mm). The assembled electrochemical cells were then transferred to a second glovebox [<1 ppm H2O, <1% O2 (MBraun)] and pressured with dry O2 [99.994% pure, <2 ppm H2O (Airgas)] to 30 psi (gauge). In each electrochemical stability test, after the cell had been held at the open circuit voltage for 2 h, a series of potentials were applied sequentially for 3 h each: 3.2, 3.4, 3.6, 3.8, 4.0, 4.2, 4.4, and 4.5 V. The current response was recorded throughout the test. All electrochemical tests were conducted employing a VMP3 potentiostat (BioLogic Science Instruments). The oxidative stabilities of [Li(G4)][salt] SILs were investigated using linear sweep voltammetry and compared to those of 1.0 M LiTFSI-G4 and 1.0 M A-G4 at a scan rate of 1 mV s−1at 80°C. Lithium metal foil (Chemetall) was used as the reference and counter electrode, and a stainless steel plate (MTI Corp.) was used as the working electrode.
Conductivity Measurements. Impedance measurements were conducted using electrochemical cells consisting of the electrolyte sandwiched between two stainless steel blocking electrodes (D = 15.5 mm). The liquid electrolyte contained one piece of a Celgard 2340 separator (thickness of 38μm, porosity of 0.45) impregnated with 100 μL of a 0.1 M sulfonimide sample in 1,2-dimethoxyethane (purchased from Acros, degassed and dried using a glass contour solvent purification system from SG Water USA, LLC). Conductivity was studied using electrochemical impedance spectroscopy (EIS) (VMP3, Bio-Logic Science Instruments) over the frequency range of 1 MHz to 0.1 Hz at a voltage amplitude of 10 mV. The bulk electrolyte conductivity, σ, is estimated from the bulk electrolyte resistance, R, obtained in the EIS measurement according to the equation
Figure 4.(a) Linear sweep voltammograms of [Li(G4)][salt] SILs compared to those of 1.0 M LiTFSI-G4 and 1.0 M A-G4 at a scan rate of 1 mV s−1at 80°C. (b) Temperature-dependent conductivity of equimolar mixture [Li(G4)][salt], compared to those of 1.0 M LiTFSI-G4 and 1.0 M A-G4.
σ = R
d A
1
where d is the thickness of the electrolyte (i.e., the thickness of the separator for the liquid electrolyte) and A is the cross-sectional area of the tested sample.
7Li and1H NMR Studies of SILs. SILs were prepared by mixing equimolar amounts of G4 and representative S-FAST Li salts in a glovebox under an Ar atmosphere. LiCl was dissolved in D2O to prepare a 0.1 M solution, which was loaded into a thin wall NMR tube. The SILs were added to a capillary sealed by a PTFE cap, which was subsequently inserted coaxially into the sample NMR tube. The 7Li and 1H NMR spectra were recorded on a Bruker 400 MHz instrument.
Diffusion NMR Spectroscopy of SILs. Diffusion NMR of7Li, 1H, and 19F was performed on a Bruker Avance 500 MHz spectrometer. Diffusion coefficients were calculated by integrating the peak intensity at various gradient strengths using the following equation:
γ δ δ
= [− Δ − ]
I I0exp D g2 2 2( /3)
where I is the peak intensity with an applied gradient, I0is the peak intensity without a gradient, D is the diffusion coefficient, γ is the gyromagnetic ratio of the nucleus of interest, g is the strength of the applied gradient,Δ is the diffusion delay, and δ is the length of the gradient. The values of Δ and δ used in the experiments were individually optimized for each sample (Figures S4 and S5).
■
ASSOCIATED CONTENT*
S Supporting InformationThe Supporting Information is available free of charge on the
ACS Publications website at DOI: 10.1021/acs.chemma-ter.9b02353.
Supplementalfigures and experimental details, including synthetic procedures, DFT calculations, and NMR, FT-IR, and TGA data (PDF)
■
AUTHOR INFORMATION Corresponding Authors *E-mail:shaohorn@mit.edu. *E-mail:jaj2109@mit.edu. ORCID Shuting Feng:0000-0001-5630-7085 Jeffrey Lopez: 0000-0002-6425-5550 Yang Shao-Horn:0000-0001-8714-2121 Jeremiah A. Johnson:0000-0001-9157-6491 Author Contributions⊥M.H. and S.F. contributed equally to this work.
Notes
The authors declare no competingfinancial interest.
■
ACKNOWLEDGMENTSThe authors acknowledge the Samsung Advanced Institute of Technology (SAIT) for funding this research. S.F. gratefully acknowledges the Link Foundation for the Link Energy Fellowship. This research used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation Grant ACI-1548562. J.L. gratefully acknowledges support by an appoint-ment to the Intelligence Community Postdoctoral Research Fellowship Program at the Massachusetts Institute of Technology, administered by the Oak Ridge Institute for Science and Education through an interagency agreement
between the U.S. Department of Energy and the Office of the Director of National Intelligence.
■
REFERENCES(1) Tarascon, J. M.; Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 2001, 414, 359−367.
(2) Chu, S.; Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 2012, 488, 294−303.
(3) Nitta, N.; Wu, F.; Lee, J. T.; Yushin, G. Li-ion battery materials: present and future. Mater. Today 2015, 18, 252−264.
(4) Manthiram, A.; Yu, X.; Wang, S. Lithium battery chemistries enabled by solid-state electrolytes. Nat. Rev. Mater. 2017, 2, 16103.
(5) Gur, T. M. Review of Electrical Energy Storage Technologies, Materials and Systems: Challenges and Prospects for Large-Scale Storage. Energy Environ. Sci. 2018, 11, 2696−2767.
(6) Wang, J.; Yamada, Y.; Sodeyama, K.; Watanabe, E.; Takada, K.; Tateyama, Y.; Yamada, A. Fire-extinguishing organic electrolytes for safe batteries. Nat. Energy 2018, 3, 22−29.
(7) Suo, L.; Borodin, O.; Gao, T.; Olguin, M.; Ho, J.; Fan, X.; Luo, C.; Wang, C.; Xu, K. Water-in-salt” electrolyte enables high-voltage aqueous lithium-ion chemistries. Science 2015, 350, 938−943.
(8) Yoshida, K.; Nakamura, M.; Kazue, Y.; Tachikawa, N.; Tsuzuki, S.; Seki, S.; Dokko, K.; Watanabe, M. Oxidative-stability enhancement and charge transport mechanism in glyme-lithium salt equimolar complexes. J. Am. Chem. Soc. 2011, 133, 13121−9.
(9) MacFarlane, D. R.; Forsyth, M.; Howlett, P. C.; Kar, M.; Passerini, S.; Pringle, J. M.; Ohno, H.; Watanabe, M.; Yan, F.; Zheng, W.; Zhang, S.; Zhang, J. Ionic liquids and their solid-state analogues as materials for energy generation and storage. Nat. Rev. Mater. 2016, 1, 15005.
(10) Liu, K.; Liu, Y.; Lin, D.; Pei, A.; Cui, Y. Materials for lithium-ion battery safety. Sci. Adv. 2018, 4, No. eaas9820.
(11) Watanabe, M.; Thomas, M. L.; Zhang, S.; Ueno, K.; Yasuda, T.; Dokko, K. Application of Ionic Liquids to Energy Storage and Conversion Materials and Devices. Chem. Rev. 2017, 117, 7190− 7239.
(12) Yoshida, K.; Tsuchiya, M.; Tachikawa, N.; Dokko, K.; Watanabe, M. Change from Glyme Solutions to Quasi-ionic Liquids f o r B i n a r y M i x t u r e s C o n s i s t i n g o f L i t h i u m B i s -(trifluoromethanesulfonyl)amide and Glymes. J. Phys. Chem. C 2011, 115, 18384−18394.
(13) Yamada, Y.; Furukawa, K.; Sodeyama, K.; Kikuchi, K.; Yaegashi, M.; Tateyama, Y.; Yamada, A. Unusual stability of acetonitrile-based superconcentrated electrolytes for fast-charging lithium-ion batteries. J. Am. Chem. Soc. 2014, 136, 5039−46.
(14) Wang, J.; Yamada, Y.; Sodeyama, K.; Chiang, C. H.; Tateyama, Y.; Yamada, A. Superconcentrated electrolytes for a high-voltage lithium-ion battery. Nat. Commun. 2016, 7, 12032.
(15) Zhang, C.; Yamazaki, A.; Murai, J.; Park, J.-W.; Mandai, T.; Ueno, K.; Dokko, K.; Watanabe, M. Chelate Effects in Glyme/ Lithium Bis(trifluoromethanesulfonyl)amide Solvate Ionic Liquids, Part 2: Importance of Solvate-Structure Stability for Electrolytes of Lithium Batteries. J. Phys. Chem. C 2014, 118, 17362−17373.
(16) Tatara, R.; Kwabi, D. G.; Batcho, T. P.; Tulodziecki, M.; Watanabe, K.; Kwon, H.-M.; Thomas, M. L.; Ueno, K.; Thompson, C. V.; Dokko, K.; Shao-Horn, Y.; Watanabe, M. Oxygen Reduction Reaction in Highly Concentrated Electrolyte Solutions of Lithium Bis(trifluoromethanesulfonyl)amide/Dimethyl Sulfoxide. J. Phys. Chem. C 2017, 121, 9162−9172.
(17) Tachikawa, N.; Yamauchi, K.; Takashima, E.; Park, J. W.; Dokko, K.; Watanabe, M. Reversibility of electrochemical reactions of sulfur supported on inverse opal carbon in glyme-Li salt molten complex electrolytes. Chem. Commun. 2011, 47, 8157−9.
(18) Dokko, K.; Tachikawa, N.; Yamauchi, K.; Tsuchiya, M.; Yamazaki, A.; Takashima, E.; Park, J.-W.; Ueno, K.; Seki, S.; Serizawa, N.; Watanabe, M. Solvate Ionic Liquid Electrolyte for Li-S Batteries. J. Electrochem. Soc. 2013, 160, A1304−A1310.
Chemistry of Materials Article
DOI:10.1021/acs.chemmater.9b02353
Chem. Mater. 2019, 31, 7558−7564 7563
(19) Yamada, Y.; Yamada, A. ReviewSuperconcentrated Electro-lytes for Lithium Batteries. J. Electrochem. Soc. 2015, 162, A2406− A2423.
(20) Yamada, Y.; Wang, J.; Ko, S.; Watanabe, E.; Yamada, A. Advances and issues in developing salt-concentrated battery electro-lytes. Nat. Energy 2019, 4, 269−280.
(21) Mandai, T.; Yoshida, K.; Ueno, K.; Dokko, K.; Watanabe, M. Criteria for solvate ionic liquids. Phys. Chem. Chem. Phys. 2014, 16, 8761−72.
(22) Ueno, K.; Tatara, R.; Tsuzuki, S.; Saito, S.; Doi, H.; Yoshida, K.; Mandai, T.; Matsugami, M.; Umebayashi, Y.; Dokko, K.; Watanabe, M. Li(+) solvation in glyme-Li salt solvate ionic liquids. Phys. Chem. Chem. Phys. 2015, 17, 8248−57.
(23) Watanabe, M.; Dokko, K.; Ueno, K.; Thomas, M. L. From Ionic Liquids to Solvate Ionic Liquids: Challenges and Opportunities for Next Generation Battery Electrolytes. Bull. Chem. Soc. Jpn. 2018, 91, 1660−1682.
(24) Kitazawa, Y.; Iwata, K.; Kido, R.; Imaizumi, S.; Tsuzuki, S.; Shinoda, W.; Ueno, K.; Mandai, T.; Kokubo, H.; Dokko, K.; Watanabe, M. Polymer Electrolytes Containing Solvate Ionic Liquids: A New Approach To Achieve High Ionic Conductivity, Thermal Stability, and a Wide Potential Window. Chem. Mater. 2018, 30, 252− 261.
(25) Ueno, K.; Yoshida, K.; Tsuchiya, M.; Tachikawa, N.; Dokko, K.; Watanabe, M. Glyme-lithium salt equimolar molten mixtures: concentrated solutions or solvate ionic liquids? J. Phys. Chem. B 2012, 116, 11323−31.
(26) Jankowski, P.; Dranka, M.; Żukowska, G. Z.; Zachara, J. Structural Studies of Lithium 4,5-Dicyanoimidazolate-Glyme Solvates. 1. From Isolated Free Ions to Conductive Aggregated Systems. J. Phys. Chem. C 2015, 119, 9108−9116.
(27) Jankowski, P.; Dranka, M.; Wieczorek, W.; Johansson, P. TFSI and TDI Anions: Probes for Solvate Ionic Liquid and Disproportio-nation-Based Lithium Battery Electrolytes. J. Phys. Chem. Lett. 2017, 8, 3678−3682.
(28) Thomas, M. L.; Oda, Y.; Tatara, R.; Kwon, H.-M.; Ueno, K.; Dokko, K.; Watanabe, M. Suppression of Water Absorption by Molecular Design of Ionic Liquid Electrolyte for Li-Air Battery. Adv. Energy Mater. 2017, 7, 1601753.
(29) Kwon, H. M.; Thomas, M. L.; Tatara, R.; Oda, Y.; Kobayashi, Y.; Nakanishi, A.; Ueno, K.; Dokko, K.; Watanabe, M. Stability of Glyme Solvate Ionic Liquid as an Electrolyte for Rechargeable Li-O2 Batteries. ACS Appl. Mater. Interfaces 2017, 9, 6014−6021.
(30) Huang, M.; Feng, S.; Zhang, W.; Giordano, L.; Chen, M.; Amanchukwu, C. V.; Anandakathir, R.; Shao-Horn, Y.; Johnson, J. A. Fluorinated Aryl Sulfonimide Tagged (FAST) salts: modular synthesis and structure-property relationships for battery applications. Energy Environ. Sci. 2018, 11, 1326−1334.
(31) Xing, L.; Borodin, O.; Smith, G. D.; Li, W. Density Functional Theory Study of the Role of Anions on the Oxidative Decomposition Reaction of Propylene Carbonate. J. Phys. Chem. A 2011, 115, 13896−13905.
(32) Gauthier, M.; Carney, T. J.; Grimaud, A.; Giordano, L.; Pour, N.; Chang, H. H.; Fenning, D. P.; Lux, S. F.; Paschos, O.; Bauer, C.; Maglia, F.; Lupart, S.; Lamp, P.; Shao-Horn, Y. Electrode-electrolyte interface in Li-ion batteries: current understanding and new insights. J. Phys. Chem. Lett. 2015, 6, 4653−72.
(33) Trasatti, S. The absolute electrode potential: an explanatory note. Pure Appl. Chem. 1986, 58, 955−966.
(34) Erlich, R. H.; Popov, A. I. Spectroscopic Studies of Ionic Solvation. X. A Study of the Solvation of Sodium Ions in Nonaqueous Solvents by 23Na Nuclear Magnetic Resonance. J. Am. Chem. Soc. 1971, 93, 5620.
(35) Schmeisser, M.; Illner, P.; Puchta, R.; Zahl, A.; van Eldik, R. Gutmann donor and acceptor numbers for ionic liquids. Chem. - Eur. J. 2012, 18, 10969−82.
(36) Ueno, K.; Murai, J.; Ikeda, K.; Tsuzuki, S.; Tsuchiya, M.; Tatara, R.; Mandai, T.; Umebayashi, Y.; Dokko, K.; Watanabe, M. Li+ Solvation and Ionic Transport in Lithium Solvate Ionic Liquids
Diluted by Molecular Solvents. J. Phys. Chem. C 2016, 120, 15792− 15802.
(37) Saito, S.; Watanabe, H.; Ueno, K.; Mandai, T.; Seki, S.; Tsuzuki, S.; Kameda, Y.; Dokko, K.; Watanabe, M.; Umebayashi, Y. Li(+) Local Structure in Hydrofluoroether Diluted Li-Glyme Solvate Ionic Liquid. J. Phys. Chem. B 2016, 120, 3378−87.
![Figure 3a shows thermogravimetric analysis (TGA) curves for [Li(G4)][A], [Li(G4)][A-SiPrF 4 ], [Li(G4)][A-SO 2 iPrF 4 ], and [Li(G4)][TFSI] as well as those of pure G4 and the pure salts](https://thumb-eu.123doks.com/thumbv2/123doknet/14688186.560663/5.938.546.784.98.728/figure-shows-thermogravimetric-analysis-curves-siprf-tfsi-salts.webp)
![Figure 4. (a) Linear sweep voltammograms of [Li(G4)][salt] SILs compared to those of 1.0 M LiTFSI-G4 and 1.0 M A-G4 at a scan rate of 1 mV s −1 at 80 °C](https://thumb-eu.123doks.com/thumbv2/123doknet/14688186.560663/6.938.102.410.313.827/figure-linear-sweep-voltammograms-salt-sils-compared-litfsi.webp)
