HAL Id: jpa-00233778
https://hal.archives-ouvertes.fr/jpa-00233778
Submitted on 1 Jan 1941
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
L’effet Raman et le pivotement des molécules dans les
cristaux. Théorie générale et vérification expérimentale
dans le cas du naphtalène
A. Kastler, A. Rousset
To cite this version:
A. Kastler, A. Rousset. L’effet Raman et le pivotement des molécules dans les cristaux. Théorie
générale et vérification expérimentale dans le cas du naphtalène. J. Phys. Radium, 1941, 2 (2),
pp.49-57. �10.1051/jphysrad:019410020204900�. �jpa-00233778�
LE
JOURNAL
DE
PHYSIQUE
-ET
LE RADIUM
L’EFFET RAMAN ET LE PIVOTEMENT DES
MOLÉCULES
DANS LES CRISTAUX.THÉORIE GÉNÉRALE
ETVÉRIFICATION EXPÉRIMENTALE
DANS LE CAS DUNAPHTALÈNE
Par A. KASTLER et A. ROUSSET.
Faculté des Sciences de Bordeaux
(1).
Sommaire. --
On montre que les raies Raman de faible fréquence, découvertes par Gross et Vuks dans le spectre de la lumière diffusée par les cristaux organiques, sont dues à des pivotements cohérents
des molécules du cristal. On calcule l’état de polarisation et l’intensité des vibrations diffusées par les
molécules pivotantes d’un cristal. Les résultats de ce calcul sont confrontés avec les mesures de polari-sation sur les spectres Raman du naphtalène et de la calcite. On rapporte, avec quelques détails, les résultats de l’étude expérimentale de la polarisation et de l’intensité des raies de faible fréquence d’un monocristal de naphtalène : ces résultats sont en excellent accord avec l’hypothèse de pivotements cohérents des molécules autour de leurs axes d’inertie. Enfin, les auteurs insistent sur les rapports
néces-saires entre l’effet du pivotement des molécules dans les cristaux et l’effet de la rotation de ces mêmes molécules dans un liquide, révélés par la diffusion moléculaire de la lumière.
SÉRIE YIII. - TOME II. 2. BVR!I MAI-JUIN 19~1.
1. Théorie
classique
de la diffusion de lalumière par une molécule
pivotante.
- SoientU,
V, W les trois axes
rectangulaires
del’ellipsoïde
d’inertie d’unemolécule;
g,g’
etg"
les réfractivitésprincipales
mesurées suivant ces mêmes axes(modèle
moléculaire deLangevin).
Nous supposons que la molécule exécute autour d’une orientation moyenne, définie par les axes fixesUo,
Vo,
Wo,
des oscilla-tionspendulaires
de faibleamplitude
(ou
pivo-tements)
defréquence
u, U, w.’
Éclairons
la molécule par la vibration lumineusesin 2 zNt
parallèle
àUo. Projetons
la vibration incidente sur chacun des axes de la moléculepivo-tante. En
multipliant l’amplitude
dechaque
projec-tion par la réfractivitécorrespondante,
nous obtenons lesprojections
du moment induit suivant ces mêmes axes. Lesamplitudes
des vibrationsdiffusées,
paral-lèles à chacun des axes fixes
U 0’ Vo,
Wo,
sontégales
auxprojections
de ce moment induit sur ces axes.la La molécule
pivote
autour deUo.
---- Aucunemodification de
fréquence
dans le moment induitqui
reste couché surUo.
(1) Exposé dans une conférence de A. Rousset devant la
Société française de Physique (séances de Pentecôte 1941)’
20 La molécule
pivote
autour deV 0 (fig. 1).
-Les
positions
des axes U et W sont àchaque
instant fixées parl’angle
oc =(;(0 sin
2Tr~. Suivant les axes de lamolécule,
lesprojections
du moment induit s’écriventd’où les
projections
de ce moment sur les axes fixesU 0’ Vo,
Wo :
Mais «o et par
conséquent
« sontpetits.
Onpeut
poser cos « =
1; sin2 « = o et sin « _ « _
aosin 2 n vt.
Il vient alors
Le moment des
fréquences
modifiées est couché sur"¥V 0;
onpeut
l’écrireLE JOURNAL DE PHYSIQUE ET LE RADIUM. - SÉRIE VIII. -
T. II. - :-i° 2. - AVRIL-MAI-JUIN 1941. 4.
30
Son
amplitude,
pour chacune des deuxfré-quences N
± P,vaut
ao(g
2013g ").
30 La molécule
pivote
autour deWo.
- Dansce
cas, les doublets de Hertz de
fréquences
N ± w sontcouchés sur
V~
et leuramplitude
vaut 2
oca(g
-g").
Ce calcul montre que les intensités des raies depivotement
varient comme le carré del’anisotropie
Fige.
optique
de lamolécule,
que seules sont efficaces lesvibra-tions excitatrices
perpendicu-laires à l’axe de
pivotement
et que le moment induit dans la molécule est
perpen-diculaire à l’axe de
pivote-ment.
Dès
1935,
l’un de nous[1]
]
avait tiré de ce calcul l’inten-sité et lapolarisation
desradiations diffusées par des
molécules
pivotantes
orientées au hasard(liquides
à molécules
polaires
parexemple);
5 l’intensitélumi-neuse
diffusée,
proportionnelle
au carré del’ampli-tude
maximum,
n’estqu’une
faible fraction duspectre
de rotation de la molécule libre de tourner.Raman et
Negundaki
[2],
enprécisant
la direction du moment induit dans le cas d’ions de même orien-tation moyenne, ont donné uneinterprétation
trèssimple
des variations d’intensité queprésentent
certaines raies Raman d’un cristal de nitrate de
sodium
quand
onchange
l’orientation du cristal parrapport
à la vibration incidente et à la direction d’observation. Ils admettent que dans lespectre
Raman du nitrate de sodium
cristallisé,
les raies de faiblefréquence (2)
185 cm-1 etg8
cm-1 sont duesà des oscillations de l’ion
N03
autour d’un axe duplan équatorial.
Ils vérifientalors,
conformément aux résultats du calculrappelés
plus
haut,
que ces raiesdisparaissent
dans lesspectres
où lesfréquences
internes io65 cm-1 et
1375
cm-1paraissent
avec lemaximum d’intensité
(3).
Ensupposant
cespivo-tements
quantifiés,
les auteurs hindous ont calculél’amplitude
maximum en fonction du momentd’inertie I et de la
fréquence v
depivotement.
On obtientimmédiatement,
pourl’amplitude
du momentdiffusé,
i2 2
V
21:I()Afin d’étendre à d’autres cristaux cette
interpré-tation des raies Raman de faible
fréquence,
nous (1) La présence dans le spectre Raman du nitrate de sodium,comme dans celui du spath calcaire, de deux raies de faible fréquence ayant les mêmes caractères de polarisation, reste à expliquer.
(1) On sait cependant (CABANNES et CANALs, Compfes
rendus, 19 3 1, 193, p. 2 8 9) que les fréquences internes 1065 cm-1 et i385 cm-1, comme les fréquences correspondantes t o8~ crn-1
et ig37 cm-1 du spath, ne présentent pas les mêmes
carac-tères de polarisation.
allons traiter le cas
plus
général
de la diffusion par les moléculespivotantes
dont les axes d’inertie sontplacés
defaçon
absolumentquelconque
parrapport
aux éléments desymétrie
ducristal,
élémentsqui
imposent
la direction des vibrations excitatrices etla direction d’observation.
Soient
U, V,
W(4)
les trois axesrectangulaires
del’ellipsoïde
d’inertie autourduquel
la molécule(ou
l’ion)
pivote
avec lafréquence
u, v ou w; g,g’
etg"
lesréfractivités
principales
de la molécule(ou
del’ion)
relatives à ces mêmes axes. Soit un
trièdre,
lié aux éléments desymétrie
ducristal,
d’axes 1, 2, 3.17ableau cles cosinus directeurs.
+
10 Le vecteur
électrique
E de l’onde incidente estparallèle
à l’axe 1. - Les --projections
Eu, Ev,
E,v sur les axesU, V,
W de la molécule sontrespectivement
proportionnelles
àal, b1
et c,. Pour chacune de cesprojections
et pour chacun despivotements
autourde
U,
V etW,
appliquons
les résultats du calcul+
précédent.
Lesamplitudes
des moments induitsMu,
*
¿-Mv et MVt- de
fréquences
lVu, iyiv,
N:.,y et leurdirection sont
données,
danschaque
cas, par le tableau ci-dessous(*) :
*
Si le vecteur
électrique
E estparallèle
à l’axe 2ou à l’axe
3,
il suffit deremplacer,
dans le tableauci-dessus, l’indice i par l’indice 2 ou 3. On obtient
alors,
pour chacune desfréquences
Raman depivotement (u,
parexemple),
le tenseur( U) qui
(4) On peut, sans ambiguïté, désigner ici et dans la suite descalculs, les directions moyennes des axes de la molécule pivotante par UVW et non plus par U 0 V 0 W o.
+
relie le vecteur
électrique
E de l’onde incidente et+
le moment induit Mu
qui
émet la raie Raman,rapportés
aux axes 1,2,
3[tous
les termes doivent êtremultipliés
par(g’
--g")].
-Les tenseurs
( V)
et(W),
relatifs à My etMw,
sedéduisent du tenseur
précédent
par unepermutation
circulaire de a,b,
c, et de g,g’, g".
2. Calcul des tenseurs dans le cas de
pivo-tements cohérents des molécules d’un cristal
monoclinique
Prenons la maille d’un cristalmonoclinique
comme lenaphtalène qui
renferme deux molécules(I
etII)
orientéessymétriquement
parrapport
auplan
desymétrie (010)
normal à l’axe binaire(axe 2).
Supposons
cohérents tous lespivotements
des molécules du cristal. Lepivotement
d’ensemble des molécules I et II autour de leur axe Uconstitue,
pour lamaille,
une vibrationsymétrique
Us
ouantisymétrique
UA
parrapport
à sonplan
desymé-trie,
suivant que lespivotements
de ces deuxmolé-cules sont
symétriques
ouantisymétriques
parrapport
à ceplan (5) .
Aupoint
de vue de sonorien-tation,
nous savons que la molécule IIpeut
êtreconsidérée comme
l’image,
parréflexion,
de la moléculeI,
dans leplan
desymétrie.
Nous obtenons son tenseur(Uil)
enchangeant,
dans le tenseurci-dessus,
lessignes
dea2, b2
et c2correspondant
auxprojections
sur l’axebinaire,
d’où la forme destenseurs
d’amplitudes
pour les deux molécules :Le
couplage
despivotements
des deux moléculesen vibration
symétrique
etantisymétrique
revient à unajustement
différent desphases
despivotements
des deux molécules I et II. La combinaisonsymé-(15) Ces pivotements sont en même temps symétriques ou
antisymétriques par rapport à l’axe binaire.
trique
est due à deuxpivotements
en concordance dephase;
les deux tenseursd’amplitudes s’ajoutent :
La combinaison
antisymétrique
est due à deuxpivotements
enopposition
dephase;
dans ce cas,une
augmentation
de z
pour laphase
change
lesigne
de tous les moments induits du tenseur(Uii) ;
les deux tenseurs
d’amplitudes
se retranchent :On obtient
ainsi,
pour les vibrationsUs
etUA,
les deux tenseurs
complémentaires
ci-dessous(6) :
Nous retrouvons pour nos
pivotements
lesrésul-tats
généraux
établis par J. Cabannes[3]
pour lesoscillations
symétriques
etantisymétriques
descristaux du
système
monoclinique.
Les deux tenseurs
d’intensités,
dont les termess’obtiennent en élevant au carré les termes
corres-pondants
des tenseursd’amplitude,
sont doncéga-lement
complémentaires
et n’ont aucun termecommun. Nous les
reporterons
dans la suite dans unmême cadre.
Les deux modes de
couplage, symétrique
etanti-symétrique, quoique
relatifs à un mêmepivotement,
mettent en
jeu
des forces derappel légèrement
diffé-rentes : par
suite,
aux vibrationssymétriques
etantisymétriques
de la maille doiventcorrespondre
desfréquences
depivotement légèrement
différentes. Dans le casgénéral
de la molécule deLangevin
àtrois réfractivités
inégales,
il faut doncprévoir
six raies « externes »
différentes,
voisines de la raieexcitatrice : Us et
UA, Vs
etV~, ivs
et’VA,
grou-pées
en trois doublets. A chacune descomposantes
d’un
doublet,
tTs
ou Lrn parexemple, correspond
l’un des tenseurscomplémentaires (vs)
ou(UA)
formé de termes soituniquement
symétriques,
soituniquement antisymétriques.
3.
Application
aunaphtalène.
- La moléculedu
naphtalène
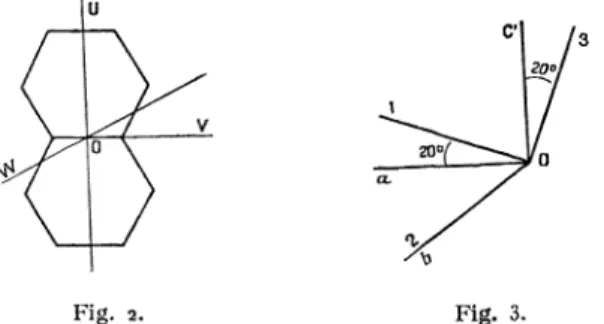
estplane. Désignons
par U songrand
axe(ou ligne joignant
les centres des noyauxbenzéniques),
par V sonpetit
axe(perpendiculaire
à U et situé dans le
plan moléculaire),
par V‘’ la normale auplan
moléculaire(fil.
2).
On connaît,par l’étude aux rayons X
(Robertson) [41,
l’orien-(6) Le tenseur (Us) ne contient que les termes Aas dont la somme des indices a + ~ est paire le tenseur; (UA) ne°
32
tation des axes U,
V,
W parrapport
au trièdre constitué par l’arête a et b de la mailleclinorhom-bique (b,
axe binaire ou axe2)
et laperpendiculaire
c’ auplan
des arêtes a et b(plan ool)
de la maille.Fig. 2. Fig. 3.
Voici le tableau des cosinus directeurs :
TABLEAU I.
On en déduit facilement les cosinus directeurs des
mêmes axes U,
V,
W parrapport
au trièdre 11 2, 3des axes de
l’ellipsoïde
des indices : les axes 1 et 3sont situés dans le
plan
desymétrie
ac’,
l’axe 3 estconfondu avec l’arête du dièdre des
plans
molécu-laires[5],
donc incliné de 2oo sur c’(fig.
3).
TABLEAU II.
’
Le monocristal de
naphtalène
utilisé dans nos recherches a la forme d’unparallélépipède rectangle
dont les arêtes sontparallèles
aux axesa, b,
c’ de Robertson(’).
Soit OXYZ un trièdre fixe dansl’espace,
OZ étant vertical. Le cristal estplacé
en 0 avec ses arêtesrespectivement parallèles
à XYZ. On éclaire lelong
de OX par un faisceau de lumièrepolarisée
suivant OY ou suivant OZ. Les vibrations diffusées dans la direction OY sontanalysées
par un biré-(7) n aurait été préférable d’étudier un parallélépipèdetaillé suivant les arêtes 1, 2, 3 de l’ellipsoïde des indices,
mais nous ne disposions pas d’un bel échantillon de ce genre.
fringent (wollaston)
de sectionprincipale
verticale,
de
façon
à donner sur la fente duspectrographe
deuximages
séparées
ducristal,
l’une i contenant les vibrations diffuséesparallèles
àOX,
l’autre 7 les vibrations diffuséesparallèles
à OZ(8).
Comme les arêtes de notre cristal ne coïncident pas exactement avec les axes des indices
principaux
(sauf
pour l’axebinaire),
labiréfringence
va introduire unecomplication :
c’est seulement dans le cas où leplan
desymétrie
est horizontal que les vibrations incidentes ou diffusées restentparallèles
aux arêtes du cristal. Si l’on éclaire(ou
si l’onobserve)
àtravers le
plan
desymétrie,
les vibrations incidentes(ou diffusées) qui
sepropagent
à travers le cristalsont
parallèles
aux axes 1 et3,
il en résultequ’en
général
les intensités et lespolarisations
expéri-mentales doivent
présenter
unléger
écart avec les valeurs calculées àpartir
des cosinusdirecteurs,
tant du Tableau 1 relatif au trièdre abc’ que du
Tableau II relatif au trièdre 1, 2, 3.
Dans les tenseurs d’intensité
ci-dessous,
nous avonsreporté
les intensités calculées en confondant le trièdre XYZ successivement avec les deux trièdres abc’ et 1, 2, 3. Danschaque
case le nombre du haut serapporte
au trièdreabc’,
celui du bas au trièdre 1, 2, 3(9).
Les intensités données par
l’expérience
doiventêtre
comprises
entre ces valeurs limites. Les inten-sitésexpérimentales
évaluéesqualitativement
surles clichés et
exprimées
par les lettres F(forte),
m
(moyenne), f (faible),
o(nulle)
ont étéreportées
dans le même cadre(1°).
Les termes
expérimentaux
ont été obtenus de la manière suivante : -. on a donné successivement au cristal les six orientations différentespossibles
parrapport
au trièdre fixe XYZ :Pour chacune de ces six
orientations,
le vecteurélectrique
de la lumière excitatrice a étédirigé
d’abordparallèlement
à l’axe OZ(E,~,),
ensuiteparallèlement
à l’axe OY(EJ.).
Les 12 cascorres-pondants
sont numérotés de 1 à 12 dans lapremière
colonne de laplanche
I.Enfin,
lebiréfringent placé
(1) Une bilame demi-onde placée devant la fente rend les deux vibrations issues du wollaston toutes deux normales
aux arêtes des prismes du spectrographe. Dans ces conditions,
les coefficients de transmission sont égaux et optima pour
ces deux vibrations.
(s) Chaque terme est à multiplier : pour le tenseur (U), par (gr -
g")2, pour le tenseur ( V), par (g -
g")2 et pour le tenseur (jV), par (g - g’)2.
(l°) Les deux tableaux complémentaires relatifs aux
pivo-tements autour d’un même axe U par exemple, sont reportés dans le même cadre; pour chacune des vibrations Us et Ua,
on ne prend évidemment que les termes non nuls des tenseurs
sur le faisceau diffusé fournit, pour chacun de ces cas, deux
spectres : spectre
i, dû à la vibrationdiffusée
parallèle
à OX,spectre
I dû à la vibrationdiffusée
parallèle
à OZ. Chacun de ces2~
spectres
correspond
à un terme déterminé(Aap)2
des tenseursd’intensité. Deux colonnes de la
planche
Iindiquent :
l’une la notation du termecorrespondant
àchaque
spectre,
l’autre son caractère desymétrie
(s,
symé-trique ;
a,antisymétrique).
Dans les deux colonnessuivantes on donne une
représentation
schématique
des
spectres
permettant
de confronter lesprévisions
théoriques
et les résultatsexpérimentaux :
l’épais-seur des traits matérialise l’intensité des raies
corres-pondantes.
Lespoints d’interrogation correspondent
à des raies non décelables à cause d’un fond continu
qui
les recouvre. Certains termes faibles[(A,,)2
de(W)
et de
( U)]
neparaissent
pastoujours
sur lesclichés,
le
temps
de poseayant
été sans doute insuffisant.Mais,
dansl’ensemble,
l’accord estfrappant
lorsqu’on
adopte
l’attribution suivante pour les six raiesvoisines de la raie excitatrice du
spectre
de diffusion dunaphtalène-cristal [6] :
Le doublet
46-5~
cm-1correspond
donc auxpivo-temenfs cohérents des molécules autour de la normale W au
plan
moléculaire,
le doublet étroit74-76
cm-1 auxpivotements
autour dupetit
axeV,
etenfin le
dou-blet 109-127 cm-1 auxpivotements
autour dugrand
axe U des molécules.
TENSKtRS D’INTENSITÉ.
L’ordre des
fréquences
est conforme à celui desmoments d’inertie
(calculés
ennégligeant
la masse des atomesd’hydrogène) :
On vérifie facilement que les raies
symétriques
(54,
76
et iogcm-’)
et les raiesantisymétriques
(45, 74
et 127cm-~1) n’apparaissent jamais
les uneset les autres dans un même
spectre.
Onpeut
remar-quer que les24
spectres
obtenus se ramènent àsix
types
différentsqui correspondent
aux six termesdifférents des tenseurs d’intensité :
L’examen des clichés montre la similitude des
spectres
d’un même groupe. Nous avonsreproduit
(pl. II)
les meilleursspectres
dechaque
type
obtenus avec unedispersion plus grande.
Nousajoutons
également
lesspectres
obtenus en lumière incidente naturellelorsque
l’axe binaire du cristal estparal-lèle au faisceau incident OX : cas
(9
+10)
et~ I 2}.
Le passage d’un de ces cas à l’autre
correspond
à larotation du cristal autour de l’axe binaire. Pendant
cette
opération,
lespectre
i contenant les raiesantisymétriques
resteinchangé [ i
=(A12)2-f-
(~23)~]’
tandis que le
spectre
I contenant les raiessymé-triques
se modifie[3],
L’intensité relative des raies
(le
doublet74-76
cm-1est le
plus
intense)
montre que la réfractivité moyenne de la molécule estdirigée
suivant sonpetit
axe V. L’étudequantitative
des intensités relatives desdifférentes raies
permettra
lacomparaison
des différences deréfractivité g
-rg’, g’ - g’r
et g
--gn
de la molécule denaphtalène.
C’estainsi,
parexemple,
que dans lespectre
i du cliché(11
+12)
dû auterme
(A12)2
+(A,,)2
les intensités des trois raiescorrespondent
à0,67
(g
pour1V A = 45 cm-i,
o@20
(g
-g"~2
pour VA== 74
cm-1 et o,38(g,
- g")2
54
PLANCHE
II
Reproduction
de
quelques
spectres
,3vecgrande dispersion
56
des clichés montre
déjà
que les différences(g
--g’)
et~g’ -- g")
sont du même ordre degrandeur.
L’étude
photométrique
nous a donné les résultatssuivants :
4.
Application
à la calcite. - Nous avonsrappelé
que Raman etNegundaki
ont vérifié leur théorie despivotements
sur les raies « externes »d’un cristal de nitrate de sodium. La maille renferme deux ions
plans
normaux à l’axe ternaire.Prenons cet axe du cristal à la fois pour l’axe W de l’ion
NO?
et pour l’axe 2 du trièdre attaché aucristal,
chaque
ion et le cristal dans son ensemblepossédant
aupoint
de vue des réfractivités lasymétrie
de révolution autour de cet axe(g
=g’).
Si les
pivotements
des deux molécules se font autourde
plusieurs
axes situés dans leurplan équatorial
et sont
incohérents,
on obtient le tenseur(U + V)
ci-dessous où tous les termes non nuls sont
égaux :
Si,
aucontraire,
cespivotements
sont cohérentset se
couplent
autour d’axesparallèles,
à la vibration« symétrique
»correspond
un tenseurnul,
à lavibra-Fig. 4.
tion
« antisymétrique » correspond
le tenseur( U
+V)
en entier. Ainsil’hypothèse
de la cohérence despivotements
des ionsN03 n’apporte
aucunchan-gement
dans lesprévisions théoriques :
elle n’a pasété
envisagée
par les auteurs hindous.De même les
pivotements
des ionsC°3"-
de la calcite donnent naissance à des raies Raman de faiblefréquence;
lapolarisation
de l’uned’elles,
283cm-l,
a étésoigneusement
étudiée parJ. Cabannes
[8]
et par -Mue D. Osborne[9].
Nousavons
reporté
sur lafigure 4
les résultatsthéoriques
déduits du tenseur( U ~
V)
dans les six casexpé-rimentaux
possibles
suivant les orientations rela-tives ducristal,
de la vibration incidente et du rayondiffusé. Les valeurs calculées du
f acteur
dedépola-risa1ion ?
sont en excellent accord avec les résultatsdes mesures de Mlle D. psborne.
L’hypothèse
despivotements
incohérents neconduit
qu’à
unefréquence
depivotement.
Or,
dans la
calcite,
comme dans le nitrate desodium,
on observe deux raies Raman de faible
fréquence
etde forte intensité : i56 et 983 cm-’ pour la
calcite,
g8
et 185 cm-’ pour le nitrate de sodium.D’après
iVhle D.Osborne,
la raie 156 cm-1présente
sensi-blement les mêmes caractères depolarisation
que la raie a 8 3 cm-1. On est donc amené à attribuer cettefréquence
à une vibration de la maille dont letenseur a la même forme que
( Lr
+V),
parexemple
à des
pivotements
cohérents des deux ions autourd’axes de leur
plan équatorial
nonplus
parallèles,
mais
rectangulaires.
Onpeut
aussi songer à despivotements
cohérents autour d’axes inclinés à à cause de lasymétrie
ternaire du cristal. Cettehypothèse
conduit à des tenseurs de mêmeforme,
mais a
priori
on doitprévoir
alors deuxfréquences
supplémentaires
suivant le mode decouplage
despivotements;
il neparaît
pasqu’un
tel dédoublementait été observé
jusqu’ici
sur l’une des deux fré-quences « externes » des ionsCOg
ouNOI.
5. Le
pivotement
des molécules dans lescristaux et
l’origine
de l’effet Cabannes-Dauredans les
liquides.
- Onsait que dans le
spectre
de la diffusion moléculaire de la lumière par unliquide,
la raie fondamentale est entourée d’un fond continudépolarisé
dontl’origine
a été attribuée à la rotation des molécules(effet Cabannes-Daure).
Onexplique
ainsi son état depolarisation,
la répar-tition des intensités avec la différence defréquences,
la variation de l’intensitéglobale
avecl’anisotropie
moléculaire et avec l’indice duliquide [1].
Cependant,
Gross et Vuks[7],
après
leurdécou-verte des raies Raman de faible
fréquence
de cer-tains cristauxorganiques (naphtalène,
para-dibromo-benzène,
etc.)
ontremarqué
que cescomposés
donnentà l’état
liquide
un effet Cabannes-Daureimportant :
ils en ont conclu que ces raies de faible
fréquence
caractérisent des liaisons intermoléculaires dans le réseau cristallin et que lespectre
continu desliquides
était encore dû à ces liaisons intermoléculaires
déformées
trèsfortement,
mais se conservant tout de même à l’intérieur de la structurequasi
cristalline desliquides.
Lespectre
continu desliquides
ne serait pas dû à la rotation desmolécules,
mais à uneffet
de vibration.Pour Sirkar et ses collaborateurs
[ 10]
aucontraire,
des
fréquences
d’oscillation linéaires du réseau cristallin nepeuvent
avoirqu’exceptionnellement
uneintensité
appréciable;
il faut donc attribuerces radiations à des oscillafions in termolécula ires
dans des groupes
f ortement polymérisés.
Ces groupesdisparaissent
à la fusion et l’effet Cabannes-Daureest dû en
majeure
partie
à la rotation des moléculesdans le
liquide.
Cettehypothèse
de Sirkar a pourorigine
la forte intensité des raies de faiblefréquence,
comparée
à l’intensité des raies Raman internes.D’après
notreinterprétation,
c’est aux forces d’orien-tation des molécules à l’intérieur ducristal,
forcesqui
admettent les éléments desymétrie
de lamaille,
qu’est
due la cohérence despivotements.
Il fautremplacer l’hypothèse
desgroupements
f ortement
polymérisés
par l’idée des domaines élémentairesde cohérence dont l’étendue reste encore à
pré-ciser.
Pour
Bhagavantam
et ses collaborateurs[1 1],
comme pourVenkàteswaran
[12]
les oscillationsdes molécules du cristal
persistent
dans leliquide
à structurequasi
cristalline,
mais cessent d’êtrequantifiées;
elles donnent un fond continu.Cepen-dant,
pour lier l’intensité de ce fond àl’anisotropie
moléculaire,
il faut supposer,qu’au
cours de cesmouvements, l’orientation des molécules varie : ils
envisagent
donc des oscillations des moléculesautour d’orientations moyennes
imposées
par le cristal ou, à sondéfaut,
imposées par la
structurequasi
cristalline dans leliquide.
Nos mesures montrent nettement que dans un cristal
organique,
mou, àpoint
de fusion bas commele
naphtalène,
lespivotements
cohérents desmolé-cules se font autour de leurs axes
principaux
pour donner naissance aux raies de faible
fréquence
découvertes par Gross et Vuks.Lorsque
latcmpé-rature
s’élève,
l’amplitude
de cespivotements
augmente
et à lafusion,
avec ladisparition
duréseau, ces
pivotements
se transforment en rotation.En
effet,
à lafusion,
les forces d’orientation duréseau cristallin
disparaissent
et avec elles les forcesde
couplage
despivotements.
Or,
la forte intensitéde la diffusion de
fréquence
modifiée,
due auxfluctua-tions d’orientation des molécules, et
qu’on
retrouvedans l’effet Cabannes-Daure des
liquides,
nepeut
s’expliquer
que par la rotationcomplète
des molé-cules : despivotements
incohérentsn’ajouteraient
à la diffusion
Rayleigh
nonchangée
delongueur
d’onde,
qu’un
faible fond continu[i].
L’intensité de l’effet de
pivotement
dans lescristaux comme celle de l’effet de rotation dans les
liquides
sont liées aux fluctuationsd’orientation,
donc à
l’anisotropie optique
des moléculesdiffu-santes. Ces deux effets doivent donc être tous les deux
particulièrement
intenses avec lescomposés
aromatiques
dontl’anisotropie optique
très élevée se révèleégalement
par unebiréfringence
remar-quable
de leurs cristaux. L’existence de radiations de faiblefréquence
dans lespectre
Raman des cristauxorganiques n’apporte
donc aucunargument
en faveur de la
persistance
à l’étatliquide
d’unestructure
quasi
cristalline,
contrairement aux conclu-sions de Gross etVuks,
et, à leursuite,
de certainsauteurs hindous.
Nous sommes reconnaissants à M. J.
Cabannes,
Professeur à la
Sorbonne,
de l’intérêtqu’il
nous atémoigné pendant
ces recherches. Nous remercionsnotre
collègue,
M.Brus,
Directeur de l’Institut duPin,
qui
a mis à notredisposition
unepartie
dumatériel
spectrographique
de son laboratoire.BIBLIOGRAPHIE.
[1] A. ROUSSET, Thèses, Paris, 1935; Annales de Physique,
1936, 5, p. 60.
[2] C. V. RAMAN et T. M. K. NEGUNDAKI, Nature, 1939,
143, p. 679.
[3] J. CABANNES, Comptes rendus, 1940, 211, p. 625. [4] J. M. ROBERTSON, Proc. Roy. Soc. London, A, 1933,
142, p. 674.
[5] H. BENEL, Thèses, Bordeaux, 1940, p. 7 et planche I.
[6] A. KASTLER et A. ROUSSET, Comptes rendus, 1941, 212,
p. 645.
[7] E. GROSS et M. VUKS, Nature, 1935, 135, p. 100, 431
et 998; Journ. de Phgs., 1935, 6, p. 457 et 1936, 7, p. 113. [8] J. CABANNES, Comptes rendus, 1929, 188, p. 1041; Tran-sactions of the Faraday Society, septembre 1929, p. 823.
[9] D. OSBORNE, Thèse d’Université, Montpellier, 1931.
[10] S. C. SIRKAR, Indian Journal of Physics, 1936, 10, p. 75;
S. C. SIRKAR et B. S. MOOKERJEE, ibid., p. 375; S. C. SIRKAR et J. GUPTA, ibid., p. 473, et 1937, 11,
p. 55 et 283; S. C. SIRKAR ibid., p. 343; S. C. SIRKAR et I. C. BISHUI, ibid., 1938, 11, p. 417; S. C. SIRKAR et J. GUPTA, ibid., 1938, 12, p. 35.
[11] S. BHAGAVANTAM, Proc. of the Indian Academy of Sciences, A, 1935, 2, p. 63; S. BHAGAVANTAM et T. VENKATARAYUDU, ibid., 1939, 9, p. 224.
[12] C. S. VENKATESWARAN, Proc. of the Indian Academy of Sciences, A, 1936, 4, p. 414; Current Science,