Synthèse de Nouvelles Nitrones du Type Pyrroline-N-Oxyde - Manuscrit
299
0
0
Texte intégral
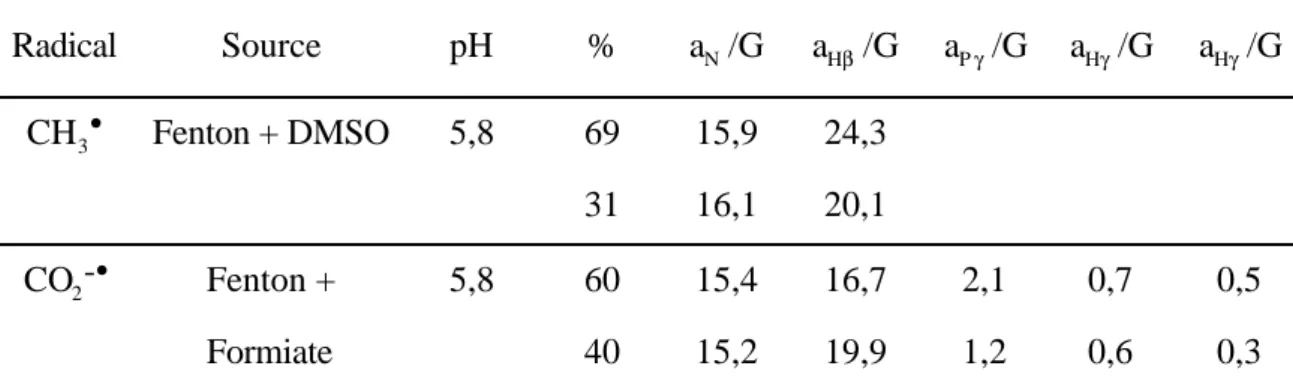
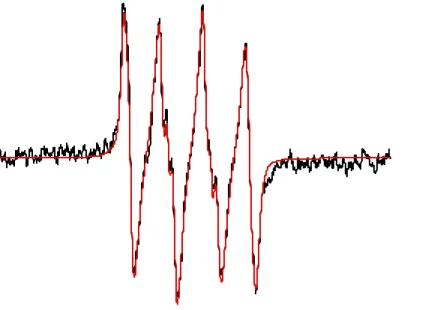
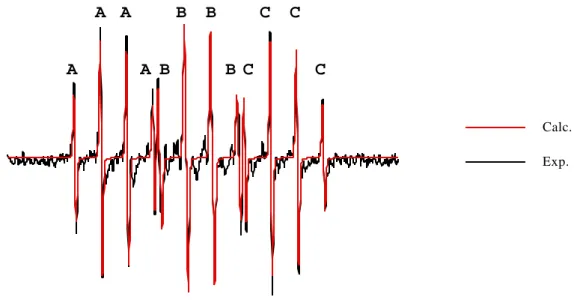
Figure

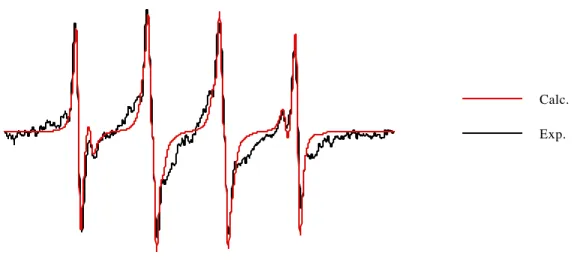


+7

Documents relatifs
Mais comme i l aurait été insatisfaisant pour nous de passer dans un laboratoire de chimie organique sans toucher un seul bécher , nous avons aussi entrepris un
Nous considérons donc que les N (E+DET:G) N:G prédicatifs du grec moderne doivent être inclus dans les tables du lexique-grammaire au même titre que les noms prédicatifs
