HAL Id: tel-00746653
https://tel.archives-ouvertes.fr/tel-00746653
Submitted on 29 Oct 2012
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
régénération hépatique et moyens d’amélioration de la
prise de greffe hépatocytaire
Panagiotis Lainas
To cite this version:
Panagiotis Lainas. Transplantation d’hépatocytes génétiquement modifiés : régénération hépatique et
moyens d’amélioration de la prise de greffe hépatocytaire. Médecine humaine et pathologie. Université
Paris Sud - Paris XI, 2012. Français. �NNT : 2012PA114842�. �tel-00746653�
Ecole Doctorale
: Innovation thérapeutique du fondamental à l’appliqué
Discipline: Sciences Chirurgicales
Année 2012
Série Doctorat N
o1190
Equipe mixte Inserm U 972
Les cellules souches: de leurs niches aux
applications thérapeutiques
THÈSE DE DOCTORAT
soutenue le 9 octobre 2012
par
Panagiotis LAÏNAS
TRANSPLANTATION D'HÉPATOCYTES GÉNÉTIQUEMENT
MODIFIÉS: RÉGÉNÉRATION HÉPATIQUE ET MOYENS
D'AMÉLIORATION DE LA PRISE DE GREFFE HÉPATOCYTAIRE
COMPOSITION DU JURY
Directeur de thèse :
Pr Ibrahim DAGHER (Hôpital Antoine-Béclère, Clamart)
Co-directeur de thèse :
Dr Anne WEBER (INSERM U972, Villejuif)
Président du jury :
Pr Dominique FRANCO (Hôpital Antoine-Béclère, Clamart)
Rapporteurs :
Pr Olivier FARGES (Hôpital Beaujon, Clichy)
Dr Nicolas FERRY (ANSM, Saint-Denis)
«
Quant au rusé Prométhée il l'attacha par des nœuds indissolubles et
douloureux ; puis il envoya contre lui un aigle aux ailes étendues qui rongeait
son foie immortel ; il en renaissait autant durant la nuit ce que l'oiseau aux
larges ailes en avait dévoré pendant le jour. »
« Théogonie
» d’Hésiode (750-700 av. J.-C.)
(D’après John Elias Skandalakis « Surgical Anatomy - the embryologic and anatomic basis of Modern Surgery », 2004 , volume II, p.1005. Permission accordée par Professeur Lee Skandalakis, Université d’Emory, Atlanta, GA, Etats-Unis d’Amérique).

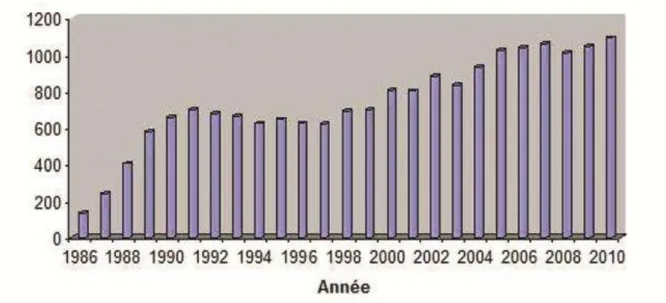
![Figure 7. Durée d’attente avant greffe hépatique selon la période d’inscription ( d’après le site de l’Agence de Biomédecine 2012 [231])](https://thumb-eu.123doks.com/thumbv2/123doknet/14232370.485688/42.892.111.727.426.955/figure-durée-attente-hépatique-période-inscription-agence-biomédecine.webp)
![Figure 10. Courbe de survie du receveur hépatique selon la période de greffe (d ’après le site de l’Agence de Biomédecine 2012 [231] )](https://thumb-eu.123doks.com/thumbv2/123doknet/14232370.485688/45.892.137.726.626.1061/figure-courbe-survie-receveur-hépatique-période-agence-biomédecine.webp)
![Figure 11. Organes et tissus pour lesquels la xénogreffe pourrait être appliquée comme thérapie potentielle (d ’après Ekser et al, Lancet 2012 [244] )](https://thumb-eu.123doks.com/thumbv2/123doknet/14232370.485688/47.892.152.691.304.1071/figure-organes-xénogreffe-pourrait-appliquée-thérapie-potentielle-lancet.webp)
![Tableau 3. Patients traités pour insuffisance hépatique aiguë par des approches de foie artificiel (d’après Stockmann et al, Ann Surg 2000 [254] )](https://thumb-eu.123doks.com/thumbv2/123doknet/14232370.485688/48.892.105.775.796.942/tableau-patients-traités-insuffisance-hépatique-approches-artificiel-stockmann.webp)