Biodegradable polylactide/hydroxyapatite nanocomposite foam scaffolds for bone tissue engineering applications
15
0
0
Texte intégral
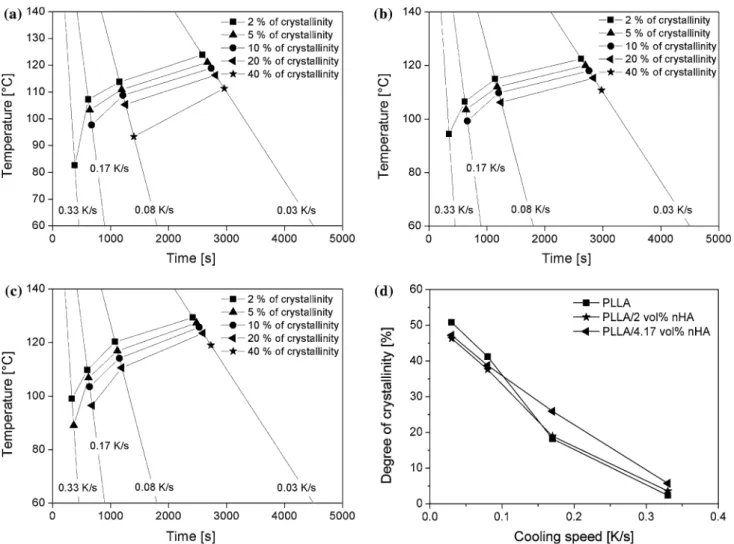
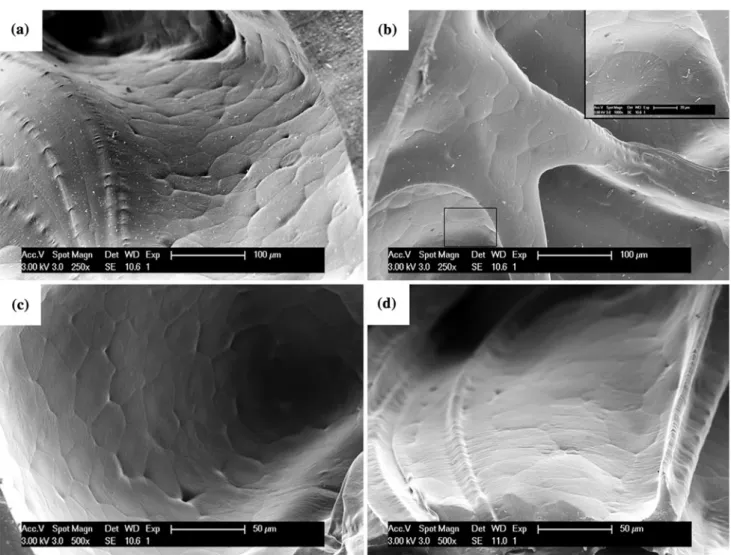
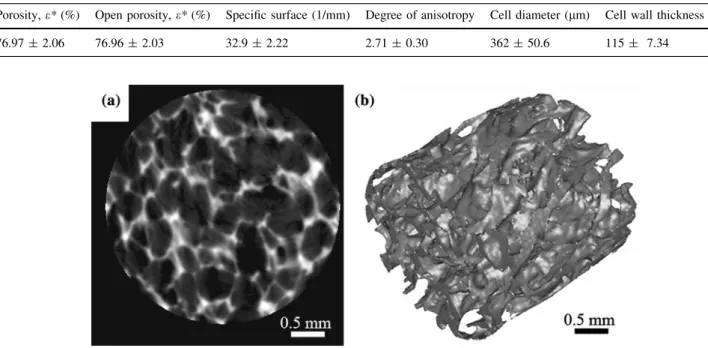
Figure
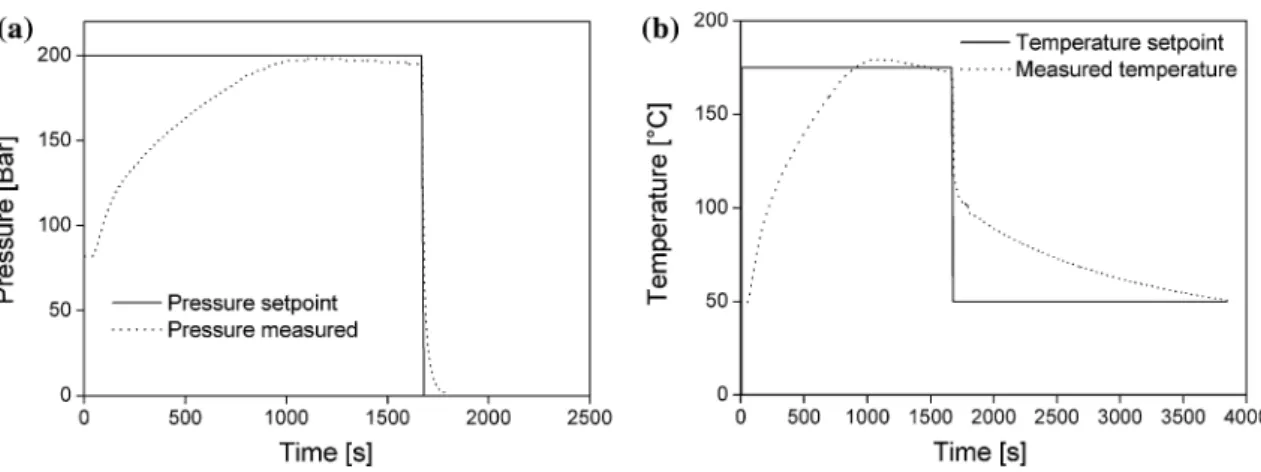
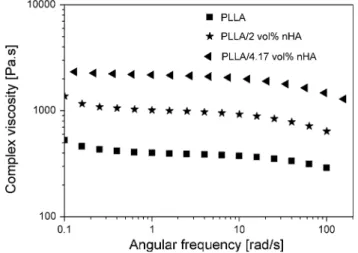

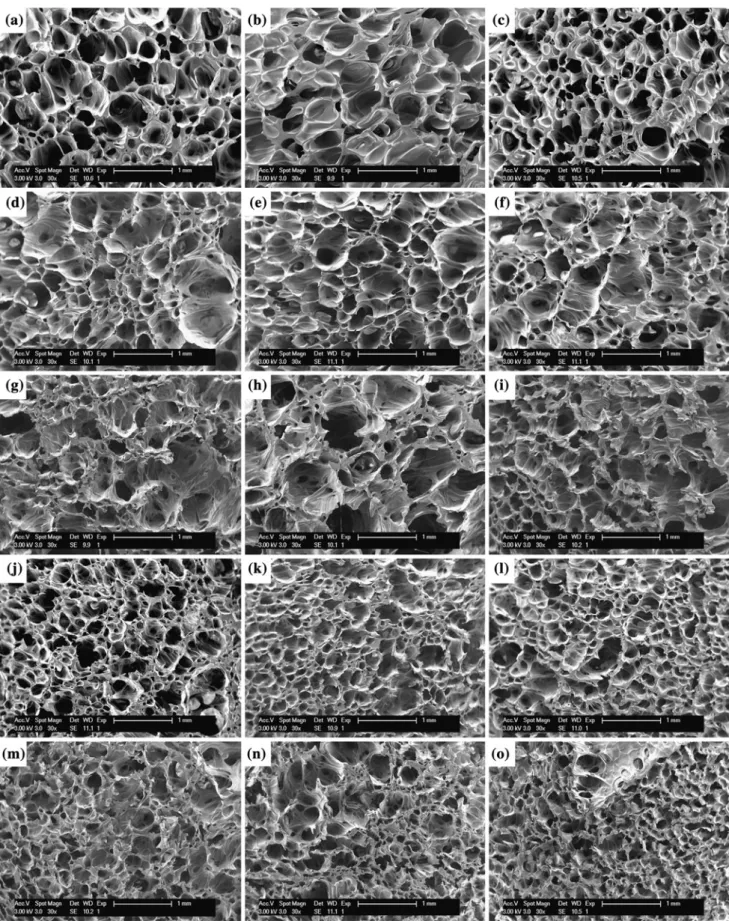
+6
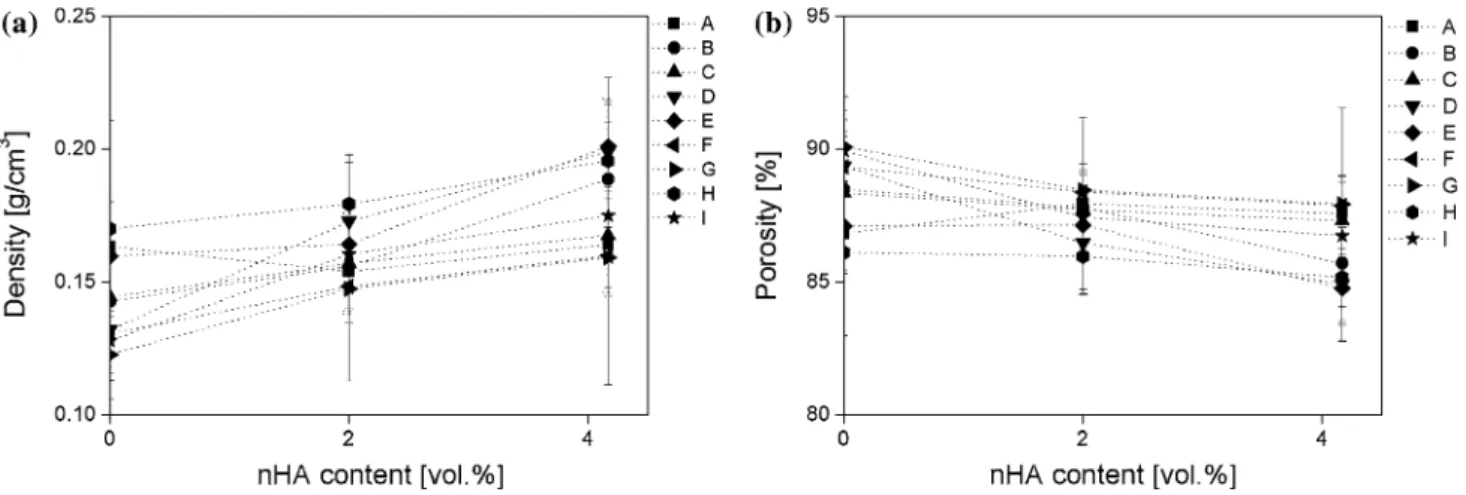

Documents relatifs