PW1/Peg3 expression regulates key properties that determine mesoangioblast stem cell competence
14
0
0
Texte intégral
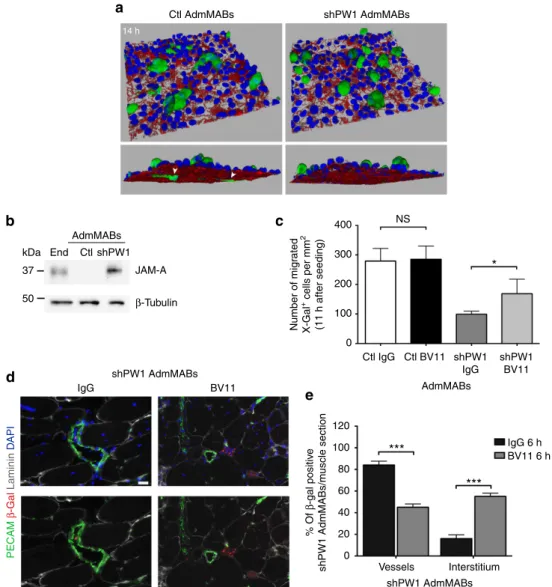
Figure
+4


Documents relatifs