Chromatin maintenance and dynamics in senescence: a spotlight on SAHF formation and the epigenome of senescent cells
Texte intégral
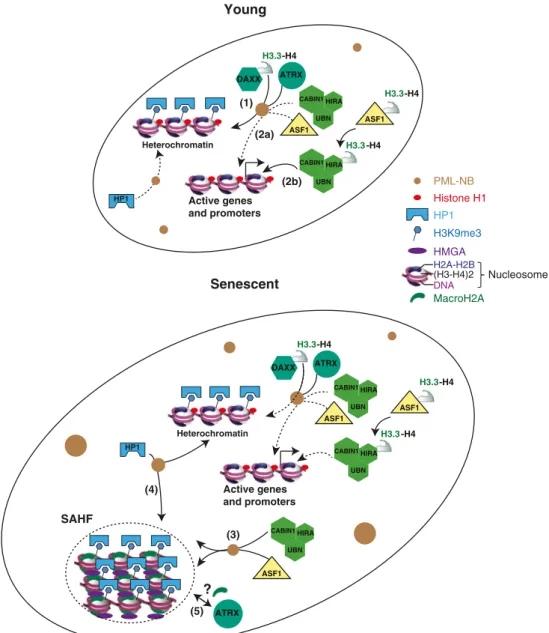
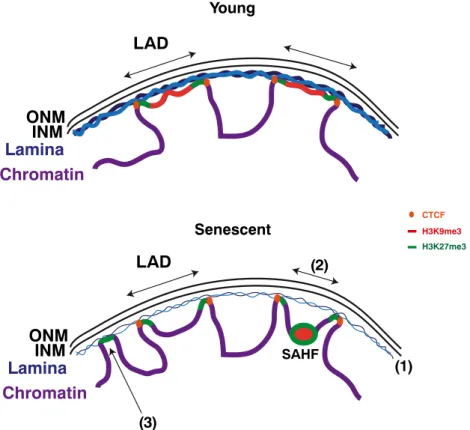
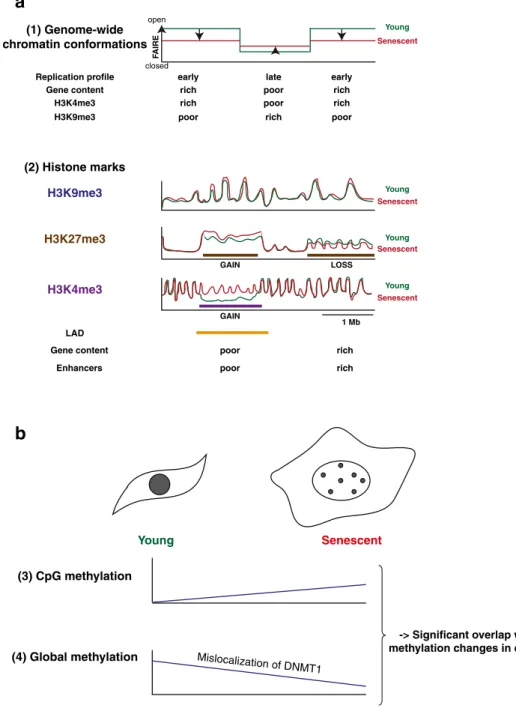
Figure

Documents relatifs
In the study of Lund et alP\ load-sensitive or load-dependent systolic function parameters were used and diastolic function was assessed from pure filling parameters which are
Il doit être en mesure de créer un cadre favorable à la captation des investissements privés dans le développement de ses ports par l’application d’un
Plus en aval, des additifs d’usages courants dans les produits alimentaires transformés exposent l’organisme de façon chronique à des NPs dont le potentiel de nocivité
An example of an artificial neural network with 3 inputs and 2 outputs (left) and the corresponding visual representations using interactive non-rectangular weighted rainbow
evaluated joining single rupture forces obtained using several AFM probes over different ex- perimental sessions. Since the total number of molecules N elastically compressed by the
Similarly, analysis of copy number alterations from 1042 cancer cell lines in the CCLE indicated 24% of cell lines harbor partial SF3B1 deletion, including 16/61 (26%) of breast
Pour réduire le problème de la dérive du facteur d’échelle, nous avons proposé deux méthodes d’intégration d’une information provenant d’un odomètre.. La première méthode
Since calibra- tion reliability increases with the number of events and the number of hydrograph points – usually peaks – available to describe each event, the aim of the present
