HAL Id: hal-03040984
https://hal.archives-ouvertes.fr/hal-03040984
Submitted on 16 Dec 2020
HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
dynamics and function of histone variants
Aline V Probst, Bénédicte Desvoyes, Crisanto Gutierrez
To cite this version:
Aline V Probst, Bénédicte Desvoyes, Crisanto Gutierrez. Similar yet critically different: the distribu-tion, dynamics and function of histone variants. Journal of Experimental Botany, Oxford University Press (OUP), 2020, 71 (17), pp.5191 - 5204. �10.1093/jxb/eraa230�. �hal-03040984�
doi:10.1093/jxb/eraa230 Advance Access Publication 11 May 2020
This paper is available online free of all access charges (see https://academic.oup.com/jxb/pages/openaccess for further details)
© The Author(s) 2020. Published by Oxford University Press on behalf of the Society for Experimental Biology.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse, distribution, and reproduction in any medium, provided the original work is properly cited.
REVIEW PAPER
Similar yet critically different: the distribution, dynamics and
function of histone variants
Aline V. Probst1, , Bénédicte Desvoyes2, and Crisanto Gutierrez2,
1 Université Clermont Auvergne, CNRS, Inserm, GReD, F-63000 Clermont-Ferrand, France
2 Centro de Biologia Molecular Severo Ochoa, CSIC-UAM, Nicolas Cabrera 1, Cantoblanco, 28049 Madrid, Spain
* Correspondence: [email protected] or [email protected]
Received 27 February 2020; Editorial decision 4 May 2020; Accepted 6 May 2020 Editor: Geraint Parry, Cardiff University, UK
Abstract
Organization of the genetic information into chromatin plays an important role in the regulation of all DNA template-based reactions. The incorporation of different variant versions of the core histones H3, H2A, and H2B, or the linker histone H1 results in nucleosomes with unique properties. Histone variants can differ by only a few amino acids or larger protein domains and their incorporation may directly affect nucleosome stability and higher order chromatin organization or indirectly influence chromatin function through histone variant-specific binding partners. Histone variants employ dedicated histone deposition machinery for their timely and locus-specific incorporation into chro-matin. Plants have evolved specific histone variants with unique expression patterns and features. In this review, we discuss our current knowledge on histone variants in Arabidopsis, their mode of deposition, variant-specific post-translational modifications, and genome-wide distribution, as well as their role in defining different chromatin states. Keywords: Arabidopsis, cell division and differentiation, chromatin, histone chaperones, histone modifications, histone variants.
Introduction
In eukaryotes, the long nuclear DNA molecules are organ-ized into chromatin. As a dynamic platform, chromatin inte-grates signaling information from the cellular environment; stores and perpetuates transient or heritable epigenetic infor-mation; and influences accessibility of the cellular machinery to DNA during transcription, replication, recombination, and repair. The first electron micrographs of chromatin structure revealed a ‘beads on a string’ organization, giving a first glimpse of the basic structural subunits of chromatin, the nucleosomes (Olins and Olins, 1974). Within a nucleosome, around 146 bp of DNA is wrapped around an octamer of core histone proteins (Box 1), two each of H3, H4, H2A, and H2B (Luger et al., 1997). One nucleosome is connected to the other via linker DNA of variable length, which can be bound by the linker histone H1 (Fyodorov et al., 2018). Histones are basic proteins,
rich in lysine and arginine residues, which facilitate inter-action with the negatively charged DNA through electrostatic interactions. They contain a characteristic histone fold domain formed by three α-helices and connected by two loop regions that mediates head-to-tail interaction with other histones to form histone dimers (Fig. 1A). During nucleosome assembly two H3–H4 dimers form a tetramer, which is then flanked by two H2A–H2B dimers.
To meet the requirement of a dynamic stage for epigenetic information, chromatin organization is modified at different levels. DNA itself can be methylated and histone proteins can undergo various post-translational modifications that are read by specific ‘reader’ proteins and play a role in gene expression regulation, higher-order chromatin formation, and DNA re-pair. A further level of chromatin complexity resides in the
nucleosome composition, which can be modulated through incorporation of different histone variants. Histone variants can differ only by a few amino acids or by larger protein do-mains. They can be incorporated at different moments during the cell cycle or only in particular cell types using similar or specific histone deposition pathways mediated by a network of proteins termed histone chaperones (Box 1). The incorpor-ation of histone variants may affect the setting of histone post-translational modifications and/or directly alter nucleosome stability, interaction between nucleosomes or interaction be-tween chromatin fibers. This review focuses on plant histone variants, with a particular emphasis on histone H3 variants for which we have gained much information in recent years con-cerning their transport, assembly, and turnover during the cell cycle, mainly from studies in the model organism Arabidopsis.
Histone variants
Histone variants have been described in all model organisms studied so far, from the unicellular yeast Saccharomyces cerevisiae to plants and animals, and for all histones but histone H4 (which is of a single type), with only a few exceptions (Long
et al., 2019). A major need for new histone deposition arises during DNA replication, when DNA molecules are dupli-cated and new histones are deposited. These histones, highly expressed during S-phase and incorporated at the replication fork, were termed replicative histone variants (Box 1) or canon-ical histones. In mammals, but also in Drosophila, the genes encoding these replicative histone variants are organized in
clusters, facilitating concerted gene regulation (Elsaesser et al., 2010). Mammalian histone gene promoters are activated during S-phase and genes within these histone clusters have particular structures: they are intronless, comprise short un-translated regions (UTRs), and have no polyadenylation sites, but instead harbor a stem-loop structure and a purine-rich his-tone downstream element. Processing of the hishis-tone precursor mRNA is achieved through action of the stem-loop binding protein (SLRP), which stabilizes the stem loop in the histone mRNA, leading to the endonucleolytic cleavage of the pre-cursor. If not bound and stabilized by SLRP, the stem loop is sufficient for histone mRNA degradation and SLRP protein itself is cell cycle regulated and degraded at the end of S-phase. Altogether, the different control points at transcriptional and post-transcriptional level contribute to the tight coupling of histone gene transcript stability with the cellular needs for new histones in mammalian cells (reviewed in Marzluff and Koreski, 2017; Mendiratta et al., 2019).
In addition to the replicative variants, non-replicative variants, also called replacement histone variants (Box 1), are polyadenylated, are expressed throughout the cell cycle, and ensure histone turnover, exchange, or de novo deposition during processes that require transient histone eviction, such as transcription (Venkatesh and Workman, 2015). Generally, re-placement histone genes contain introns. Finally, some histone variants are only expressed in specific tissues.
In most green lineage species, histone gene clusters have not been reported and all histone genes are transcribed into polyadenylated transcripts. In addition, the presence or absence of introns seems not be a faithful criterion to differentiate
Box 1. Definitions of histone variants and chaperones Core histones
Histones H3, H4, H2A, and H2B, which form the histone octamer. Core histones are characterized by a histone fold domain consisting of three α-helices linked by loop regions (L1 and L2) and flexible tail regions that protrude from the nucleosome structure.
Linker histone variants
Linker histone H1, consisting of a central globular domain with more flexible N- and C-terminal tail regions. H1 binds to nucleosomal DNA as well as to linker DNA around the DNA entry and exit sites and is thought to facilitate the formation of higher order chromatin structures.
Histone chaperones
Proteins or protein complexes that bind non-nucleosomal histones to allow histone transport, storage, or nucleosome assembly. Histone chaperones can bind histones through different binding modules; some are able to bind different core histones, some interact specifically with one histone type, while others are even able to distinguish between histone variants.
Replicative histone variants
Core histone variants that are predominantly expressed during S-phase and deposited during DNA replication in a DNA-synthesis-dependent manner.
Replacement histone variants
Histone variants that are expressed throughout the cell-cycle and are deposited in a DNA-synthesis-independent manner to ensure histone turnover and replacement during processes such as transcription.
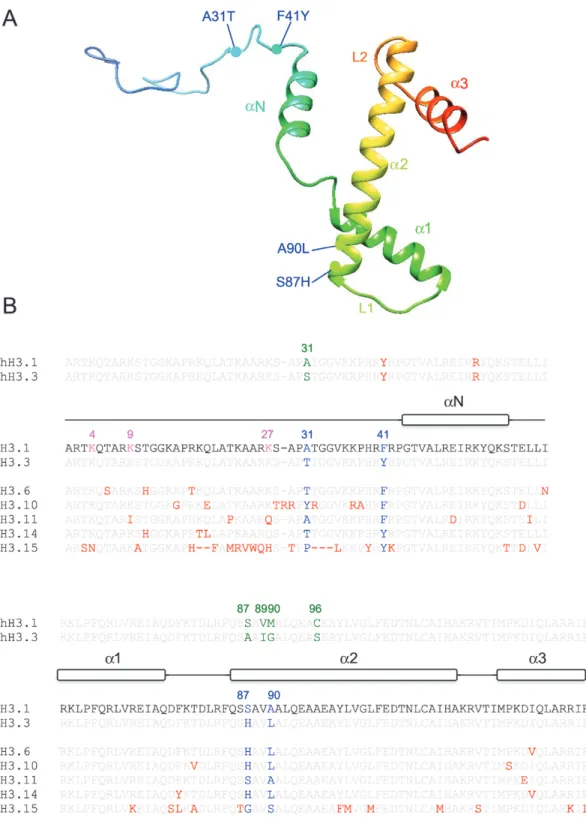
Fig. 1. Structural features of H3 proteins. (A) 3D model of Arabidopsis histone H3.1 using PHYRE2 (Kelley et al., 2015) and Chimera. The position of the
four amino acids differing between the replicative variant H3.1 and the replacement variant H3.3 is indicated by colored circles. In each case, the first residue corresponds to H3.1. Positions 31 and 41 are situated in the histone tail, and positions 87 and 90 in the histone fold domain consisting of three α-helices (α1, α2 and α3) connected by two loops (L1 and L2). (B) Alignment of Arabidopsis H3.1, H3.3, and five atypical H3 proteins namely H3.6, H3.10, H3.11, H3.14, and H3.15 as well as human H3.1 and H3.3 (hH3.1; hH3.3). Positions of the three α-helices constituting the histone fold domain (α1, α2, and α3) as well as the N-terminal α-helix (αN) were obtained from HistoneDB 2.0 (Draizen et al., 2016) are indicated. The five amino acids (A31S, S87A, V89I, M90G, and C96S) that differ between human H3.1 and H3.3 are indicated in green; the four amino acids that differ between Arabidopsis H3.1 and H3.3 (A31T, F41Y, S87H, and A90L) are indicated in blue. Additional differences in amino acids compared with Arabidopsis H3.1 are indicated in red. Gaps are indicated by dashes. While H3.6, H3.10, and H3.14 carry the H3.3 signature (H87 and L90), H3.11 carries the H3.1-specific signature (S87 and A90). Note that H3.11 lacks two critical lysine residues in the N-terminal tail (namely K9 and K27, colored pink in H3.1). The differences are likely to impact on the methylation status in the genomic regions, where these variants are incorporated. Similarly, the differences in amino acids adjacent to K27 in H3.10 may impact the function of the K27 histone methyltransferases. H3.15 is highly divergent, lacking K4 and K27 (colored pink in H3.1), but has so far only been shown to be very weakly expressed.
between replicative and replacement histone variants, as none of the H2B protein-encoding genes presents introns in Arabidopsis, while all H2A encoding genes do. The correl-ation seems only true for H3 protein-encoding genes, where the replicative histone genes are intronless, while most genes encoding replacement variants comprise introns (Chaubet
et al., 1992). As of today, much remains to be understood of how replicative histone gene transcription is controlled in plants to meet the significant need for new histones during S-phase. Tight control of histone gene transcription certainly contributes to the non-random distribution of histone variants in the genome (see below) and failure to control histone gene expression during the cell cycle may have repercussions on his-tone dynamics and epigenetic plasticity.
Characteristics of H3 variants in
Arabidopsis
Genes encoding Arabidopsis histone H3 variants
The Arabidopsis genome contains 15 histone H3 genes termed HISTONE THREE RELATED 1–15. Five genes (HTR1, HTR2, HTR3, HTR9, and HTR13) show S-phase-specific expression and encode identical proteins with charac-teristics similar to the mammalian replicative histone variant H3.1 (Fig. 1; Table 1), suggesting that they are deposited in a DNA-synthesis-dependent manner (Chaubet et al., 1992;
Okada et al., 2005; Jiang and Berger, 2017). Histone H3.1 dif-fers by only four amino acids from the histone variant H3.3 encoded by three genes (HTR4, HTR5, and HTR8) expressed in all tissues including non-proliferating cells, suggesting DNA-synthesis independent deposition of H3.3 (Chaubet
et al., 1992; Okada et al., 2005). The amino acid differences are situated within the second α-helix of the histone fold domain (positions S(H3.1)87H(H3.3) and A90L), within the N-terminal tail (position 31, A31T), and at position 41 (F41Y). When considering the amino acids that distinguish H3.1 from H3.3 in different species, these can be situated at different positions and involve different sets of amino acids, strongly suggesting that histone H3 variants appeared inde-pendently in different lineages (Waterborg and Robertson, 1996; Talbert et al., 2012). For example, the phenylalanine at position 41 found in Arabidopsis H3.1 is not present in yeast or vertebrate H3 histones (Fig. 1). Interestingly, this amino acid is part of a short protein stretch critically situated at the entry/ exit site of the DNA gyres from the nucleosome and therefore has the potential to affect histone–DNA interactions. With re-gard to plant evolution, it is interesting to note that in mosses and some green algae H3.1 and H3.3 variants differ only at positions 31 and 87, while at positions 41 and 90 we find tyrosine and leucine as in angiosperm H3.3 (Lu et al., 2018). Phenylalanine (F) 41 appeared first in ferns (Lu et al., 2018), and differences at position 90 (leucine replaced by alanine or serine) are found only in gymnosperm and angiosperm H3.1 proteins. It can therefore be hypothesized that the more an-cient histones were of the H3.3 type and that specific histones dedicated to DNA-synthesis-dependent deposition evolved later. Furthermore, even when we consider that H3 variants
Table 1. List of core and linker histone genes in Arabidopsis
Histone H3 Gene Histone H3 H3.1 At5g65360, HTR1 At1g09200, HTR2 At3g27360, HTR3 At5g10400, HTR9 At5g10390, HTR13 H3.3 At4g40030, HTR4 At4g40040, HTR5 At5g10980, HTR8 H3.6 At1g13370, HTR6 H3.7 At1g75610, HTR7 H3.10 At1g19890, HTR10 H3.11 At5g65350, HTR11 CenH3 At1g01370, HTR12 H3.14 At1g75600, HTR14 H3.15 At5g12910, HTR15 Histone H4 H4 At3g46320 At5g59690 At2g28740 At1g07820 At3g53730 At5g59970 At3g45930 At1g07660 Histone H2A
H2A.1 At5g54640, HTA1
H2A.2 At4g27230, HTA2
H2A.10 At1g51060, HTA10
H2A.13 At3g20670, HTA13
H2A.X.3 At1g54690, HTA3
H2A.X.5 At1g08880, HTA5
H2A.W.6 At5g59870, HTA6
H2A.W.7 At5g27670, HTA7
H2A.W.12 At5g02560, HTA12
H2A.Z.4 At4g13570, HTA4
H2A.Z.8 At2g38810, HTA8
H2A.Z.9 At1g52740, HTA9
H2A.Z.11 At3g54560, HTA11
Histone H2B H2B.1 At1g07790, HTB1 H2B.2 At5g22880, HTB2 H2B.3 At2g28720, HTB3 H2B.4 At5g59910, HTB4 H2B.5 At2g37470, HTB5 H2B.6 At3g53650, HTB6 H2B.7 At3g09480, HTB7 H2B.8 At1g08170, HTB8 H2B.9 At3g45980, HTB9 H2B.10 At5g02570, HTB10 H2B.11 At3g46030, HTB11 Histone H1 H1.1 At1g06760, H1.1 H1.2 At2g30620, H1.2 H1.3 At2g18050, H1.3
For histone H1, H2A, and H2B, each gene encodes a unique histone variant, while all H4-encoding genes encode an identical H4 protein. Several genes encode H3.1 and H3.3 proteins.
have appeared several times in evolution, the positions where the amino acid changes occur are very similar (31, 87, 89, and 90 in mammals/Drosophila, 31, 41, 87, and 90 in plants). This implies selective pressure for differences to occur exactly at these positions within the histone structure. Interestingly, in mammals the different amino acids in the histone fold domain allow specific recognition by chaperone proteins (see below) and determine their mode of deposition, either replication-dependent or -inreplication-dependent (Tagami et al., 2004; Elsässer et al., 2012; Ricketts et al., 2015). Therefore an intriguing hypothesis would be that the different histone deposition pathways de-pendent on or indede-pendent of DNA synthesis have been an evolutionary driving force in the appearance of H3.1 and H3.3 variants. The contribution of histone gene expression patterns reinforced by the different gene structures might also have contributed to sub-functionalization of H3 variants.
In addition to H3.1- and H3.3-encoding genes, the Arabidopsis genome also has five genes, HTR6, HTR10, HTR11, HTR14, and HTR15, encoding atypical H3 vari-ants named H3.6, H3.10, H3.11, H3.14, and H3.15 (Fig. 1B), respectively, while HTR7 was classified as pseudogene. H3.6, H3.10, and H3.14 carry the characteristic residues H87 and L90 present in histone H3.3, while H3.11 presents the H3.1 signatures F41, S87, and A90. HTR11 could be derived from H3.1 encoding genes, as it is also intronless. Further differences are present in these atypical H3 variants, but the functional impact of these changes has yet to be explored. While human nucleosomes containing H3.1 or H3.3 have nearly identical
structures (Tachiwana et al., 2011), a single amino acid differ-ence in human H3.6 renders nucleosomes with this variant unstable (Taguchi et al., 2017). It is of interest to note that H3.11 lacks the lysines at positions 9 and 27, which when methylated are involved in silencing of transposable elements and Polycomb-mediated gene repression, respectively.
Finally, HTR12 encodes the Arabidopsis CenH3 variant that localizes to a specific subset of the centromeric 180 bp repeats and specifies the centromere (Nagaki et al., 2003). It is the most divergent H3 histone, showing reduced identity to H3 in the histone fold domain, differences in the Loop1 region, which are thought to result in altered contacts with nucleosomal DNA, and no sequence similarity in their long N-terminal tail (Malik and Henikoff, 2003). CenH3 protein sequences vary substantially, even between closely related spe-cies, which has been suggested to involve adaptive evolution of CenH3 proteins with centromeric repeats (Talbert et al., 2002;
Simon et al., 2015).
Genome-wide distribution of histone H3 variants
In whole seedlings both the replicative H3.1 and the re-placement H3.3 are expressed, but their genome-wide dis-tribution differs substantially (Fig. 2). At the chromosome scale, H3.1 is found enriched in heterochromatic regions, while H3.3 is enriched at chromosome arms (Ingouff et al., 2010; Stroud et al., 2012; Wollmann et al., 2012). H3.3 ac-cumulates in the 3′ regions of transcribed genes and H3.3
H2A.W H3.1 H3.3 H2A.X H2A.Z H2A.Z H2A, H2A.X H3.1, H3.3 H3.1, H3.3 TSS TTS H3.1 H2A.Z CS2 CS1 CS3 CS6 CS4 CS4 TSS TTS CS2 CS5 CS4 CS4 H3.3 H3.3 H3.3 cenH3 CS8CS9
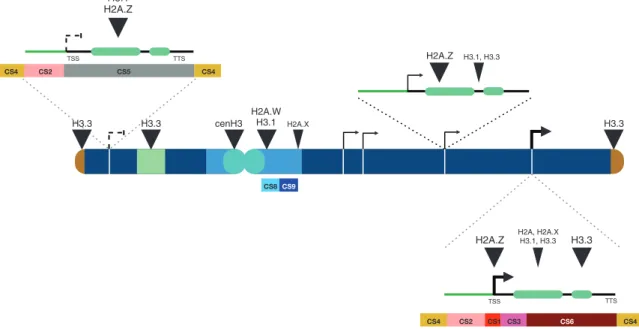
Fig. 2. Preferential location of H3 and H2A histone variants along an Arabidopsis chromosome. Telomeres (brown) and nucleolar organizer regions
(NORs, dark green) are enriched in histone H3.3, while pericentromeric regions (light blue) are enriched in H3.1 and H2A.W variants and contain lower levels of H3.3, H2A, and H2A.X. CenH3 is localized exclusively at centromeric repeats (cyan). Highly expressed genes (thick arrows; promoters, green line; exons, light green bars; introns, 5′ and 3′ UTRs, black lines; TSS, transcription start site; TTS, transcription termination site) are enriched in H2A.Z at their TSS and H3.3 at the 3′ end of the gene, while genes expressed at low levels (thin arrows) show H2A.Z all over their gene body, lower H3.3 levels, and enrichment in H3.1. Genes repressed by the Polycomb machinery (left on short arm) are enriched in H3.1 and H2A.Z across the gene bodies. Strong enrichment is designated by a large triangle, lower enrichment by a smaller triangle. Chromatin states, as described by Sequeira-Mendes et al. (2014), appear below the schematic representation, with examples of a highly expressed gene and the Polycomb repressed gene: TSS (CS1, red), proximal promoter (CS2, salmon), 5′ end of expressed genes (CS3, pink), rest of transcribed gene (CS6, brown), intergenic region (CS4, dark yellow), repressed Polycomb-regulated gene (CS5, grey). Pericentric heterochromatin contains two types, AT-rich (CS8, light blue) and GC-rich (CS9, dark blue). Genes with intermediate expression levels are more complicated to ascribe a particular state, but still they tend to contain the same organization as the very active genes, only with less active marks.
occupancy positively correlates with transcriptional activity (Stroud et al., 2012; Wollmann et al., 2012), but is also found enriched at regulatory regions (Shu et al., 2014), telomeres (Vaquero-Sedas and Vega-Palas, 2013), and rDNA repeats (Shi et al., 2011; Duc et al., 2017). Considering that H3.3 has evolved independently in plants and animals, it is interesting to note that in both kingdoms H3.3 is enriched in gene-rich regions (Mito et al., 2005; Goldberg et al., 2010) indicating evolutionary convergence. However, in contrast to plants, where H3.3 enrichment is particular high in the 3′ end of the gene and depleted from the transcription start sites (TSS), mammalian H3.3 is found around the TSS in both active and repressed genes and along gene bodies of tran-scriptionally active genes (Goldberg et al., 2010).
Post-translational modifications of histone H3 variants Given the differences in primary amino acid sequence be-tween H3.1 and H3.3, it can be asked to what extent these two histone variants undergo differential post-translational modi-fications. Indeed, mass-spectrometry analysis conducted in mammals (Loyola et al., 2006) and plants (Johnson et al., 2004) identified preferential enrichment of H3.1 and H3.3 variants in certain histone modifications. Using purification of tagged histone variants from HeLa cells (Loyola et al., 2006), a pref-erential enrichment of H3.1 in the repressive marks histone H3 lysine-9 methylation (H3K9me3) and H3K27me3 was observed, while H3K36me3 and Histone H3 lysine-9 acetyl-ation (H3K9ac) were preferentially enriched on the replace-ment variant H3.3. Similarly in Arabidopsis, more H3K27me1 and H3K27me3 is found on H3.1 compared with H3.3 and the reverse is true for mono-, di-, and trimethylated forms of H3K36 (Johnson et al., 2004). It can be argued that these dif-ferences are solely the consequence of the differential enrich-ment of histone variants along the genome, but at least for one modification, H3K27me1, enriched in heterochromatin (Mathieu et al., 2005; Jacob et al., 2009), the amino acid se-quence of the H3 variant determines the methylation state when this modification is set by the histone methyltransferases ARABIDOPSIS TRITHORAX-RELATED PROTEIN 5 (ATXR5) and ATXR6 (Jacob et al., 2009). A single amino-acid difference, namely the threonine at position 31 in H3.3 compared with alanine in H3.1, confers specificity of H3.1 over H3.3 as the threonine causes steric hindrance when it is encountered by the ATXR5/6 methyltransferases preventing methylation of the replacement variant H3.3 (Jacob et al., 2014). ATXR5 and 6 contain a PROLIFERATING CELL NUCLEAR ANTIGEN (PCNA) interacting protein box and have therefore been hypothesized to function directly at the replication fork and to methylate de novo deposited H3.1 his-tones. The perpetuation of H3K27me1 methylation at the rep-lication fork via H3.1 methylation is also required to propagate the H3K27me3 mark, which is maintained through Polycomb group proteins tethered to the replication fork via PCNA (Jiang and Berger, 2017).
Taken together, accumulating evidence suggests that the amino acid differences between histone H3 variants can be of
functional importance for downstream processes such as post-translational modifications.
Handling histone H3 variants
Due to their basic nature, histones can undergo unspecific interactions with other proteins and cellular nucleic acids. To avoid such unscheduled interactions, histone proteins are ac-companied from their synthesis to their deposition by specific proteins or protein complexes called histone chaperones (De Koning et al., 2007).
Histone H3 dynamics during DNA replication
DNA replication is a critical moment during the cell cycle that challenges epigenetic inheritance, but also presents a window of opportunity to initiate changes in chromatin structure. The replication of the genomic DNA requires not only access to the DNA template and therefore transient eviction of histones, but also considerable de novo histone deposition to ensure mainten-ance of nucleosomal density. Most of the parental histones are recycled to allow faithful copying of chromatin marks, which permits preservation of epigenetic information present on these parental histones (Reverón-Gómez et al., 2018). The dis-tribution of parental H3–H4 tetramers at the replication fork is mostly random with a slight bias towards the lagging strand in yeast (Yu et al., 2018a), a phenomenon that requires leading strand polymerase epsilon (Pol ε) and its binding partners Dpb3 and Dpb4. Likewise in mammals, subunits of Pol ε have been shown to function as histone chaperones and to ensure the balance between parental recycling and new histone de-position (Bellelli et al., 2018). Mutants in the Arabidopsis Pol ε have been shown to affect repression of genes and transposons (Pedroza-Garcia et al., 2016; del Olmo et al., 2016), but whether this is due to imbalanced recycling of histones together with their parental marks or general defects in nucleosomal occu-pancy remains to be established. Nevertheless, the catalytic subunit of Pol ε binds to components of the Polycomb com-plex thereby establishing a link between DNA replication and histone modifications (Pedroza-Garcia et al., 2016; del Olmo
et al., 2016; Jiang and Berger, 2017).
Recycling of parental histones happens concomitantly with de novo histone deposition that is ensured through the action of the evolutionarily highly conserved histone chaperone complex CHROMATIN ASSEMBLY FACTOR 1 (CAF-1) (Fig. 3). In plants, CAF-1 consists of the subunits FASCIATA1 (FAS1), FAS2, and MULTICOPY SUPPRESSOR OF IRA (MSI1), the first two likely being specific to the CAF-1 com-plex, while MSI1 is also part of chromatin remodeling or Polycomb complexes (Hennig et al., 2005). CAF-1 is recruited to newly replicated DNA via interaction between its largest subunit (FAS1) and PCNA (Jiang and Berger, 2017). Seminal experiments in mammalian cells using epitope-tagged histone H3.1 and H3.3 proteins revealed that subunits of the CAF-1 complex were precipitated together with H3.1, but not with H3.3 (Tagami et al., 2004). In line with the peak in H3.1 ex-pression during S-phase, this demonstrated that Arabidopsis
CAF-1 deposits H3.1 at the replication fork. Similar to what was observed in other organisms, CAF-1 incorporates pref-erentially the replicative histone variant H3.1, as fas mutants show reduced H3.1 incorporation (Otero et al., 2016; Jiang and Berger, 2017; Benoit et al., 2019). However, in mutant contexts of CAF-1 knockout, H3.1 incorporation is not com-pletely abolished. Not all non-nucleosomal H3.1 is therefore degraded but partially ends up being incorporated via other pathways. Surprisingly, plants lacking a functional CAF-1 complex were described nearly 20 years ago and, contrary to mouse (Houlard et al., 2006) or Drosophila (Klapholz et al., 2009), these are viable and give rise to progeny, but show many phenotypes in part linked to deficient meristem maintenance (Kaya et al., 2001). Nevertheless, the fact that plants deficient in replication-coupled histone deposition can be identified points to important plasticity in histone handling in plants.
For histones, being readily available for deposition requires not only elevated expression during S-phase, but also the pres-ence of a non-nucleosomal histone pool. Histone chaperones Anti Silencing Factor 1 (ASF1) and Nuclear Autoantigenic Sperm protein (NASP) have been shown to be involved in binding to newly synthesized histones and in coordin-ating their nuclear import as H3–H4 dimers (Campos et al., 2010). Indeed, ASF1 binds histone dimers (English et al., 2006;
Natsume et al., 2007) and hands over histones to the CAF-1
complex. Arabidopsis encodes two highly similar ASF1 pro-teins that can complement each other’s function, as single mu-tants show no particular phenotype (Zhu et al., 2011). Double mutants are viable, but show defects in gametophytic develop-ment (Min et al., 2019) and altered transcriptional response to heat stress compared with wild type plants (Weng et al., 2014).
Deposition of replacement histone variants
In mammals, the replacement histone variant H3.3 is in-corporated through dedicated deposition pathways involving the histone chaperone complex HIRA (Ray-Gallet et al., 2002) and Alpha Thalassemia-mental Retardation X-linked (ATRX)–DAXX (Goldberg et al., 2010) (Fig. 3). As in mam-mals, the Arabidopsis HIRA complex consists of three sub-units, HIRA, CABIN, and UBINUCLEIN (Nie et al., 2014;
Duc et al., 2015), and HIRA can be immunoprecipitated from plants expressing tagged histone H3.3 (Nie et al., 2014). Loss of HIRA has little impact on plant phenotype, but this pathway of histone deposition is essential in a situation when replication-coupled deposition is impaired and its absence re-sults in reduced nucleosome occupancy both in genes and in heterochromatin (Duc et al., 2015). Given that deletion of all three H3.3 genes is lethal (Wollmann et al., 2017), the only subtle impact on plant phenotype in hira mutants suggested
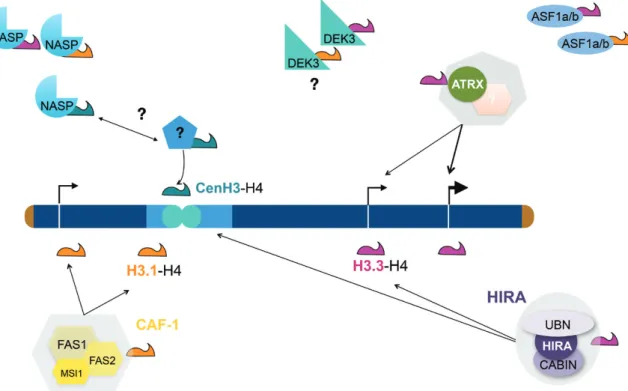
Fig. 3. Scheme illustrating our current model of de novo histone deposition in plants. The CHROMATIN ASSEMBLY FACTOR 1 (CAF-1, yellow)
consisting of FASCIATA 1 (FAS1), FAS2, and MULTICOPY SUPPRESSOR OF IRA (MSI1) (Kaya et al., 2001) deposits the replicative variant H3.1 (orange) in a DNA synthesis-dependent manner throughout the genome (Jiang and Berger, 2017; Benoit et al., 2019). The replacement variant H3.3 (magenta) is deposited by the HIRA complex (UBINUCLEIN 1/2, HIRA, and CALCINEURIN BINDING PROTEIN 1 (CABIN1)) (Nie et al., 2014; Duc et al., 2015). It likely functions both in euchromatin (dark blue) and in heterochromatin (light blue) and it is important for rescuing nucleosomal occupancy when DNA synthesis-dependent histone deposition is impaired (Duc et al., 2015). H3.3 can also be deposited via another pathway implicating ATRX, which may function with a yet unknown partner. The small histone chaperone ANTI SILENCING FACTOR 1 (ASF1) (Zhu et al., 2011) is thought to transport H3–H4 dimers and hand them over to the HIRA and CAF-1 complexes. Whether the two paralogues ASF1a and ASF1b would preferentially hand histone dimers over to CAF-1 or HIRA in plants is not known. NUCLEAR AUTOANTIGENIC SPERM PROTEIN (NASP) not only escorts H3.1 and H3.3 (Maksimov et al., 2016) but also binds CenH3 (Le Goff et al., 2020). NASP may hand over CenH3 to its actual deposition factor, which still remains to be identified. Finally, DEK3 binds histone H3 (Waidmann et al., 2014).
the existence of alternative pathways of replacement histone deposition. Genetics as well as molecular analysis showed that the plant orthologue of ATRX is involved in a complementary pathway of histone deposition and that its loss reduces H3.3 occupancy (Duc et al., 2017; Wang et al., 2018). In the mam-malian ATRX–DAXX heterodimer, the DAXX protein brings specificity to H3.3 over H3.1 as it favors the glycine residue at position 90 in H3.3 over the methionine in H3.1 (Elsässer
et al., 2012). No homologue of DAXX has been identified in plants. Arabidopsis ATRX itself binds histones (Duc et al., 2017), but whether it partners with a yet unknown histone binding protein remains to be determined. Finally, DEK pro-teins have also been suggested to be involved in histone depos-ition. DEK3 (one of the four DEK proteins in Arabidopsis) can interact with H3 and H4 and affects nucleosome occupancy at a few loci (Waidmann et al., 2014). DEK3 is enriched at the 3′ end of genes similar to H3.3, suggesting that it may have a preference for H3.3 over H3.1 variants (Brestovitsky et al., 2019, Preprint). While replacement histone variant deposition seems to employ evolutionarily highly conserved pathways, the centromeric variant CenH3 uses distinct deposition machin-eries in different species such as Holliday junction recognition protein (HJURP) in humans (Dunleavy et al., 2009; Foltz et al., 2009) or the fly-specific protein CAL1 in Drosophila (Phansalkar
et al., 2012). The existence of plant-specific CenH3 assembly proteins can therefore be expected. Contrary to mammalian CenH3, which is deposited during early G1 (Jansen et al., 2007), plant CenH3 is deposited in the G2 phase of the mi-totic cell cycle (Lermontova et al., 2006) and CenH3 levels are reduced in mutants of the KINETOCHORE NULL2 (KNL2) homologue (Lermontova et al., 2013). While so far no CenH3 deposition factor could be identified in plants, recent evidence shows that Arabidopsis NASP functions as a CenH3 escort factor (Le Goff et al., 2020). Finally, histone exchange and recycling also occurs in a co-transcriptional manner that involves also SPT6 and the FACILITATES CHROMATIN TRANSCRIPTION (FACT) complex (Duroux et al., 2004;
Grasser, 2020).
In summary, most histone chaperone complexes are evolu-tionarily highly conserved, suggesting conserved H3 variant deposition modes, but given the convergent evolution of his-tone variants, it can be expected that some of these complexes have adopted plant-specific characteristics.
Specific functions for different H3 variants?
Plants with strongly reduced expression of H3.1-encoding genes show developmental defects that resemble caf1 mutants (Kaya et al., 2001), likely due to H3.1 deposition coupled to DNA synthesis and the fact that H3.1 can be modified dir-ectly at the replication fork (Jiang and Berger, 2017). These phenotypes cannot be rescued by expressing H3.3 under con-trol of an H3.1 promoter, clearly demonstrating that correct expression timing or simply H3 availability is not critical for rescue, but that the difference in amino-acid sequences be-tween these two variants is essential (Jiang and Berger, 2017). Similarly, simultaneous knockout of all three H3.3 encoding
genes was found to cause lethality (Wollmann et al., 2017), and plants with reduced H3.3 levels show altered phenotypes as well as changes in gene expression patterns. In the absence of a clear correlation between H3.3 enrichment and gene ex-pression changes, this questions a direct impact of H3.3 on transcription. Nevertheless, depletion of H3.3 led to invasion of gene bodies with linker histone H1 and reduced gene body DNA methylation (Wollmann et al., 2017).
H3.1 and H3.3 variant dynamics during cell proliferation and differentiation
The spatiotemporal regulation of gene expression is intimately coordinated with chromatin composition, including different histone variants containing a variety of post-translational modi-fications. Chromatin changes are also typical of cells at dif-ferent cell cycle stages and during difdif-ferentiation (Pfluger and Wagner, 2007; Kundaje et al., 2015). Based on this information, Otero et al. hypothesized that changes in the H3.1/H3.3 ratio along the root longitudinal axis (Birnbaum et al., 2003) could be associated with different cell proliferation rates (Hofmann, 2016; Otero et al., 2016). Indeed, they demonstrated that the H3.1/H3.3 ratio is an excellent proxy of the proliferative status of a cell. Moreover, they observed that a massive replacement of H3.1 with H3.3 occurs in root meristem cells during the last G2 phase, which is longer than in other dividing cells, prior to entering the differentiation stage. This is strikingly remin-iscent of what has been reported to occur during the last cell cycle of the Drosophila embryo, which is unique in having a longer G2 phase (Blythe and Wieschaus, 2015). These findings underscore the relevance of the H3.1/H3.3 balance in cell proliferation since the same occurs in other plant tissues where cells are actively proliferating and exit to differentiation, e.g. the stomatal lineage. Furthermore, a similar process also occurs in cells undergoing the endocycle, thus allowing the identifica-tion of cells in their last stages before initiating differentiaidentifica-tion (Otero et al., 2016).
Specific H3 variants at the gametophytic stage
Important histone H3 variant dynamics have been reported during both male and female gametophyte development: not all H3 genes are expressed at this developmental stage (Ingouff
et al., 2007, 2010) and nuclei-specific H3 gene expression pat-terns were observed. The male vegetative nucleus expresses the H3.3 genes HTR5 and HTR8 as well as HTR14 encoding the atypical H3.14 histone variant (Okada et al., 2005; Ingouff
et al., 2010). In contrast, the germ cell nucleus contains H3.3 (from the HTR5 gene), the centromeric CenH3, and the pollen-specific H3.10. In this way, enrichment in H3.10 and H3.14 correlates with opposing chromatin states: the small sperm nuclei present highly condensed chromatin while the vegetative nuclei are characterized by large nuclei with rather decondensed chromatin lacking conspicuous heterochromatin structures. In the female gametophyte, the egg cell expresses CenH3, high levels of HTR5, and low levels of HTR8, whereas the central cell shows the reverse pattern (low HTR5 and high HTR8 gene expression), plus a small amount of H3.1 and
H3.14 (Ingouff et al., 2010). Despite these specific expression patterns, the knockout of the HTR10 gene is fertile (Okada
et al., 2005), suggesting that it rather has a role in fine tuning histone replacement and maybe gene expression during this particular developmental stage.
Taken together, accumulating evidence suggest not only that replicative and replacement H3 histone variants are re-quired for correct and timely deposition of histones at different moments during the cell cycle to ensure nucleosome occu-pancy, but also that the incorporation of specific variants with subtle differences in primary amino acid sequences has func-tional consequences. The exact role of these dynamics in pos-sible transcriptome changes, reprogramming, or transmission of post-translational marks is an exciting question to explore.
H2A and H2B variants
Further variability in nucleosome composition is achieved through incorporation of different H2A and H2B variant proteins. The Arabidopsis genome encodes several H2A pro-teins, four replicative H2A variants, four H2A.Z, two H2A.X, and three H2A.W variants, which are specific for the plant kingdom (Table 1). Differences in the primary amino acid sequences between the H2A variants can be noted in their C-terminal motifs (Kawashima et al., 2015). As the H2A C-terminal tail is located at the DNA entry/exit site of the nucleosome, these differences may impact nucleosomes sta-bility. H2A.Z and H2A.W also differ from H2A and H2A.X in their Loop1 region (Kawashima et al., 2015). Compared to the other H2A variants, H2A.Z also carries a distinct and highly conserved docking domain (comprising ~40 amino acids in a region extending from the α2-helix to the C-terminal tail) that interacts with histone H3 and that has similarities to the corresponding domain in other eukaryotes (Kawashima et al., 2015). These distinctive features confer specific characteristics to the nucleosomes, e.g. an H2A.Z–H2B dimer is displaced more rapidly than a dimer containing other H2A variants (Osakabe et al., 2018). Furthermore, the H2A.W C-terminal tail makes contacts with linker DNA and protects it from di-gestion by MNase. Interestingly and in contrast to animals and yeast, plant nucleosomes are homotypic, i.e. they contain only one type of H2A variant (Osakabe et al., 2018). Even within the H2A.W variant family consisting of H2A.W.6, H2A.W.7, and H2A.W.12, the majority of nucleosomes contain either H2A.W.6 or H2A.W.7 suggesting either that specific path-ways ensure deposition of these different proteins or that the small differences between the variants are sufficient to desta-bilize certain histone variant combinations and to favor others. Using tagged variants representative of each of the four his-tone variant groups, Yelagandula et al. (2014) characterized their genome-wide distribution. While H2A.X was enriched over the entire genome, H2A and H2A.Z were depleted from pericentromeric heterochromatin, which is instead enriched in the H2A.W variant (Fig. 2). At the gene level, highly expressed genes are particularly enriched in H2A.Z at the TSS and 5′ end of genes, while inactive genes or those expressed at low levels
are enriched in H2A.Z over their gene body (Coleman-Derr and Zilberman, 2012; Yelagandula et al., 2014).
H2A.Z deposition plays a dual role in transcriptional regu-lation depending on the genomic location where it is placed, a situation contrary to that of H2A.Z in animal cells. It can be associated with gene activation when it is enriched at the +1 nucleosome relative to the transcriptional start site (TSS) to-gether with increased H3K36ac (Mahrez et al., 2016), and with gene repression when located within gene bodies (Sura et al., 2017) (Fig. 2), which depends on mono-ubiquitination carried out by PRC1 Polycomb complexes (Gómez-Zambrano et al., 2019). Nucleosomes containing H2A.Z appear to have specific structural features that can mediate a transcription-independent response to ambient temperature (Kumar and Wigge, 2010). The replacement of canonical H2A by the H2A.Z variant has been revealed as a widespread chromatin feature with implications for multiple aspects of plant physiology. These include, for example, several developmental phase transitions (Jarillo and Piñeiro, 2015), metabolism (Nützmann and Osbourn, 2015; Yu et al., 2016), immunity (Berriri et al., 2016), or stress responses ( Asensi-Fabado et al., 2017), including changes in ambient temperature (Cortijo et al., 2017; Tasset et al., 2018; del Olmo et al., 2019).
The H2A.X histone variant plays a crucial role in DNA repair in both plants and mammals. In the presence of double strand breaks, the Ataxia Telangiectasia Mutated (ATM) kinase and to a lesser extent the Ataxia Telangiectasia Mutated and Rad3-related (ATR) kinase (Friesner et al., 2005) phosphor-ylate the C-terminal domain of H2A.X (γ-H2A.X) at the conserved SQ motif. This is a key signal to recruit other factors necessary to trigger the DNA damage response (van Attikum and Gasser, 2009). H2A.X phosphorylation can occur across relatively large genomic regions that can span up to 1 Mb in mammalian cells and around 100 kb in plants, allowing the formation of foci that can be visualized under the micro-scope by specific antibodies recognizing the phosphorylated form of H2A.X. In plants, ATR also phosphorylates H2A.X to signal DNA replication stress (Amiard et al., 2010). A rather surprising finding was that DNA repair in heterochromatin needs a specialized H2A variant, H2A.W.7. This was identified in a proteomic analysis searching for phosphorylated proteins detected after DNA damage. In this study, H2A.W.7 peptides were retrieved phosphorylated at the SQ motif, a consensus for the ATM/ATR kinases (Roitinger et al., 2015).
H2A.W variants were found earlier to be an exclusively het-erochromatic mark needed for proper deposition of H3K9me2 and cytosine methylation, and for transposable element silen-cing (Yelagandula et al., 2014). Remarkably, H2A.W vari-ants are typical of land plvari-ants and all contain the consensus SPKK motif (Yelagandula et al., 2014). The H2A.W.6 variant promotes interchromatin fiber associations dependent on the SPKK motif in its C-terminus (Yelagandula et al., 2014). This is consistent with the progressive increase in the enrich-ment of H3.1 and H2A.W.6 that occur during chromocenter maturation (Benoit et al., 2019). The current view is that in Arabidopsis H2A.W plays a role analogous to the HP1 and KAP1 proteins in mammalian cells (Lorković et al., 2017;
Lorković and Berger, 2017).
Several H2B variants are present in plants (e.g. 11 different variants in Arabidopsis, which are termed H2B.1–H2B.11;
Table 1), but our knowledge of their genome-wide distribution and possible function in chromatin organization is sparse. An H2B variant specifically expressed in pollen has been reported in lily (Ueda et al., 2000) and several post-translational modi-fications have been determined for H2B variants isolated from Arabidopsis cell cultures (Bergmüller et al., 2007). Furthermore, in rice, the Switch (SWI)/Sucrose Non-Fermentable (SNF)-2 ATPase BRHIS1 was found to interact specifically with the mono-ubiquitinated form of rice H2B.7, illustrating the pos-sibility that certain proteins are able to distinguish between closely related H2B variants. However, not much is known of whether these variants are enriched preferentially at transcrip-tionally active or silent regions or whether they show prefer-ential association with specific H2A variants.
Deposition of H2A variants
In comparison with the H3–H4 tetramer, H2A–H2B di-mers are exchanged more rapidly and to date much less is known concerning their mode of deposition. The Arabidopsis genome encodes four different NUCLEOSOME ASSEMBLY PROTEINs 1 (NAP1s) as well as two NAP1-RELATED PROTEINs (NRPs), which bind H2A–H2B dimers (Dong
et al., 2005; Zhu et al., 2006). While NAP1s are mainly lo-calized in the cytoplasm, NRPs are lolo-calized in the nucleus. Surprisingly, loss of all NAPs and NRPs in a sextuple mutant has no impact on plant growth under standard growth condi-tions, but affects gene expression and renders the plant more susceptible to DNA-damaging agents (Liu et al., 2009; Zhou
et al., 2016). To what extend loss of NAPs and NRPs (Zhou
et al., 2015b) disturbs the genome-wide distribution of all or a specific set of H2A variants and their dynamics remains to be determined. During transcription and replication, H2A–H2B dimers are also handled by the FACT complex (Lolas et al., 2010; Grasser, 2020).
H2A.Z deposition relies on the activity of SWR1c (after the yeast SWi2/snf2-Related 1 (SWR1) complex), a SWI/ SNF chromatin remodeling complex (reviewed in Wang et al., 2019) that contains 11 subunits identified by various combin-ations of mass spectrometry approaches and conserved with those in the yeast complex (Potok et al., 2019; Sijacic et al., 2019; Nie et al., 2019). One of its components, INOSITOL REQUIRING 80 (INO80), has been widely studied in its co-ordinated role with ETHYLENE INSENSITIVE 6 (EIN6)/ RELATIVE OF EARLY FLOWERING 6 (REF6) in con-trolling the level of H3K27me3 and H2A.Z deposition at the 5′UTR of ETHYLENE INSENSITIVE 2 (EIN2), a key regulator of ethylene signaling (Zander et al., 2019). SWR1c complexes are recruited to the genomic sites of H2A.Z ex-change by the SWR1 COMPLEX 4 (SWC4) factor ( Gómez-Zambrano et al., 2018) and can interact with other chromatin modifiers such as BRAHMA (Torres and Deal, 2019) and the methyl-CpG-BINDING DOMAIN 9 (MBD9) (Potok et al., 2019; Sijacic et al., 2019; Nie et al., 2019).
Histone H1 variants
Linker histones have a conserved tripartite structure consisting of a short N-terminal tail, a central globular (GH1) domain, and a long intrinsically disordered C-terminal tail. The globular domain binds to the nucleosome dyad and the C-terminal tail makes contacts with linker DNA (Zhou et al., 2015a). Human histone H1 has been reported to play a role in modulating chromatin structure by interacting with SWI/SNF ATP-dependent chromatin remodeling complexes (Ramachandran
et al., 2003). Likewise, plant SWI/SNF complexes and histone H1 are important for nucleosome positioning (Jerzmanowski, 2007). In fact, members of the SWI family of chromatin remodeling complexes, such as maize CHB101, an Arabidopsis SWI3D homologue, are required for providing proper nucleo-some density around the transcriptional start sites of genes that respond to abiotic stress (Yu et al., 2018b).
In humans and mouse, 11 different H1 variants have been identified (Fyodorov et al., 2018), while the Arabidopsis genome encodes three variants: H1.1, H1.2, and H1.3 (Table 1). H1.3 differs from H1.1 and H1.2 by a shorter N- and C-terminal tail, lacking (S/T)PXK motifs involved in DNA binding. H1.3 con-sequently shows higher mobility within chromatin. The main variants H1.1 and H1.2 are highly abundant chromatin com-ponents and are enriched in heterochromatin and gene bodies anti-correlating with gene expression (Rutowicz et al., 2015) in agreement with studies suggesting that H1 binding is hindered by the presence of H3.3 (Braunschweig et al., 2009; Wollmann
et al., 2017). Consistent with these properties, the main H1 vari-ants are necessary but not sufficient for proper heterochromatin compaction, although not for transcriptional silencing of trans-posable elements (Rutowicz et al., 2019; Benoit et al., 2019). Furthermore, H1.1 and H1.2 are required for efficient depos-ition of H3K27me3 and regulation of a subset of target genes relevant for flowering, lateral root formation, and seed develop-ment (Rutowicz et al., 2019). Although the main role of histone H1.1 and H1.2 is in chromatin compaction, H1.3 is necessary for proper response to abiotic stress. Under non-stressed con-ditions H1.3 is expressed only in a few cell types such as guard cells, but is strongly induced by low light, drought, and ABA (Rutowicz et al., 2015). H1.3 therefore occurs in both con-stitutive (but guard-cell specific) and stress-inducible histone pools. Upon induction, H1.3 is thought to compete with H1.1 and H1.2 for the same binding sites (Rutowicz et al., 2015), but may induce altered chromatin features given its different structure and shorter chromatin residence time. The role of H1.3 in stress response was confirmed by transcriptomic ana-lysis of h1.3 mutants that were impaired in normal growth, sto-matal closure, and other adaptive responses to light and drought (Rutowicz et al., 2015). Arabidopsis H1 proteins undergo many post-translational modifications (Kotliński et al., 2016), and the way H1.3 functions may depend upon specific ones. However, the identification of these modifications is not trivial, not only because they may occur in a limited number of cells or gen-omic locations to modify gene expression, but also because they occur extremely fast upon stress, as demonstrated for H1.3 (Rutowicz et al., 2015).
Histone variants and the chromatin
landscape
Histone variants show specific localization in the genome ac-cording to their expression pattern and modes of deposition. This together with epigenetic modifications contributes sub-stantially to the structural and functional complexity of chro-matin. An initial effort to define chromatin states (CSs) and reduce this combinatorial complexity was made in Arabidopsis and identified active, repressed, silent, and intergenic regions (Roudier et al., 2011). Genome-wide studies also including DNA features and histone H3 and H2A variants as well as nucleosomal occupancy reported the identification of nine CSs (Sequeira-Mendes et al., 2014). It is important to note that these CSs co-localize with functional genomic elem-ents (Fig. 2) such as TSS (CS1), proximal promoters (CS2), 5′ end of transcribed genes (CS3), intergenic regions (CS4), Polycomb chromatin (CS5), 3′ end of transcribed genes (CS6), long coding sequences (CS7), AT-rich heterochromatin (CS8), and GC-rich heterochromatin (CS9).
The presence of histone variants constituted a key feature for defining CSs. Thus, histone H3.3 is enriched in proximal promoters (CS2), particularly ≤500 bp upstream from the TSS, and at the 3′ end of more active genes (CS6). These regions are characterized by accessible chromatin as revealed by DNase I footprinting (Shu et al., 2012). Some distal regulatory regions, e.g. enhancers, as well as TTS and the 5′ end of active genes are enriched in H2A.Z. Intriguingly, while most permissive post-translational modifications are poorly enriched at large transcribed genes, H2A.Z is maintained, including the region around the TTS. H2A.Z is also enriched across the gene body, but not at the TSS or 5′ end of genes, in inactive genes, e.g. Polycomb-regulated genes, or genes expressed at low levels. On the contrary, this variant is depleted from heterochromatin (Sequeira-Mendes et al., 2014; Sequeira-Mendes and Gutierrez, 2015). H3.3 is highly depleted in Polycomb chromatin (CS5) and heterochromatin (CS8 and 9), but in turn, the latter is highly enriched in the replicative histone H3.1.
Therefore, it is clear that the presence of different histone variants at specific locations of different genomic elements con-fers key functional features including not only gene expression regulation, but also the activity of DNA replication origins (Vergara and Gutierrez, 2017; Sequeira-Mendes et al., 2019).
Perspectives
Given the functional and structural constraints of nucleosome organization, histones are some of the most conserved proteins. Even small differences in the amino acid sequence are expected to have an enormous impact on nucleosomal structure and to affect transcriptional output (Nacev et al., 2019). As the histones need to fit together as compatible bricks, it can be expected to find preferential combinations of different histone variants within the same nucleosome that together may have a stronger or different impact on nucleosome stability as the presence of one of the variants alone. As an example, nucleosomes that con-tain both H3.3 and H2A.Z are particularly unstable compared with H3.1–H2A.Z combinations (Jin and Felsenfeld, 2007; Jin
et al., 2009), but little information is available concerning pref-erential combinations of histone variants in plant nucleosomes. The observation that many histone variants have emerged in-dependently in plants and animals and may have only a few differences in the primary amino acid sequence at nearly iden-tical positions underscores the structural constraints within the nucleosome. This further suggests that these differences are of functional significance in determining both mode of depos-ition and functional output and that we may learn more by comparing histone variants between species as well as by ana-lysing potential co-evolution with their binding partners and deposition machinery. It also implies that much remains to be discovered of how histone variants are deposited at the right place and right time and what the functional consequences are of their locus-specific deposition. Dynamics of histone variants likely participate in all developmental or environmental re-sponses that involve major changes in chromatin structure and gene expression, such as circadian rhythm (Tong et al., 2020), transcriptional responses, and chromatin reorganization in re-sponse to stress (Zhu et al., 2013; Probst and Mittelsten Scheid, 2015) or developmental transitions such as flowering (Jarillo and Piñeiro, 2015). Gaining further insights into these and re-lated questions will require studies at different levels, including structural, molecular, cellular, genetic, and developmental, and will be enriched with novel technological advances. Therefore, exciting avenues to be explored appear ahead.
Acknowledgements
We apologize to all colleagues whose work could not be cited due to space limitations and thank S. Amiard and C. Tatout for critical reading of the manuscript. AP acknowledges networking support from European Cooperation in Science and Technology Action CA16212 (http://www.cost. eu/COST_Actions/ca/CA16212) and support from the 16-IDEX-0001 – CAP 20-25 challenge 1. Research in the CG’s laboratory is supported by grants RTI2018-094793-B-I00 (Ministerio de Ciencia y Universidades and Fondo Europeao de Desarrollo Regional) and ERC-2018-AdG_833617 (European Research Council Advanced Grant, European Union), and by institutional grants from Banco de Santander and Fundacion Ramon Areces to the Centro de Biologia Molecular Severo Ochoa.
References
Amiard S, Charbonnel C, Allain E, Depeiges A, White CI, Gallego ME.
2010. Distinct roles of the ATR kinase and the Mre11-Rad50-Nbs1 complex in the maintenance of chromosomal stability in Arabidopsis. The Plant Cell
22, 3020–3033.
Asensi-Fabado MA, Amtmann A, Perrella G. 2017. Plant responses to
abiotic stress: the chromatin context of transcriptional regulation. Biochimica et Biophysica Acta. Gene Regulatory Mechanisms 1860, 106–122. Bellelli R, Belan O, Pye VE, Clement C, Maslen SL, Skehel JM, Cherepanov P, Almouzni G, Boulton SJ. 2018. POLE3-POLE4 is a
his-tone H3-H4 chaperone that maintains chromatin integrity during DNA repli-cation. Molecular Cell 72, 112–126.e5.
Benoit M, Simon L, Desset S, Duc C, Cotterell S, Poulet A, Le Goff S, Tatout C, Probst AV. 2019. Replication-coupled histone H3.1 deposition
determines nucleosome composition and heterochromatin dynamics during Arabidopsis seedling development. New Phytologist 221, 385–398. Bergmüller E, Gehrig PM, Gruissem W. 2007. Characterization of
post-translational modifications of histone H2B-variants isolated from Arabidopsis
thaliana. Journal of Proteome Research 6, 3655–3668.
Berriri S, Gangappa SN, Kumar SV. 2016. SWR1 chromatin-remodeling
complex subunits and H2A.Z have non-overlapping functions in immunity and gene regulation in Arabidopsis. Molecular Plant 9, 1051–1065.
Birnbaum K, Shasha DE, Wang JY, Jung JW, Lambert GM, Galbraith DW, Benfey PN. 2003. A gene expression map of the Arabidopsis root. Science 302, 1956–1960.
Blythe SA, Wieschaus EF. 2015. Zygotic genome activation triggers
the DNA replication checkpoint at the midblastula transition. Cell 160,
1169–1181.
Braunschweig U, Hogan GJ, Pagie L, van Steensel B. 2009. Histone
H1 binding is inhibited by histone variant H3.3. The EMBO Journal 28,
3635–3645.
Brestovitsky A, Ezer D, Waidmann S, et al. 2019. DEK influences the
trade-off between growth and arrest via H2A.Z-nucleosomes in Arabidopsis. bioRxiv 829226. [Preprint].
Campos EI, Fillingham J, Li G, et al. 2010. The program for processing
newly synthesized histones H3.1 and H4. Nature Structural & Molecular Biology 17, 1343–1351.
Chaubet N, Clement B, Gigot C. 1992. Genes encoding a histone
H3.3-like variant in Arabidopsis contain intervening sequences. Journal of Molecular Biology 225, 569–574.
Coleman-Derr D, Zilberman D. 2012. Deposition of histone variant
H2A.Z within gene bodies regulates responsive genes. PLoS Genetics 8,
e1002988.
Cortijo S, Charoensawan V, Brestovitsky A, Buning R, Ravarani C, Rhodes D, van Noort J, Jaeger KE, Wigge PA. 2017. Transcriptional
regulation of the ambient temperature response by H2A.Z nucleosomes and HSF1 transcription factors in Arabidopsis. Molecular Plant 10, 1258–1273. De Koning L, Corpet A, Haber JE, Almouzni G. 2007. Histone
chap-erones: an escort network regulating histone traffic. Nature Structural & Molecular Biology 14, 997–1007.
del Olmo I, López JA, Vázquez J, Raynaud C, Piñeiro M, Jarillo JA.
2016. Arabidopsis DNA polymerase ϵ recruits components of Polycomb repressor complex to mediate epigenetic gene silencing. Nucleic Acids Research 44, 5597–5614.
del Olmo I, Poza-Viejo L, Piñeiro M, Jarillo JA, Crevillén P. 2019. High
ambient temperature leads to reduced FT expression and delayed flowering in Brassica rapa via a mechanism associated with H2A.Z dynamics. The Plant Journal 100, 343–356.
Dong A, Liu Z, Zhu Y, Yu F, Li Z, Cao K, Shen WH. 2005. Interacting
proteins and differences in nuclear transport reveal specific functions for the NAP1 family proteins in plants. Plant Physiology 138, 1446–1456.
Draizen EJ, Shaytan AK, Mariño-Ramírez L, Talbert PB, Landsman D, Panchenko AR. 2016. HistoneDB 2.0: a histone database with variants—
an integrated resource to explore histones and their variants. Database
2016, baw014.
Duc C, Benoit M, Détourné G, et al. 2017. Arabidopsis ATRX
modu-lates H3.3 occupancy and fine-tunes gene expression. The Plant Cell 29,
1773–1793.
Duc C, Benoit M, Le Goff S, Simon L, Poulet A, Cotterell S, Tatout C, Probst AV. 2015. The histone chaperone complex HIR maintains
nucleo-some occupancy and counterbalances impaired histone deposition in CAF-1 complex mutants. The Plant Journal 81, 707–722.
Dunleavy EM, Roche D, Tagami H, Lacoste N, Ray-Gallet D, Nakamura Y, Daigo Y, Nakatani Y, Almouzni-Pettinotti G. 2009.
HJURP is a cell-cycle-dependent maintenance and deposition factor of CENP-A at centromeres. Cell 137, 485–497.
Duroux M, Houben A, Růzicka K, Friml J, Grasser KD. 2004. The
chro-matin remodelling complex FACT associates with actively transcribed re-gions of the Arabidopsis genome. The Plant Journal 40, 660–671. Elsaesser SJ, Goldberg AD, Allis CD. 2010. New functions for an old
variant: no substitute for histone H3.3. Current Opinion in Genetics & Development 20, 110–117.
Elsässer SJ, Huang H, Lewis PW, Chin JW, Allis CD, Patel DJ. 2012.
DAXX envelops a histone H3.3-H4 dimer for H3.3-specific recognition. Nature 491, 560–565.
English CM, Adkins MW, Carson JJ, Churchill ME, Tyler JK. 2006.
Structural basis for the histone chaperone activity of Asf1. Cell 127,
495–508.
Foltz DR, Jansen LE, Bailey AO, Yates JR 3rd, Bassett EA, Wood S, Black BE, Cleveland DW. 2009. Centromere-specific assembly of CENP-a
nucleosomes is mediated by HJURP. Cell 137, 472–484.
Friesner JD, Liu B, Culligan K, Britt AB. 2005. Ionizing
radiation-dependent gamma-H2AX focus formation requires ataxia telangiectasia mutated and ataxia telangiectasia mutated and Rad3-related. Molecular Biology of the Cell 16, 2566–2576.
Fyodorov DV, Zhou BR, Skoultchi AI, Bai Y. 2018. Emerging roles of
linker histones in regulating chromatin structure and function. Nature Reviews. Molecular Cell Biology 19, 192–206.
Goldberg AD, Banaszynski LA, Noh KM, et al. 2010. Distinct factors
control histone variant H3.3 localization at specific genomic regions. Cell
140, 678–691.
Gómez-Zambrano Á, Crevillén P, Franco-Zorrilla JM, et al. 2018.
Arabidopsis SWC4 Binds DNA and recruits the SWR1 complex to modu-late histone H2A.Z deposition at key regulatory genes. Molecular Plant 11,
815–832.
Gómez-Zambrano Á, Merini W, Calonje M. 2019. The repressive role of
Arabidopsis H2A.Z in transcriptional regulation depends on AtBMI1 activity. Nature Communications 10, 2828.
Grasser KD. 2020. The FACT histone chaperone: tuning gene
transcrip-tion in the chromatin context to modulate plant growth and development. Frontiers in Plant Science 11, 85.
Hennig L, Bouveret R, Gruissem W. 2005. MSI1-like proteins: an escort
service for chromatin assembly and remodeling complexes. Trends in Cell Biology 15, 295–302.
Hofmann NR. 2016. Last exit to differentiation: histone variants as
sign-posts. The Plant Cell 28, 1235.
Houlard M, Berlivet S, Probst AV, Quivy JP, Héry P, Almouzni G, Gérard M. 2006. CAF-1 is essential for heterochromatin organization in
pluripotent embryonic cells. PLoS Genetics 2, e181.
Ingouff M, Hamamura Y, Gourgues M, Higashiyama T, Berger F. 2007.
Distinct dynamics of HISTONE3 variants between the two fertilization prod-ucts in plants. Current Biology 17, 1032–1037.
Ingouff M, Rademacher S, Holec S, Šoljić L, Xin N, Readshaw A, Foo SH, Lahouze B, Sprunck S, Berger F. 2010. Zygotic resetting of the
HISTONE 3 variant repertoire participates in epigenetic reprogramming in
Arabidopsis. Current Biology 20, 2137–2143.
Jacob Y, Bergamin E, Donoghue MT, et al. 2014. Selective methylation
of histone H3 variant H3.1 regulates heterochromatin replication. Science
343, 1249–1253.
Jacob Y, Feng S, LeBlanc CA, Bernatavichute YV, Stroud H, Cokus S, Johnson LM, Pellegrini M, Jacobsen SE, Michaels SD. 2009. ATXR5
and ATXR6 are H3K27 monomethyltransferases required for chromatin structure and gene silencing. Nature Structural & Molecular Biology 16,
763–768.
Jansen LE, Black BE, Foltz DR, Cleveland DW. 2007. Propagation of
centromeric chromatin requires exit from mitosis. The Journal of Cell Biology
176, 795–805.
Jarillo JA, Piñeiro M. 2015. H2A.Z mediates different aspects of
chro-matin function and modulates flowering responses in Arabidopsis. The Plant Journal 83, 96–109.
Jerzmanowski A. 2007. SWI/SNF chromatin remodeling and linker
his-tones in plants. Biochimica et Biophysica Acta 1769, 330–345.
Jiang D, Berger F. 2017. DNA replication-coupled histone modification
maintains Polycomb gene silencing in plants. Science 357, 1146–1149. Jin C, Felsenfeld G. 2007. Nucleosome stability mediated by histone
vari-ants H3.3 and H2A.Z. Genes & Development 21, 1519–1529.
Jin C, Zang C, Wei G, Cui K, Peng W, Zhao K, Felsenfeld G. 2009.
H3.3/H2A.Z double variant-containing nucleosomes mark ‘nucleosome-free regions’ of active promoters and other regulatory regions. Nature Genetics 41, 941–945.
Johnson L, Mollah S, Garcia BA, Muratore TL, Shabanowitz J, Hunt DF, Jacobsen SE. 2004. Mass spectrometry analysis of Arabidopsis
histone H3 reveals distinct combinations of post-translational modifications. Nucleic Acids Research 32, 6511–6518.
Kawashima T, Lorković ZJ, Nishihama R, Ishizaki K, Axelsson E, Yelagandula R, Kohchi T, Berger F. 2015. Diversification of histone H2A
variants during plant evolution. Trends in Plant Science 20, 419–425. Kaya H, Shibahara KI, Taoka KI, Iwabuchi M, Stillman B, Araki T.
2001. FASCIATA genes for chromatin assembly factor-1 in Arabidopsis maintain the cellular organization of apical meristems. Cell 104, 131–142.