Special Report
Consensus criteria for diagnosis, staging, and treatment
response assessment of T-cell prolymphocytic leukemia
Philipp B. Staber,1Marco Herling,2-4Mar Bellido,5Eric D. Jacobsen,6Matthew S. Davids,6Tapan Mahendra Kadia,7Andrei Shustov,8 Olivier Tournilhac,9Emmanuel Bachy,10Francesco Zaja,11Kimmo Porkka,12Gregor Hoermann,13,14Ingrid Simonitsch-Klupp,15 Claudia Haferlach,16Stefan Kubicek,17,18Marius E. Mayerhoefer,19,20Georg Hopfinger,21Ulrich Jaeger,1and Claire Dearden221Division of Hematology and Hemostaseology, Department of Internal Medicine I, Medical University of Vienna, Vienna, Austria;2Department of Internal Medicine,
Center for Integrated Oncology Aachen-Bonn-Cologne-Duesseldorf, and3Excellence Cluster for Cellular Stress Response and Aging-Associated Diseases,
University of Cologne, Cologne, Germany;4Center for Molecular Medicine, University of Cologne, Cologne, Germany;5Hematology Department, University
Medical Center Groningen, University of Groningen, Groningen, The Netherlands;6Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA;
7Department of Leukemia, The University of Texas, MD Anderson Cancer Center, Houston, TX;8Seattle Cancer Care Alliance, University of Washington, Seattle,
WA;9Centre Hospitalier Universitaire Estaing, Clermont-Ferrand, France;10Department of Hematology, Hospices Civils de Lyon, Lyon, France;11S.C. Ematologia
Azienda Sanitaria Universitaria Integrata, Trieste, Italy;12Department of Hematology, Helsinki University Hospital Comprehensive Cancer Center, Helsinki, Finland;
13Central Institute of Medical and Chemical Laboratory Diagnostics, University Hospital Innsbruck, Innsbruck, Austria;14Department of Laboratory Medicine and
15Clinical Institute of Pathology, Medical University of Vienna, Vienna, Austria;16Munich Leukemia Laboratory, Munich, Germany;17CeMM Research Center for
Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria;18Christian Doppler Laboratory for Chemical Epigenetics and Anti-Infectives, CeMM
Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria;19Department of Biomedical Imaging and Image-Guided Therapy,
Medical University of Vienna, Vienna, Austria;20Department of Radiology, Memorial Sloan Kettering Cancer Center, New York, NY;21Division of Blood and Marrow
Transplantation, Department of Medicine I, Medical University of Vienna, Vienna, Austria; and22The Royal Marsden Hospital, NHS Foundation Trust, London,
United Kingdom
T-cell prolymphocytic leukemia (T-PLL) is a rare, mature T-cell neoplasm with a heterogeneous clinical course. With the advent of novel treatment options that will potentially change the management of patients with T-PLL, it has become necessary to produce consensus guidelines for the design and conduct of clinical trials. The T-PLL International Study group (TPLL-ISG) set out to
define standardized criteria for diagnosis, treatment in-dication, and evaluation of response. These criteria will facilitate comparison of results from clinical trials in T-PLL, and will thus support clinical decision making, as well as the approval of new therapeutics by healthcare authorities. (Blood. 2019;134(14):1132-1143)
Introduction
In 1973, T-PLL had been recognized as distinct from chronic lymphocytic leukemia (CLL) by its specific clinical presentation, the morphological characteristic of affected lymphoid cells, and the poor clinical outcome.1Modern oncology has witnessed an
increasing number of new effective anticancer drugs; however, so far, they have limited benefit for patients with T-PLL. With an overall incidence of only 0.6/Mio in the general population, T-PLL is a very rare disease.2,3Because of the rapidly progressing
clinical course and often uncertain diagnosis, it has been a dif-ficult disease to approach systematically. Case reports, single-center retrospective studies, and small trials are difficult to compare because of poorly defined and inconsistent diagnos-tic criteria and response evaluation.4-9So far, T-PLL has been
managed according to criteria for CLL, as described by the in-ternational workshop on CLL (IWCLL).10-13T-PLL is biologically
and clinically very different from CLL, and the IWCLL criteria are not always appropriate. Furthermore, modifications of the IWCLL criteria adopted by investigators have not been uniform. Thus, there is an unmet need for disease-specific criteria for the establishment of diagnosis and response evaluation to provide a basis to compare results and to systematically advance treat-ment strategies for T-PLL. To define T-PLL-specific consensus
standards for the design and conduct of clinical trials, the T-PLL International Study Group (TPLL-ISG) assembled in May 2017 in Vienna, Austria. In the following months, the group agreed on a blueprint consensus paper that aims to provide definitions to be used in routine clinical practice and upcoming studies, even-tually paving the way for harmonization of data that will come from future clinical trials.
Diagnosis of T-PLL
The 2017 World Health Organization classification defines T-PLL as aggressive T-cell leukemia of proliferating small to medium-sized T-lymphocytic cells that, despite their postthymic origin and mature phenotype, are called T-prolymphocytes.1,14T-PLL
includes the formerly used category of T-cell CLL.
T-PLL represents 2% of chronic leukemias in adults.2,3The largest
observational studies that described clinicopathologic features at presentation included between 38 and 119 T-PLL cases, in-dicating series with more than 50 T-PLL cases should be con-sidered to be very large.2,3,15,16Clinical presentation includes
B-symptoms, hepato-splenomegaly, and usually excessive lym-phocytosis above 1003 109/L. Nodal and extranodal presentation
is also frequent, including skin, pleural or peritoneal effusions in around 25% of patients, and central nervous system in-volvement in less than 10% of patients.2,14 Periorbital,
con-junctival edema and peripheral edema are frequently observed in patients with T-PLL.2
T-PLL is a molecularly and clinically distinct disease that has to be discriminated from other leukemic T-cell neoplasia demon-strating overlapping features, such as S ´ezary syndrome, T-cell large granular lymphocytic leukemia, adult T-cell lymphocytic leukemia, or other (nodal) mature T-cell tumors with marked leukemic presentation.3,16,17To verify the correct diagnosis, it is
necessary to evaluate peripheral blood smear (morphology; Figure1), immunophenotype, and genetic features (Figure 2), although in most of the cases, the diagnosis can be established using only morphology and immunophenotype of peripheral blood lymphocytes. Bone marrow evaluation is required for treatment evaluation (confirmation of a remission), but is not usually required to confirm the diagnosis. Table1 lists rec-ommended assessments at diagnosis and during the course of the disease. The diagnosis of T-PLL is established if all 3 major criteria are met or if the first 2 major criteria and 1 minor criterion are met (TCL1-family negative T-PLL; Table2). As discussed in more detail here, the first 2 major criteria are T-PLL-defining characteristics.2,3,17The third major criterion,
alterations of TCL1A, TCL1B (TML1), or MTCP, is present in more than 90% of cases; however, it is not present in 100%, and therefore at least 1 of the less common genetic features or T-PLL typical site involvements has to be present.3
Blood and bone marrow
The diagnosis of T-PLL requires the presence of clonal pro-lymphocytic T cells in the peripheral blood or bone marrow, which can present in 3 morphologic variants1,2,14,18-20: most
commonly (75%), T-PLL cells are medium-sized with a high nuclear/cytoplasmic ratio, moderately condensed chromatin,
a single visible nucleolus, and a slightly basophilic cytoplasm without granules but typically demonstrating cytoplasmic pro-trusions (blebs); in 20%, T-PLL present as a small cell variant with condensed chromatin and a nucleolus that is invisible by light microscopy; and in about 5% of cases, T-PLL demonstrates irregular nuclei similar to the cerebriform nuclei found in S ´ezary cells of mycosis fungoides (cerebriform variant). These variants are not associated with distinct clinical presentations or with outcomes.
The clonality of these T lymphocytes should be confirmed by polymerase chain reaction (PCR; or next-generation sequencing [NGS]-based assessment) of the clonal rearrangement of T-cell receptor genes (TRB and TRG) or byflow cytometry.20
However, it is to the physician’s discretion to judge whether an explicit test for clonality might be redundant in certain situations. Serologic or PCR-based testing for HTLV (human T lymphotropic virus) type 1 is negative (to distinguish from adult T-cell lymphocytic leukemia).2,14,17-19
Infrequently, asymptomatic patients are diagnosed as part of the evaluation of an abnormal laboratory test result. A bone marrow examination is not generally required to establish the diagnosis of T-PLL; however, it may be important to elucidate unclear cytopenias. Evaluation of the bone marrow (aspirate and biopsy) is mandatory to confirm a complete remission (CR) and complete remission with incomplete re-covery (CRi).
Immunophenotype
T-PLL cells are postthymic T cells that coexpress mature T-lymphoid markers and demonstrate cytoplasmatic expression of TCL1 oncogenes as a frequent hallmark (Table 3; Figure 2).3,14
Immunophenotyping for differential diagnosis to related dis-eases are listed in Table 3.
A
C
B
D
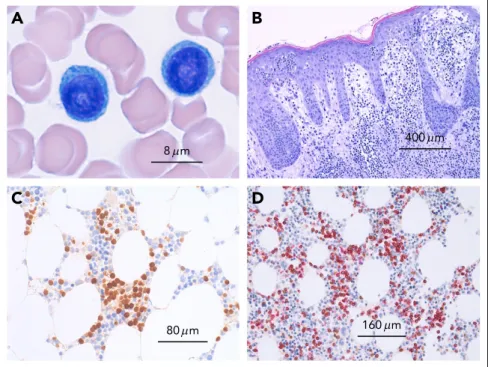
8 m 80 m 400 m 160 m Figure 1. Microscopy of T-cell prolymphocytic leu-kemia.(A) Peripheral blood smear showing 2 medium-sized atypical lymphocytes with round nuclei and prominent nucleoli and abundant basophilic cytoplasms without granules. (B) Cutaneous involvement in T-PLL. Dense dermal infiltrates with marked epidermotropism simulating mycosis fungoides. (C) Bone marrow infil-tration by T-PLL with an interstitial infilinfil-tration pattern depicted by an immunohistologic stain with an anti-body against TCL1. (D) T-cell origin of TCL-11cells
confirmed by immunohistology by double staining with CD5 (red) and TCL1 (brown).
Genetics
T-cell receptor genes (TRB or TRG) are clonally rearranged in T-PLL cells, which should be confirmed (by PCR/ NGS or flow cytometry20). Patients with T-PLL commonly demonstrate complex
karyotypes (in 70%-80%) with recurrent genetic features that help establish the diagnosis.2,21-23Therefore, chromosome banding
analysis andfluorescence in situ hybridization (FISH) can help to distinguish T-PLL from other T-lymphoproliferative disorders. Rearrangements involving TCL1 (T-cell leukemia/lymphoma1) family genes TCL1A, MTCP1 (mature T-cell proliferation), or TCL1B (alias TCL1/MTCP1-like 1 [TML1]), are relatively specific for T-PLL and are present in more than 90% of cases, either as inv(14)(q11q32) or t(14;14)(q11;q32) (involving TCL1A or TCL1B), or t(X;14)(q28;q11) (involving MTCP1; mature T-cell proliferation). Detection of aberrant TCL1 protein expression viaflow cytometry or immunohistochemistry is more sensitive than cytogenetics and also represents a diagnostic hallmark.3,22,24,25Few cases are
ob-served in which neither a rearrangement nor an overexpression of TCL1A, TCL1B, or MTCP1 is detected, but which otherwise carry typical clinical, cytomorphological, and molecular criteria of T-PLL (clonal expansion of cells with a prolymphocytic T-cell phenotype). These cases should be collected for a more comprehensive sci-entific analysis and labeled as TCL1-family negative T-PLL. Deletions of or missense mutations at the ataxia telangiectasia mutated (ATM) locus at 11q23 are found in up to 80% to 90% of
T-PLL cases.16,26-28Patients with ataxia telangiectasia who harbor
ATM germ line mutations have an increased risk of develop-ing T-PLL.27,28Other chromosomal or genetic abnormalities of
sporadic T-PLL include chromosome 8 (idic(8)p11), t(8;8)(p11-12; q12), and trisomy 8q),29deletion in 12p,30and abnormalities in
chromosome6 in 33% of cases.31Deletions of the TP53 gene are
observed in 31% of cases, and TP53 mutations in 14% of cases in T-PLL.32
NGS has identified additional recurrent genomic abnormalities, many involving JAK/STAT signaling (up to 75% of cases).33-35
JAK3 mutations demonstrated a significant negative effect, and are so far the only genetic alteration of putative prognostic sig-nificance in T-PLL.32
Other common genetic lesions are haploinsufficiency for the CDKN1B tumor suppressor gene (half of cases30,36) and
tions in the epigenetic regulators EZH2 (deletions, 18%; muta-tions, 13%), TET2 (17%), and BCOR (9%), none of which are specific for T-PLL.35Taken together, assessment of clonal TCR
rearrangement, cytogenetics, and FISH are relevant genetic tests to establish the diagnosis T-PLL. Genetic sequencing is currently not a diagnostic requirement; however, it may provide information regarding the underlying pathogenesis of T-PLL or might help to identify relevant prognostic subgroups.
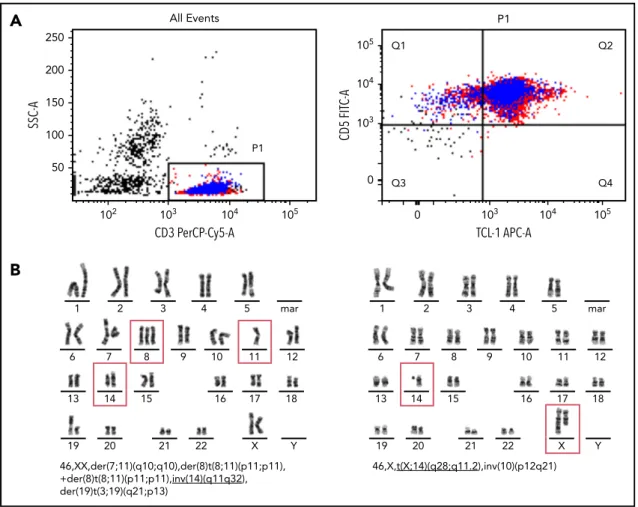
50 P1 All Events CD3 PerCP-Cy5-A SSC-A 102 103 104 105 100 150 200 250 mar 13 14 15 16 17 18 1 2 3 4 5 6 7 8 9 10 11 12 19 20 21 22 X Y 46,XX,der(7;11)(q10;q10),der(8)t(8;11)(p11;p11), +der(8)t(8;11)(p11;p11),inv(14)(q11q32), der(19)t(3;19)(q21;p13) Q1 Q2 Q3 Q4 CD5 FIT C-A TCL-1 APC-A P1 0 103 104 105 0 103 104 105 3 4 5 mar 10 9 13 14 15 16 17 18 1 2 6 7 8 11 12 19 20 21 22 X Y 46,X,t(X;14)(q28;q11.2),inv(10)(p12q21)
A
B
Figure 2. Immunophenotypic markers of T-PLL and typical chromosome banding analysis.(A) Flow cytometry of CD31, CD51, cytoplasmic TCL11T-PLL cells. (B) Two
representative T-PLL chromosome banding analyses.
Serum markers
Similar to other lymphoproliferative disorders, serum lactate dehydrogenase (LDH) and b 2 microglobulin (B2MG) may reflect disease burden in T-PLL. In a retrospective study of 119 patients with T-PLL, LDH higher than 1668 U/L and B2MG higher than 8 mg/L were associated with inferior overall survival (OS).15High LDH levels were also associated with a shorter OS in
multivariate analysis.15Elevated B2MG together with
lymphade-nopathy and organomegaly were suggestive of bulky disease.15
Both LDH and B2MG might therefore indicate tumor mass, and should be used in prospective clinical trials to validate their relative value in disease staging and further patient management.
Clinical staging and indication for
treatment
Most patients with T-PLL present with an elevated white blood cell count (typically.100 3 109/L), splenomegaly, hepatomegaly,
and generalized lymphadenopathy.2,17However, up to 20% to
30% of patients with T-PLL demonstrate initially stable or slowly progressive disease.37These 2 different disease states are relevant
for the clinical management of patients. Criteria for initiating treatment in a clinical trial and in general practice are the same as those that define active disease. Because there is no evidence that asymptomatic patients with T-PLL with inactive disease (“inactive T-PLL”) would benefit from immediate treatment, it should be
Table 1. Recommended tests for patients with T-PLL
Assessments General practice Trials
To establish diagnosis
History, physical examination Always Always
Blood count and differential count Always Always
Bone marrow aspirate and biopsy When clinically indicated Desirable
Biopsy or aspirate of suspected involved site When clinically indicated Desirable
Immunophenotyping of blood lymphocytes Always Always
T-cell receptor rearrangement (TRB, TRG) Always Always
chromosome banding analysis Desirable Desirable
Fluorescence in situ hybridization (FISH) Desirable Desirable
Testing for HTLV Always* Always*
Before treatment start and at response assessment
CIRS,† physical examination, performance status Always Always
Blood count and differential count Always Always
Bone marrow aspirate and biopsy‡ Always Always
Serum chemistry Always Always
Infectious disease status (HBV, HCV, HIV, HSV)† Always Always
Serum lactate dehydrogenase Always Always
b2-microglobulin Desirable Always
CT scan of neck, chest, abdomen, pelvis Always Always
PET scans Not generally indicated Not generally indicated
Chest radiograph Not generally indicated Not generally indicated
Abdominal ultrasound Not generally indicated Not generally indicated
Next-generation sequencing, MRD Not generally indicated Desirable
CIRC, cumulative illness rating scale; HBV, hepatitis B virus; HCV, hepatitis C virus; HSC, herpes simplex viruses; HTLV, human T-cell leukemia-lymphoma virus. *In patients from endemic countries.
†Before treatment start. ‡To confirm a remission.
Table 2. Requirements to establish the diagnosis of T-PLL
Major criteria Minor criteria (at least 1 required)
• .5 3 109/L cells of T-PLL phenotype in peripheral blood or bone marrow • Abnormalities involving chromosome 11 (11q22.3; ATM) • T-cell clonality (by PCR for TRB/TRG, or by flow cytometry) • Abnormalities in chromosome 8: idic(8)(p11), t(8;8), trisomy 8q • Abnormalities of 14q32 or Xq28 OR expression of TCL1A/B, or MTCP1* • Abnormalities in chromosome 5, 12, 13, 22, or complex karyotype
• Involvement of T-PLL specific site (eg, splenomegaly, effusions)
*Cases without TCL1A, TCL1B, or MTCP1 rearrangement or their respective overexpression are collected as TCL1-family negative T-PLL.
restricted to patients with active or symptomatic disease (“active T-PLL”). For inactive T-PLL (indicating an early phase and avoiding the misleading term“indolent”), a period of observation is re-commended unless there is disease progression or development of disease-related symptoms (Table 4). Note, however, that within 1 to 2 years, nearly all of inactive T-PLL convert into active disease. During the observation period, blood counts should be performed at monthly intervals along with a clinical examination. A blood lymphocyte doubling time less than 8.5 months has been associated with a worse prognosis.22
Provided the initial T-PLL cell count is greater than 30 000/mL, lymphocyte doubling time of less than 6 months or an increase of more than 50% within 2 months indicate active T-PLL (in analogy to CLL as 1 criterion among others).12
Table 1 lists the recommended assessments before a treatment is commenced. An evaluation of comorbidities as established for patients with CLL by using the cumulative illness rating scale is recommended before the start of treatment, as it might be
relevant for treatment choice, dosing, and assessments of re-sponse to therapy.38Treatment is indicated for patients with active
T-PLL and any of the listed disease-related symptoms (Table 4). This recommendation is generally for primary treatment decisions, as well as for second- and subsequent-line decisions; however, recurrent disease rarely presents as inactive T-PLL.2,3,5,17
De
finition of response, relapse, and
refractory disease
Response evaluation needs to monitor the actual state of dis-ease, as well as the performance status. Assessments include a careful physical examination, blood tests, radiology, and confirmation of remission by evaluation of bone marrow (both aspirate and trephine; Table 1). Because response kinetics and recovery of normal cells depend on characteristics and modal-ities of specific treatment strategies, a general recommendation on adequate timing of response, assessments cannot be made. Therefore, timing needs to be evaluated individually for che-motherapeutics, which are administered in a defined treatment period; for antibodies; and for small molecule inhibitors, the latter of which are often more continuously administered. For continuous therapeutic strategies, response assessment should be performed at predefined times and treatment interruptions are not considered to be required for response assessment. In analogy to CLL, 2 groups of parameters should be assessed to define a response to a given therapy.12 Group A parameters
evaluate constitutional symptoms and direct parameters of T-PLL (tumor load), group B parameters evaluate indirect effects of T-PLL cells on the function of the hematopoietic system (Table 5). No specific response criteria for imaging tests such as computed tomography (CT) are currently established for T-PLL. Although adoption of the iwCLL12or Lugano39criteria that are used for
response assessment in patients with CLL and patients with lymphoma might be an option, both systems rely on bidimen-sional measurements (ie, the sum of the products of long- and short-axis diameters) of up to 6 target lesions, which is a time-intensive approach that may be impractical for clinical use. Recently, the International Working Group proposed RECIL,
Table 3. Immunophenotype of T-PLL and the most relevant differential diagnosis
Differential diagnosis Immunophenotype
T-PLL cyTCL11(.90%), CD31(.80%), CD41(60%), CD51(100%), CD71(.90%), CD81(15%), CD41CD81(25%) T-ALL Tdt1, CD1a1 Leukemic PTCL cyTLC12
T-cell large granular lymphocytic leukemia
CD81, CD571, CD161
S ´ezary syndrome CD72, CD41, CD251 Adult T-cell lymphocytic
leukemia
CD41, CD251, HTLV11
Percentage of patients with T-PLL expressing specific markers.19
Table 4. Criteria for staging and indication of treatment in T-PLL
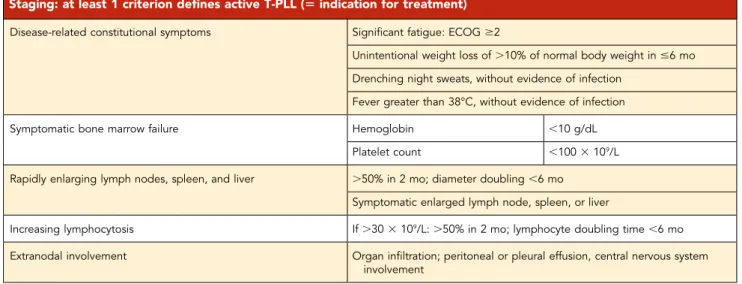
Staging: at least 1 criterion defines active T-PLL (5 indication for treatment)
Disease-related constitutional symptoms Significant fatigue: ECOG $2
Unintentional weight loss of.10% of normal body weight in #6 mo Drenching night sweats, without evidence of infection
Fever greater than 38°C, without evidence of infection
Symptomatic bone marrow failure Hemoglobin ,10 g/dL
Platelet count ,100 3 109/L
Rapidly enlarging lymph nodes, spleen, and liver .50% in 2 mo; diameter doubling ,6 mo Symptomatic enlarged lymph node, spleen, or liver
Increasing lymphocytosis If.30 3 109/L:.50% in 2 mo; lymphocyte doubling time ,6 mo Extranodal involvement Organ infiltration; peritoneal or pleural effusion, central nervous system
involvement
a variation of RECIST 1.1, as a simple alternative to the Lugano criteria for response assessment in lymphomas.40Contrary to the
Lugano (and IWCLL) criteria, RECIL relies on unidimensional measurements (ie, the sum of long-axis diameters, SLD) of just up to 3 target lesions. This recommendation is based on the analysis of 47 828 imaging measurements from 2983 individual patients with adult and pediatric lymphoma enrolled in 10 multicenter clinical trials, which showed both a high correlation between unidimensional measurements and bidimensional areas and that the use of 3 instead of 6 target lesion does not have a relevant effect on the response category to which patients are assigned.40Because of their ease of use, we therefore propose to
use the RECIL criteria for response assessment in T-PLL, based on changes in lesion size on CT or magnetic resonance imaging (Table 5). [18F]FDG-PET-based response assessment, which is
increasingly used in lymphoma, cannot be recommended at present, because there is practically no evidence with regard to the FDG avidity (ie, the glucose metabolism) of T-PLL. Other PET radiotracers, such as the CXCR4 tracer [68Ga]Ga-Pentixafor,
which has shown promise in CLL, also require further evaluation.41
In line with the diagnostic workup of T-PLL, a number of different methods are used to detect remaining tumor cells when assessing the response to treatment. Immunophenotypic studies (flow cytometry or immunohistochemistry) are applied in addition to morphology. Furthermore, genetic analysis (TCR clonality testing, FISH, chromosome banding analysis) can be used to demonstrate the absence of a genetic aberration detected at diagnosis. These genetic tests show an analytical sensitivity that is roughly compa-rable to that of morphology and immunophenotyping in T-PLL, and can be applied complementarily to discriminate neoplastic from normal T cells.42In addition, highly sensitive molecular tests
(clone-specific real-time PCR or high-sensitive NGS analyses of TCR rearrangements) have been described for analysis of minimal re-sidual disease (MRD) in T-PLL,43 and individual chromosomal
breakpoints involving the TCL1 or MTCP1 locus could represent other targets for MRD assessment (by clone-specific real-time PCR or NGS) in T-PLL. With the current treatment options, the clinical relevance of low-level MRD in patients achieving CR remains un-clear, and the value of standardized high-sensitive MRD testing needs to be assessed in forthcoming studies.
Best objective response rate (ORR) can be defined as a determined response with no additional improvement during at least 4 weeks of a continued or 8 weeks of an interval therapy. Given the acute nature of active T-PLL, an established response in peripheral blood (CR or CRi) requires confirmation by a bone marrow examination (aspirate and trephine), but does not necessarily need to be sustained for a certain period of time (in contrast to CLL). Other parameters are assessing the duration of responses in addition to ORR. Assessments to establish a CR, such as CT scans and bone marrow examination, should be performed after the patient has responded in the pe-ripheral blood. Follow-up visits including blood count, chemistry, and physical examination after the end of any (continued or time-limited) therapy should be performed every 4 to 6 weeks.
With the advent of novel continuous treatment, stabilization of a prior rapidly progressing disease can be considered as ben-eficial for patients. We therefore propose inclusion of the term disease control rate which involves CR, CRi, partial remission (PR), and stable disease (SD). In the same way we would like to introduce the term disease control time.
CR
CR requires all the following criteria (Table 5).
1. Absence of lymphadenopathy
For previously enlarged lymph nodes ($1.5 cm long-axis di-ameter), regression of the long-axis diameters to ,1.0 cm (RECIL-2017) on CT or magnetic resonance imaging. Once re-sponse is established, further imaging should only be performed when disease progression is apparent by clinical examination or blood evaluation.
2. Absence of splenomegaly and hepatomegaly
Absence of splenomegaly or hepatomegaly by physical exami-nation and by CT scan or ultrasound. In analogy to criteria for CLL a cutoff of 13 cm in craniocaudal length defines splenomegaly.12,44
At this time, there are no clear-cut criteria for hepatomegaly be-cause of significant size differences among healthy individuals.
3. Absence of disease-related constitutional symptoms 4. Peripheral blood: Lymphocyte count<4 3 109/L
5. Clearance of phenotypic T-PLL cells in bone marrow (<5% of mononuclear cells)
A bone marrow aspirate and biopsy are required to confirm CR. They should be performed if clinical and laboratory results listed in Table 5 indicate that CR may be achieved. For CR, the cytological or histological evaluation of a technically adequate sample needs to document a bone marrow with T-PLL less than 5% of nucleated cells (assessed by immunohistochemistry orflow cytometry). In clinical trials aiming at evaluating deep remissions,flow cytometry and genetic analyses (FISH for gene rearrangements detected at diagnosis or PCR for clonality analysis of TCR gene rearrange-ment) may be applied to detect MRD. However, for the defi-nition of CR, tests with a particularly high analytical sensitivity (,1%; see MRD) are not needed.
6. Absence of other specific site involvement
If previously involved, absence of pleural or peritoneal effusions, skin infiltrates, and involvement of central nervous system, or other extra-medullary sites.
7. Complete recovery of bone marrow function
1. Platelets$100 3 109/L (untransfused)
2. Hemoglobin$11.0 g/dL (untransfused, independent of Erythropoietin)
3. Neutrophils$1.5 3 109/L (independent of growth factor)
Complete remission with incomplete marrow
recovery
These are patients achieving a CR with incomplete marrow re-covery (CRi; fulfilling all criteria for CR in group A), but fail CR criteria in group B because of persistent anemia, thrombocy-topenia, or neutropenia that are unrelated to T-PLL but, rather, to treatment toxicities. CRi has to be assessed prospectively in clinical trials to evaluate whether there is a prognostic signifi-cance when compared with regular CR and to PR.
PR
For a PR, at least 2 parameters of group A and 1 parameter of group B need to improve as specified if previously abnormal
Table 5. De fi nition of response after treatment of patients with T-PLL Group and parameter CR (all met) PR (‡ 2 in A and ‡ 1 in B ) S D (all met) PD (‡ 1 in A or B met) Group A Lymph nodes long-axis diameters to , 1.0 cm D ecrease $ 30% in SLD Change of 2, 30% to 1 # 20% Increase . 20% in S LD Spleen Spleen size , 13 cm Decrease $ 50% in vertical length beyond normal from b aseline Change of 2 49% to 1 49% b eyond normal from b aseline Increase $ 50% in vertical length beyond normal from baseline Constitutional symptoms None Any Any Any Circulating lymphocyte count , 4 3 10 9/L # 30 3 10 9/L and d ecrease $ 50% from baseline . 30 3 10 9/L or change o f2 49% to 1 49% Increase $ 50% from baseline Marrow T -PLL cells , 5% of mononuclear cells Any Any Any Any other speci fic site involvement* None Any Any Any Group B Platelet count $ 100 3 10 9/L $ 100 3 10 9/L or increase $ 50% from baseline Change of 2 49% to 1 49% Decrease of $ 50% from baseline Hemoglobin $ 11.0 g/dL (untransfused) $ 11 g/dL or increase $ 50% from baseline , 11.0 g/dL or , 50% from b aseline, or change , 2 g /dL Decrease of $ 2 g /dL from b aseline Neutrophils $ 1.5 3 10 9/L $ 1.5 3 10 9/L or increase $ 50% from baseline Change of 2 49% to 1 49% Decrease of $ 50% from baseline CR, all o f the criteria ha ve to be met); CR i, all C R crite ria of group A a re met but at least 1 in B is not achie ved; PR, at least 2 parameters o f g roup A and 1 o f g roup B need to improv e if p revious ly abnormal; PD, at least 1 o f the criteria of group A o r g roup B has to be met; SD, all the criteria have to be met, constitutional symptoms alone do not de fine PD; SLD, sum o f long-ax is diameters o f u p to 3 target lesions. *Pleural or peritone al effusion, skin in filtration, central nervous system involvement.
(Table 5). Only 1 parameter needs to improve if only 1 within each group was abnormal. Constitutional symptoms related to T-PLL should be noted because their presence for more than a month precludes a CR, but are irrespective to distinguish from other responses.
1. Decrease of lymphadenopathy
For previously enlarged lymph nodes ($1.5 cm long-axis di-ameter), at least 30% regression in the SLD of up to 3 target lesions, but not CR (RECIL-2017).
2. Decrease of splenomegaly
Regression of splenomegaly by 50% or less in vertical length beyond normal (13 cm) from baseline compared with baseline.
3.<30 3 109/L circulating lymphocyte count in peripheral
blood and decrease by 50% or more 4. Any result other than CR in bone marrow
Bone marrow aspirate and biopsy is performed if clinical and laboratory results indicate that a CR (PB and CT clearance) may have been achieved.
5. Partial recovery of bone marrow function of at least 1 of following parameters
1. Platelets$100 3 109/L (untransfused) or by 50% or more
compared with baseline
2. Hemoglobin$11.0 g/dL or by 50% or more compared with baseline (untransfused, independent of Erythropoietin) 3. Neutrophils$1.5 3 109/L or by 50% or more compared with
baseline (independent of growth factor)
SD
For SD, patients have not achieved a remission (CR or PR) and do not demonstrate progressive disease (PD). This state needs to be sustained for at least 3 months. Because a stabilization of a prior rapidly progressing disease over a certain period can be considered as beneficial for some patients, it should therefore not be called nonresponse or treatment failure. However, ORR only considers CR, CRi, and PR. If SD is included in the re-sponse measurements, it needs to be explicitly stated, as in the proposed term disease control rate and disease control time, both of which include CR, CRi, PR, and SD.
ORR
ORR is defined as the proportion of patients who have a CR, CRi, or PR, to therapy.
Disease control rate
Disease control rate is a composite of ORR and SD and is useful for continuous therapies that have tumorostatic effects.
PD
For PD, 1 or more parameters of group A are present. For group B, parameters (marrow impairment) need to be directly attrib-utable to increased T-PLL marrow infiltration and unrelated to autoimmune cytopenias or treatment toxicities. For clarification, a bone marrow aspirate and biopsy may be required (Table 5). Progressive lymphadenopathy is indicated by a more than 20% increase in the SLD of up to 3 target lesions; for small lymph nodes measuring less than 1.5 cm after therapy, a minimum
increase of 5 mm and a long-axis diameter of more than 1.5 cm, or appearance of a new lesion (RECIL-2017).
Progression-free survival
Progression-free survival (PFS) is defined as duration between thefirst treatment day and the time of first sign of progression or death from any cause.
Event-free survival
Event-free survival is defined as the interval between the first treatment day to the time offirst sign of disease progression or start of new treatment or withdrawal from trial because of tox-icities or death, whichever occursfirst.
Time to next treatment
Time to next treatment is defined as interval between the first treatment day and the patient starting an alternative treatment of T-PLL.
OS
OS in a clinical trial is defined as the interval from the first treatment day to death from any cause.
Relapse
Relapse is defined as disease progression (PD; Table 5) after a CR, CRi, PR, or SD has been achieved and documented.
Refractory disease, treatment failure
Refractory disease and treatment failure are defined as disease progression (Table 5) without a previous response or disease control (CR, CRi, PR, or SD) having been achieved and documented.
MRD
MRD is defined by the detection of a small number of remaining T-PLL cells in PB or BM, using highly sensitive methods in a patient achieving CR or CRi. The clinical relevance of MRD assessment in T-PLL as well as the technical method remains to be established in forthcoming studies.
Current treatment of T-PLL
Very few trials have been conducted and published on T-PLL. Initial attempts with CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) or CHOP-like regimens have been particularly discouraging.45Table 6 summarizes the data of the
most relevant clinical studies. The bestfirst-line treatment to achieve a CR is IV alemtuzumab (anti-CD52) with a ORR higher than 90% and PFS between 8 and 11 months.4,46,47Despite the
high OR, patients will relapse, with a median duration of remission for responders of less than 2 years. For this reason, patients achieving a complete remission should be considered for con-solidation therapy with an allogeneic stem cell transplant. About a third of patients may achieve long-term survival with this ap-proach, although TRM and relapse rates remain high.48,49 In
a prospective study from the European Society for Blood and Marrow Transplantation, patients who received conditioning with at least 6 Gy total body irradiation had a lower relapse rate.50,51
Autologous stem cell transplant is an option, particularly for patients without a donor, and has been shown to prolong PFS, but with no long-term survivors.48 There is no evidence to support
alemtuzumab (or other) maintenance therapy.
Table 6. Summary of most relevant clinical studies on T-PLL Regimen Trial Disease status n ORR, % CR, % PR, % PFS, mo OSa, mo Reference Pentostatin Single center, retrospective Pretreated 56 45 9 3 6 6 9 5 5 Alemtuzumab, IV Single center, retrospective Pretreated 15 73 60 13 6 8 46 Alemtuzumab, IV Multicenter, prospective Pretreated 39 76 60 16 7 1 0 4 7 Alemtuzumab, IV Multicenter, retrospective Untreated 4 7 5 7 5 0 4.5 7 .5 52 Pretreated 72 50 37.5 12.5 Pentostatin 1 alemtuzumab, IV Single center, prospective Pretreated 13 69 62 8 7 .8 10.2 56 Alemtuzumab, IV Single center, prospective Untreated 32 91 81 10 (67%) 1 2 m o (37%) 48 mo 4 Alemtuzumab, sc Untreated 9 3 3 3 3 0 (67%) 1 2 m o (33%) 48 mo Alemtuzumab, IV Pretreated 45 74 60 14 (26%) 1 2 m o (18%) 48 mo FMC 1 alemtuzumab, IV Multicenter, prospective Untreated 16 92 48 44 11.5 17.1 5 Pretreated 9 Bendamustine Multicenter, retrospective Untreated 6 5 5.3 2 0 3 3.3 5 8.7 7 Pretreated 9 Alemtuzumab, IV Single center, retrospective Untreated 42 81 61 20 11 15 15 Alemtuzumab, IV 1 pentostatin Untreated 13 82 73 9 4.3 10.4 Alemtuzumab, IV Pretreated 15 46 46 — 31 5 Alemtuzumab, IV 1 pentostatin Pretreated 5 7 5 5 0 2 5 2 .6 2.6 FMC 1 alemtuzumab, sc Multicenter, prospective Untreated 13 68.7 25.8% CR, 6.25% CRi 36.6 7.5 1 1.5 8 Pretreated 5 sc, subcutaneous.
Recommendation: IV alemtuzumab induction therapy for 10 to 12 weeks (to achieve best response), followed by consolidation with HSCT where feasible.
After relapse, the prognosis of T-PLL is dismal. Salvage regimens (Table 6) can achieve an ORR 50% to 76%, but only short du-ration and an OS of 6 to 9 months.7,15,47,52There are preliminary
in vitro and in vivo data to suggest that novel agents such as BCL2-inhibitors, HDAC inhibitors, and JAK3 inhibitors may have a therapeutic effect in TPLL.6,32,35,53,54Clinical trials testing these
novel therapeutic approaches are badly needed.
Discussion
The consensus criteria proposed here for diagnosis and treat-ment response evaluation in T-PLL are applicable for both daily clinical use and clinical studies. Although some of our recom-mendations are based on small to medium-sized clinical studies and observations on T-PLL itself, some have been extrapolated from other disease entities based on consensus evaluation. Historically, criteria for CLL, as defined by the IWCLL, have been used. However, CLL is very different from T-PLL and there is a need for the introduction of disease-specific criteria. The T-PLL- specific criteria presented here differ from the IWCLL criteria particularly in the following aspects:
1. The diagnosis of T-PLL relies on presence of clonal mature T cells with characteristic prolymphocytic morphology and distinct cytogenetics.
2. Instead of clinical staging according to Binet or Rai in CLL, we considered 2 disease states, inactive and active, as clinically more appropriate in T-PLL. At diagnosis, nearly all patients with T-PLL present with generalized disease as per highly elevated white blood cell count, splenomegaly, hepato-megaly, and generalized lymphadenopathy. So far, there are no reported observations or studies indicating that the number of areas of lymphoid tissues that are affected or whether hepatosplenomegaly might correlate with disease state or course or outcome. In comparison with other leu-kemias, especially acute leukemias (acute myeloid leukemia, acute lymphoblastic leukemia), staging criteria are not con-sidered relevant and are therefore not established. Although the proposed phase-based distinction of T-PLL somewhat resembles the established system used in chronic myeloid leukemia, T-PLL does not typically show classical t-lympho-blasts-associated transformation. Nevertheless, an inactive (chronic) phase of T-PLL is usually followed by an active (acute) phase within 1 to 2 years. Neither cell-phenotypic changes (blastic transformation) nor other measurable changes other than altered disease kinetics and symptoms are detected on the transition into the active state of T-PLL.
However, entering this active stage of disease requires treat-ment initiation (similar to Binet C or Rai III/IV stage in CLL). 3. Because T-PLL is clinically closer related to acute
leuke-mias, we propose to confirm a response, such as a CR, with a bone marrow examination. However, as for acute leukemias, a clinical response that is confirmed in the bone marrow does not require to be maintained for a certain time.
4. We recommend the use of the recently proposed RECIL-2017 criteria for radiologic response assessment based on unidi-mensional changes (ie, SLD) of just up to 3 target lesions in CT or magnetic resonance imaging.40RECIL-2017 are easier to
use and appear more practical for both retrospective and prospective clinical studies and are nevertheless validated for lymphatic disease.
Ultimately, it will be critical and challenging to match the T-PLL consensus criteria as presented here in the consensus criteria to current and novel treatments. This will be an ongoing effort to optimize patient management and outcome in this rare leuke-mia. Future studies in T-PLL should not only evaluate the ac-curacy of these criteria but also try to establish the role of molecular assessments in prognostication, response pre-diction, and evaluation of depth of responses (minima residual disease) to better guide our treatments in this for the most part still-fatal disease.
Authorship
Contribution: P.B.S. organized the consensus meeting of the T-PLL International Study Group in May 2017 in Vienna, provided thefirst draft, and composed the last version of the manuscript; all authors of the manuscript are members of the T-PLL International Study Group; all authors discussed all recommendations until a consensus was reached, and all authors contributed equally to write and agree on the article.
Conflict-of-interest disclosure: The authors declare no competing fi-nancial interests.
ORCID profiles: P.B.S., 0000-0001-6729-7708; O.T., 0000-0002-9438-621X; K.P., 0000-0003-4112-5902; S.K., 0000-0003-0855-8343; U.J., 0000-0001-9826-1062.
Correspondence: Philipp B. Staber, Medical University of Vienna, De-partment of Medicine I, Division of Hematology & Hemostaseology, W ¨ahringer G ¨urtel 18-20, A-1090 Vienna, Austria; e-mail: philipp.staber@ meduniwien.ac.at.
Footnotes
Submitted 2 March 2019; accepted 26 June 2019. Prepublished online as Blood First Edition paper, 10 July 2019; DOI 10.1182/blood.2019000402. There is a Blood Commentary on this article in this issue.
R E F E R E N C E S
1. Catovsky D, Galetto J, Okos A, Galton DA, Wiltshaw E, Stathopoulos G. Prolymphocytic leukaemia of B and T cell type. Lancet. 1973; 2(7823):232-234.
2. Matutes E, Brito-Babapulle V, Swansbury J, et al. Clinical and laboratory features of 78 cases of T-prolymphocytic leukemia. Blood. 1991;78(12):3269-3274.
3. Herling M, Khoury JD, Washington LT, Duvic M, Keating MJ, Jones D. A systematic approach to diagnosis of mature T-cell leu-kemias reveals heterogeneity among WHO categories. Blood. 2004;104(2):328-335. 4. Dearden CE, Khot A, Else M, et al. Alemtuzumab
therapy in T-cell prolymphocytic leukemia:
com-paring efficacy in a series treated intravenously
and a study piloting the subcutaneous route. Blood. 2011;118(22):5799-5802.
5. Hopfinger G, Busch R, Pflug N, et al.
Sequential chemoimmunotherapy offludarabine,
mitoxantrone, and cyclophosphamide induction followed by alemtuzumab consolidation is effective in T-cell prolymphocytic leukemia. Cancer. 2013;119(12):2258-2267.
6. Boidol B, Kornauth C, van der Kouwe E, et al. First-in-human response of BCL-2 inhibitor venetoclax in T-cell prolymphocytic leukemia. Blood. 2017;130(23):2499-2503.
7. Herbaux C, Genet P, Bouabdallah K, et al. Bendamustine is effective in T-cell prolym-phocytic leukaemia. Br J Haematol. 2015; 168(6):916-919.
8. Pflug N, Cramer P, Robrecht S, et al. New
lessons learned in T-PLL: results from
a prospective phase-II trial with
fludarabine-mitoxantrone-cyclophosphamide-alemtuzumab induction followed by alemtuzumab mainte-nance. Leuk Lymphoma. 2019;60(3):649-657.
9. Gandhi V, Tam C, O’Brien S, et al. Phase I trial
of nelarabine in indolent leukemias. J Clin Oncol. 2008;26(7):1098-1105.
10. Cheson BD, Bennett JM, Rai KR, et al. Guidelines for clinical protocols for chronic lymphocytic leukemia: recommendations of the National Cancer Institute-sponsored working group. Am J Hematol. 1988;29(3): 152-163.
11. Cheson BD, Bennett JM, Grever M, et al. National Cancer Institute-sponsored Working Group guidelines for chronic lymphocytic leukemia: revised guidelines for diagnosis and treatment. Blood. 1996;87(12):4990-4997. 12. Hallek M, Cheson BD, Catovsky D, et al. iwCLL
guidelines for diagnosis, indications for treatment, response assessment, and sup-portive management of CLL. Blood. 2018; 131(25):2745-2760.
13. Hallek M, Cheson BD, Catovsky D, et al; In-ternational Workshop on Chronic Lympho-cytic Leukemia. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111(12):5446-5456. 14. Swerdlow SH, Campo E, Pileri SA, et al. The
2016 revision of the World Health
Organiza-tion classification of lymphoid neoplasms.
Blood. 2016;127(20):2375-2390.
15. Jain P, Aoki E, Keating M, et al. Characteristics, outcomes, prognostic factors and treatment of patients with T-cell prolymphocytic leukemia (T-PLL). Ann Oncol. 2017;28(7):1554-1559. 16. Schrader A, Crispatzu G, Oberbeck S, et al.
Actionable perturbations of damage respon-ses by TCL1/ATM and epigenetic lesions form the basis of T-PLL. Nat Commun. 2018;9(1):697. 17. Dearden C. Management of prolymphocytic
leukemia. Hematology Am Soc Hematol Educ Program. 2015;2015:361-367.
18. Ravandi F, O’Brien S. Chronic lymphoid
kemias other than chronic lymphocytic leu-kemia: diagnosis and treatment. Mayo Clin Proc. 2005;80(12):1660-1674.
19. Laribi K, Lemaire P, Sandrini J, Baugier de Materre A. Advances in the understanding and management of T-cell prolymphocytic leukemia. Oncotarget. 2017;8(61): 104664-104686.
20. Kotrova M, Novakova M, Oberbeck S, et al. Next-generation amplicon TRB locus
se-quencing can overcome limitations of
flow-cytometric Vb expression analysis and confirms
clonality in all T-cell prolymphocytic leukemia cases. Cytometry A. 2018;93(11):1118-1124.
21. Pekarsky Y, Hallas C, Croce CM. Molecular basis of mature T-cell leukemia. JAMA. 2001; 286(18):2308-2314.
22. Herling M, Patel KA, Teitell MA, et al. High TCL1 expression and intact T-cell receptor
signaling define a hyperproliferative subset of
T-cell prolymphocytic leukemia. Blood. 2008; 111(1):328-337.
23. D ¨urig J, Bug S, Klein-Hitpass L, et al. Combined single nucleotide polymorphism-based genomic mapping and global gene
expression profiling identifies novel
chromo-somal imbalances, mechanisms and candi-date genes important in the pathogenesis of T-cell prolymphocytic leukemia with inv(14)(q11q32). Leukemia. 2007;21(10): 2153-2163.
24. Sun Y, Tang G, Hu Z, et al. Comparison of
karyotyping, TCL1fluorescence in situ
hybridisation and TCL1 immunohistochemis-try in T cell prolymphocytic leukaemia. J Clin Pathol. 2018;71(4):309-317.
25. Sugimoto J, Hatakeyama T, Narducci MG,
Russo G, Isobe M. Identification of the TCL1/
MTCP1-like 1 (TML1) gene from the region next to the TCL1 locus. Cancer Res. 1999; 59(10):2313-2317.
26. Brito-Babapulle V, Catovsky D. Inversions and tandem translocations involving chromosome 14q11 and 14q32 in T-prolymphocytic leu-kemia and T-cell leuleu-kemias in patients with ataxia telangiectasia. Cancer Genet Cytoge-net. 1991;55(1):1-9.
27. Stilgenbauer S, Schaffner C, Litterst A, et al. Biallelic mutations in the ATM gene in T-prolymphocytic leukemia. Nat Med. 1997; 3(10):1155-1159.
28. Vorechovsk ´y I, Luo L, Dyer MJ, et al. Clustering of missense mutations in the ataxia-telangiectasia gene in a sporadic T-cell leukaemia. Nat Genet. 1997;17(1):96-99. 29. Pekarsky Y, Hallas C, Isobe M, Russo G,
Croce CM. Abnormalities at 14q32.1 in T cell malignancies involve two oncogenes. Proc Natl Acad Sci USA. 1999;96(6): 2949-2951.
30. Hetet G, Dastot H, Baens M, et al. Recurrent molecular deletion of the 12p13 region, cen-tromeric to ETV6/TEL, in T-cell prolymphocytic leukemia. Hematol J. 2000;1(1):42-47. 31. Costa D, Queralt R, Aymerich M, et al. High
levels of chromosomal imbalances in typical and small-cell variants of T-cell prolympho-cytic leukemia. Cancer Genet Cytogenet. 2003;147(1):36-43.
32. Stengel A, Kern W, Zenger M, et al. Genetic characterization of T-PLL reveals two major biologic subgroups and JAK3 mutations as prognostic marker. Genes Chromosomes Cancer. 2016;55(1):82-94.
33. Bergmann AK, Schneppenheim S, Seifert M, et al. Recurrent mutation of JAK3 in T-cell prolymphocytic leukemia. Genes Chromo-somes Cancer. 2014;53(4):309-316. 34. Kiel MJ, Velusamy T, Rolland D, et al.
Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood. 2014;124(9):1460-1472. 35. L ´opez C, Bergmann AK, Paul U, et al. Genes
encoding members of the JAK-STAT pathway
or epigenetic regulators are recurrently mu-tated in T-cell prolymphocytic leukaemia. Br J Haematol. 2016;173(2):265-273. 36. Bellanger D, Jacquemin V, Chopin M, et al.
Recurrent JAK1 and JAK3 somatic mutations in T-cell prolymphocytic leukemia. Leukemia. 2014;28(2):417-419.
37. Garand R, Goasguen J, Brizard A, et al. Indolent course as a relatively frequent pre-sentation in T-prolymphocytic leukaemia.
Groupe Français d’H ´ematologie Cellulaire. Br
J Haematol. 1998;103(2):488-494. 38. Goede V, Fischer K, Busch R, et al.
Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med. 2014;370(12):1101-1110. 39. Cheson BD, Ansell S, Schwartz L, et al.
Refinement of the Lugano Classification
lym-phoma response criteria in the era of immu-nomodulatory therapy. Blood. 2016;128(21): 2489-2496.
40. Younes A, Hilden P, Coiffier B, et al.
International Working Group consensus response evaluation criteria in lymphoma (RECIL 2017). Ann Oncol. 2017;28(7): 1436-1447.
41. Mayerhoefer ME, Jaeger U, Staber P, et al. [68Ga]Ga-Pentixafor PET/MRI for CXCR4 Im-aging of Chronic Lymphocytic Leukemia: Preliminary Results. Invest Radiol. 2018;53(7): 403-408.
42. Br ¨uggemann M, White H, Gaulard P, et al. Powerful strategy for polymerase chain reaction-based clonality assessment in T-cell malignancies Report of the BIOMED-2 Con-certed Action BHM4 CT98-3936. Leukemia. 2007;21(2):215-221.
43. Sellner L, Br ¨uggemann M, Schlitt M, et al. GvL effects in T-prolymphocytic leukemia: evi-dence from MRD kinetics and TCR repertoire analyses [published correction appears in Bone Marrow Transplant. 2017;52(4):656]. Bone Marrow Transplant. 2017;52(4): 544-551.
44. Cheson BD, Fisher RI, Barrington SF, et al; United Kingdom National Cancer Research Institute. Recommendations for initial evalu-ation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the
Lugano classification. J Clin Oncol. 2014;
32(27):3059-3068.
45. Catovsky D. Prolymphocytic leukaemia. Nouv Rev Fr Hematol. 1982;24(6):343-347. 46. Pawson R, Dyer MJ, Barge R, et al. Treatment
of T-cell prolymphocytic leukemia with human CD52 antibody. J Clin Oncol. 1997;15(7): 2667-2672.
47. Dearden CE, Matutes E, Cazin B, et al. High remission rate in T-cell prolymphocytic leu-kemia with CAMPATH-1H. Blood. 2001;98(6): 1721-1726.
48. Krishnan B, Else M, Tjonnfjord GE, et al. Stem cell transplantation after alemtuzumab in T-cell prolymphocytic leukaemia results in longer survival than after alemtuzumab alone: a multicentre retrospective study. Br J Haematol. 2010;149(6):907-910. 49. Guillaume T, Beguin Y, Tabrizi R, et al.
Allogeneic hematopoietic stem cell trans-plantation for T-prolymphocytic leukemia:
a report from the French society for stem cell transplantation (SFGM-TC). Eur J Haematol. 2015;94(3):265-269.
50. Wiktor-Jedrzejczak W, Dearden C, de Wreede L, et al; EBMT Chronic Leukemia Working Party. Hematopoietic stem cell transplantation in T-prolymphocytic leukemia: a retrospective study from the European Group for Blood and Marrow Transplantation and the Royal Marsden Consortium. Leukemia. 2012;26(5): 972-976.
51. Wiktor-Jedrzejczak W, Drozd-Sokolowska J, Eikema DJ, et al. EBMT prospective ob-servational study on allogeneic hemato-poietic stem cell transplantation in
T-prolymphocytic leukemia (T-PLL) [published online ahead of print 21 January 2019]. Bone Marrow Transplant. doi:10.1038/s41409-019-0448-x.
52. Keating MJ, Cazin B, Coutr ´e S, et al. Campath-1H treatment of T-cell prolym-phocytic leukemia in patients for whom at least one prior chemotherapy regimen has failed. J Clin Oncol. 2002;20(1): 205-213.
53. Andersson EI, P ¨utzer S, Yadav B, et al. Discovery of novel drug sensitivities in T-PLL by high-throughput ex vivo drug testing and
mutation profiling. Leukemia. 2018;32(3):
774-787.
54. Schrader A, Braun T, Herling M. The dawn of a new era in treating T-PLL. Oncotarget. 2019; 10(6):626-628.
55. Mercieca J, Matutes E, Dearden C, MacLennan K, Catovsky D. The role of pentostatin in the treatment of T-cell malignancies: analysis of response rate in 145 patients according to disease subtype. J Clin Oncol. 1994;12(12): 2588-2593.
56. Ravandi F, Aribi A, O’Brien S. Phase II study
of alemtuzumab in combination with pentostatin in patients with T-cell neoplasms. J Clin Oncol. 2009;27(32): 5425-5430.