A dual-mode rechargeable lithium–bromine/oxygen fuel cell
Texte intégral
Figure

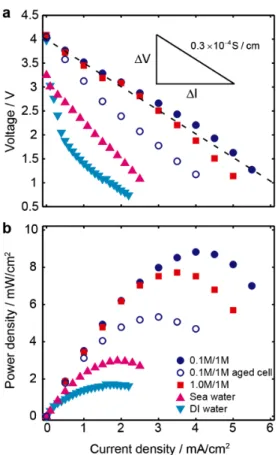
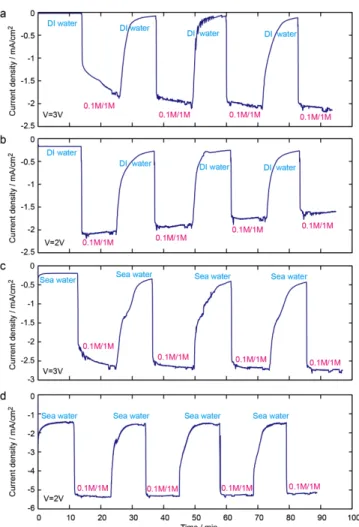

Documents relatifs
The salt used in commercial Li-ion batteries is almost exclusively lithium hexafluorophosphate (LiPF 6 ) [4], because its solutions in dipolar aprotic organic
Current density/voltage curves of monolithic dual membrane cell (● - measured) and central membrane (■ - calculated) in electrolyzer and in fuel cell operation mode at 700 o C.. The
Under the same conditions, the three following experiments (entries 2-4) gave significantly different results: conversion varied from 31 to 71% and selectivity for 2
Based on this study, two solutions are proposed, a sliding mode algebraic observer for oxygen and nitrogen partial pressures and a novel robust adaptive-gain Second Order Sliding
To control current by hydrogen flow rate, when supplying a superconducting coil by means of a single fuel cell, it is neces- sary to operate the cell in its current source working
Documenter les occupations humaines et leur environnement Fouille 2014 400 m² ouverts 41 m² fouillés manuellement Fouille 2015 775 m² ouverts 11 m² fouillés manuellement 145
For a given positive integer k, we provide multiple characterizations of relations realized by a union of k sequential WA over an infinitary finitely generated group: a
As indicated in table 1, the existing and solid components of the union model are dramatically different to those of the newly created liquid democracy model: their
