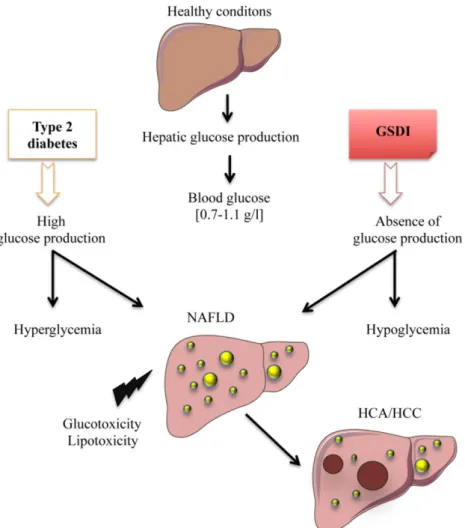
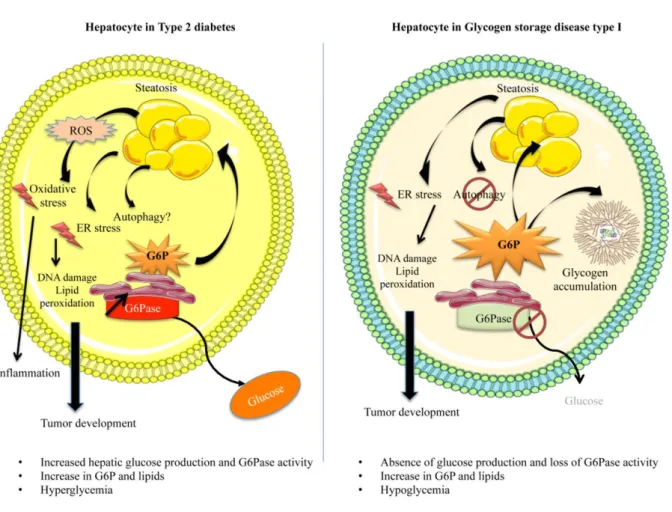
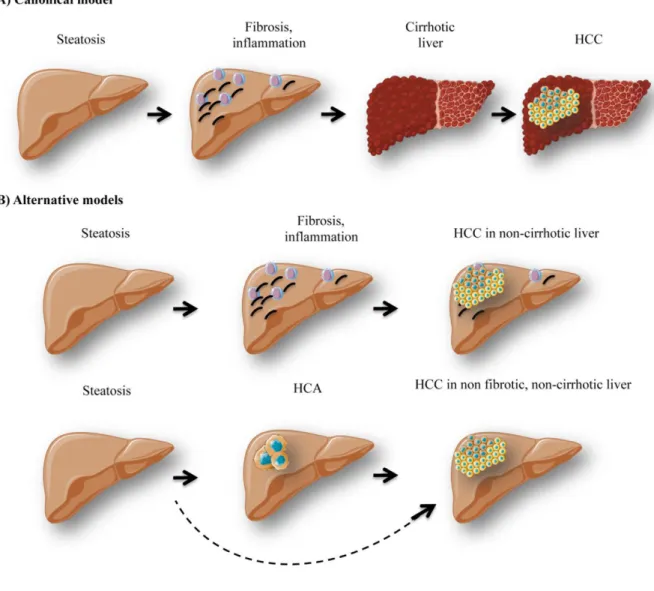
Hepatic stress associated with pathologies characterized by disturbed glucose production
Texte intégral
Figure
Documents relatifs
such as acetyl-CoA carboxylase-alpha (ACACA), fatty acid synthase (FASN), fatty acid elongase-6 (ELOVL6) and stearoyl-CoA desaturase (SCD), involved in fatty acid (FA) biosynthesis
Pour communiquer directement avec un auteur, consultez la première page de la revue dans laquelle son article a été publié afin de trouver ses coordonnées.. Si vous n’arrivez pas
Anatomical (black dotted line) and functional averaged profiles (blue line for upper profile and green line for lower profile (±SEM) were represented together with the STAP
The law of large numbers states that if the sample size n is large, the sample mean rarely deviates from the mean of the distribution of X, which in statistics is called the
methods that extend classical scheduling analysis to special classes of distributed systems, this approach applies existing analysis techniques in a modular manner: the single mod-
Le contrat peut désormais être signé avec tous les jeunes de 16 à 25 ans de niveau de formation inférieur au baccalauréat ; il est également ouvert aux jeunes de 16 à 25 ans
Pour clore, l’analyse de l’apport lexical dans l’œuvre de Gracq nous permet d’effectuer les interprétations suivantes : au début, l’apport de nouvelles unités
maltaromaticum DSM20730 a Pediocin sensitive strain (Hiu et al ., 1984) Lb plantarum LMAX Natural pediocin producer , isolated from HOLDBAC Listeria Danisco L. lactis NZ9000_pSec::
