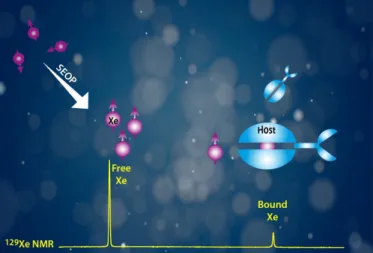
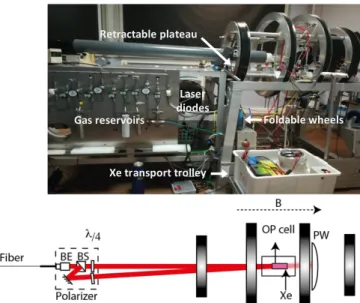
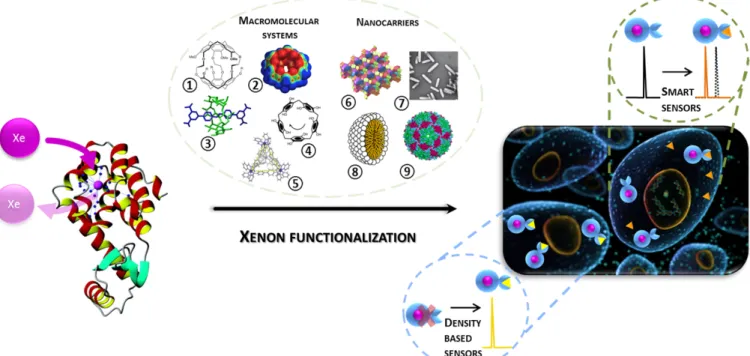
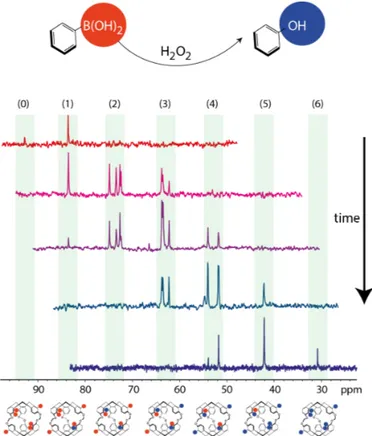
Xe NMR-based sensors: biological applications and recent methods
Texte intégral
Figure

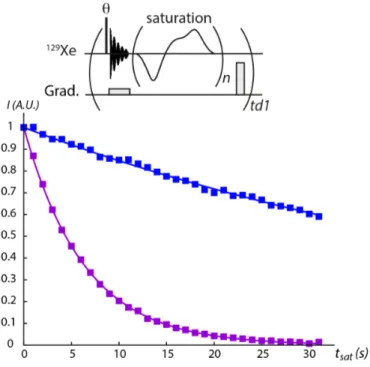
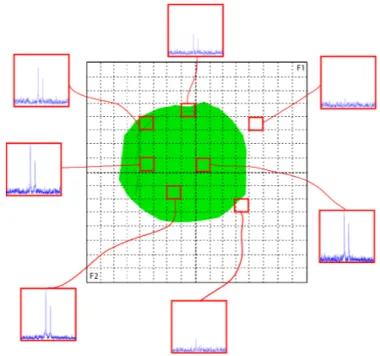
Documents relatifs
A GEANT3 simulation of a full PET design made of LXe-TPC modules has been developed and the first estimations of the performances from a realistic detector are very promising: good
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des
Dans la figure 8 nous avons tracé le diagramme de rayonnement pour un réseau rectiligne à 10 éléments dans le plan H avec un rapport a=6.. Nous constatons que les premiers
To the best of our knowledge, we report the first case of a successful control of recurrent ventricular arrhythmias in the setting of a malignant ER syndrome by adding theophylline to
Using a mild protein restriction model in Balb/C mice consuming 6% of their total energy intake as soy protein (LP-SOY), and for which we observed a significant
In this study, we investigated the abilities of the combination of evidential clustering and prostate zonal atlases to drive a segmentation process for separating the
The second major contribution is the development of a novel diffuse optics based three dimensional deep tissue blood flow imaging method, called speckle contrast opti- cal
La mention explicite des médicaments dange- reux, l’accès restreint à ces médicaments, l’amélioration de l’accès à l’information, la double vérification indépen-
