Developing a Cloud-Based Secure Computation
Platform for Genomics Research
by
Shreyan Jain
Submitted to the Department of Electrical Engineering and Computer
Science
in partial fulfillment of the requirements for the degree of
Master of Engineering in Electrical Engineering and Computer Science
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
May 2020
c
○ Massachusetts Institute of Technology 2020. All rights reserved.
Author . . . .
Department of Electrical Engineering and Computer Science
May 15, 2020
Certified by . . . .
Bonnie Berger
Simons Professor of Mathematics and Professor of EECS
Thesis Supervisor
Certified by . . . .
Hyunghoon Cho
Schmidt Fellow, Broad Institute
Thesis Supervisor
Accepted by . . . .
Katrina LaCurts
Chair, Master of Engineering Thesis Committee
Developing a Cloud-Based Secure Computation Platform for
Genomics Research
by
Shreyan Jain
Submitted to the Department of Electrical Engineering and Computer Science on May 15, 2020, in partial fulfillment of the
requirements for the degree of
Master of Engineering in Electrical Engineering and Computer Science
Abstract
Recent advances in genomic sequencing technologies and big data analytics present a golden opportunity for bioinformatics, making it possible to efficiently analyze hun-dreds of thousands of individual genomes and identify statistically significant genetic determinants of disease. However, most research institutes lack sufficient genomic data to detect fine-grained signals crucial for understanding complex human diseases, and data sharing is often impractical due to strict privacy protections. By lever-aging a cryptographic technique known as secure multiparty computation (MPC), researchers can securely cooperate on large-scale genomic studies without revealing sensitive data to any collaborators.
In this thesis, we propose a public cloud-based computational framework that implements MPC for an essential genomic analysis workflow known as genome-wide association study (GWAS). By productionizing secure GWAS tools in an easy-to-use interface that abstracts away the technical challenges involved with implementing and running a protocol on several independent, geographically separated machines, we hope to enable researchers around the world to launch meaningful genomic studies with minimal overhead. We hope such efforts will prove instrumental towards the broader aim of establishing a single general-purpose platform for secure genomics research.
Thesis Supervisor: Bonnie Berger
Title: Simons Professor of Mathematics and Professor of EECS Thesis Supervisor: Hyunghoon Cho
Acknowledgments
First and foremost, I would like to thank my advisors Bonnie Berger and Hoon Cho for their invaluable support and supervision throughout every step of this project. Without their guidance, this thesis would not have been possible.
Second, I would like to thank David Bernick at the Broad Institute for advising me about system design best practices. Thanks also to the other members of the Berger Lab for their support throughout the year, and a special thank you to Patrice Macaluso for helping me with all the administrative aspects of my project.
Lastly, I would like to thank my parents and my sister for their never-ending support throughout my four years at MIT.
Contents
1 Introduction 13
1.1 Genome Wide Association Studies . . . 14
1.2 Genomic Privacy Protections . . . 15
1.3 Our Approach . . . 16
1.4 Overview . . . 18
2 Background 19 2.1 Secure Multi-Party Computation . . . 19
2.2 MPC for Genomics Research . . . 21
2.3 Secure GWAS Codebase . . . 23
2.4 Existing Public Cloud Approaches . . . 26
3 Implementing a More Scalable Secure GWAS Pipeline 29 3.1 Overview of Codebase . . . 29
3.1.1 MPC Library . . . 30
3.1.2 Client Programs . . . 32
3.1.3 Usage . . . 34
3.2 Supporting Multiple Data-Sharing Parties . . . 36
3.2.1 Parameter File Changes . . . 38
3.2.2 Updated Client Programs . . . 39
3.3 Synchronized Multi-Threading to Improve Scalability . . . 40
3.3.1 Support for Parallel Inter-Party Communication . . . 42
3.3.3 Chunked Data Streaming and Analysis . . . 46
4 Designing a Public Cloud Platform for GWAS 49 4.1 Design Goals . . . 49 4.2 Architecture . . . 50 4.2.1 Software Specifications . . . 51 4.2.2 Endpoints . . . 52 4.3 Workflow . . . 52 5 Experimental Results 61 5.1 Verifying Accuracy of Updated GWAS Pipeline . . . 61
5.2 Synchronized Multi-Threading Enables Huge MPC Speedups . . . 64
5.3 Final GWAS Pipeline Achieves Practical Runtimes . . . 68
5.3.1 Experiments on Toy Dataset . . . 69
5.3.2 Experiments on Real World Dataset . . . 76
6 Conclusion 81 6.1 Future Work . . . 82
6.1.1 A Production-Level Cloud Platform Design . . . 82
6.1.2 Leveraging Cloud Services to Improve Performance . . . 85
List of Figures
2-1 Secure Computation of 𝑓 (𝑥, 𝑦) = 𝑥𝑦 . . . 20
2-2 Network Setup for Secure GWAS Protocol . . . 24
3-1 Modified Network Setup for Updated Protocol . . . 37
4-1 Public Cloud GWAS Platform Design . . . 51
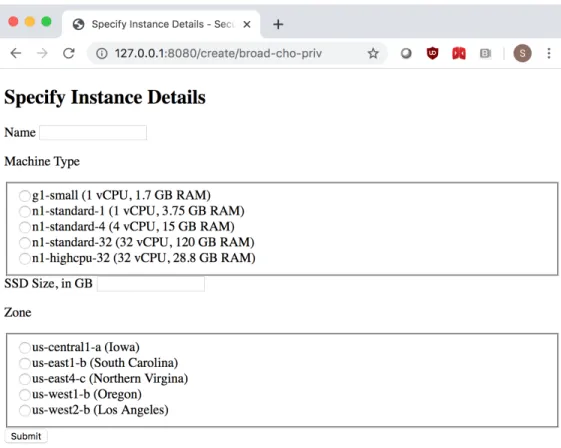
4-2 Selecting a Compute Instance . . . 54
4-3 Configuring a New GWAS-enabled Instance . . . 55
4-4 Uploading Secret GWAS Dataset . . . 56
4-5 Uploading Shared Config File . . . 57
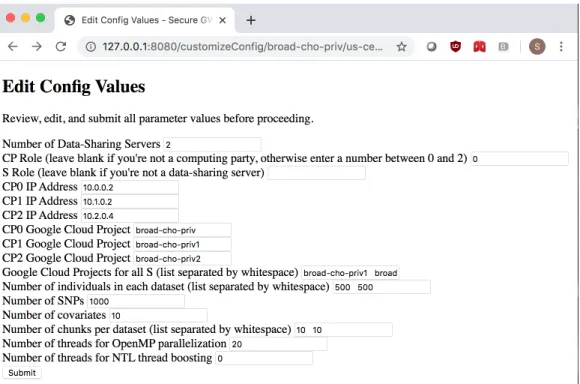
4-6 Editing GWAS Parameter Values . . . 58
4-7 Initiating Secure GWAS Protocol . . . 59
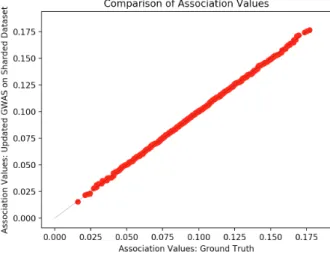
5-1 Verifying GWAS Correctness on Multiple Datasets . . . 63
5-2 Verifying GWAS Correctness using Multiple Threads . . . 63
5-3 Verifying GWAS Correctness on Lung Dataset . . . 78
List of Tables
5.1 Benchmarking Results for Parallel MPC Routines . . . 65
5.2 Comparison of Parallelization Strategies for MPC Routines . . . 68
5.3 Evaluating Different High-Level Optimizations . . . 70
5.4 Effect of Parallelization on GWAS Runtime . . . 72
5.5 Effect of Dataset Size on GWAS Runtime . . . 73
5.6 Effect of Cloud Network Configuration on GWAS Runtime . . . 75
Chapter 1
Introduction
Ever since the start of the Human Genome Project in 1990, modern genomics research has revolutionized the way we understand, diagnose, and treat human disease. At every step in the road, further progress in genomics research has been unlocked by leveraging current technological advancements to improve our ability to sequence, map, and analyze human genomes at scale. Improvements in sequencing technologies alone, for example, have made it possible to sequence one person’s entire genome in a few days for a cost of $1000, as compared to the ten years and $1 billion it took for the first such sequencing effort [18]. As a result, researchers now have access to repositories containing hundreds of thousands of individual sequenced genomes, a treasure trove of data that can be mined to uncover countless new gene-disease correlations [25]. These discoveries can, in turn, be used to develop effective genetic screening and diagnosis tools and identify opportunities for novel therapies.
In this thesis, we aim to further expand genomics research capabilities by taking advantage of two cutting-edge technological trends. In particular, we utilize secure computation to alleviate concerns over data sharing, and we leverage cloud computing to improve the scalability of cryptographic techniques. Through the combination of these two approaches, we provide researchers with a platform that allows them to conduct larger-scale genomic studies than were previously possible in a practically efficient manner. We hope such efforts will help address the obstacles researchers face today in their attempts to understand the genetic basis of disease.
1.1
Genome Wide Association Studies
The earliest genetic approaches to pathology focused on rare disorders caused by mutations within a single gene [3]. In the case of such diseases, which include cystic fibrosis and Huntington’s disease, variants of the gene of interest have a significant effect on phenotype, manifesting in simple patterns of inheritance that can be easily studied within families exhibiting the trait. Techniques such as linkage analysis -which analyzes co-inheritance patterns of the disease and multiple genetic markers within affected families - have proven incredibly effective at localizing the specific genes responsible for many such rare diseases [21].
By contrast, these same approaches have yielded far less promising results when applied to more common diseases such as heart disease and cancer, whose genetic mechanisms are significantly more complex than that of rare diseases [3]. In particu-lar, complex diseases are caused by many different genetic variants that are relatively common across the larger population but have much smaller effect sizes [22]. As a result, effectively tracing the genetic origin of complex diseases requires an approach known as the genome-wide association study (GWAS), in which genetic markers are analyzed at the level of the entire population rather than individual families.
In a GWAS study, millions of small genetic variations known as single-nucleotide polymorphisms (SNPs) must be analyzed across a large pool of individual genomes to identify specific SNPs with a statistically significant correlation with the phenotype (disease) of interest [26]. Although the statistical methodology underpinning GWAS techniques is well-known and empirically effective [17], the results of GWAS studies are often unreliable due to a variety of practical computational considerations. First, real-world GWAS studies often use sample sizes that are too small to draw statistically significant conclusions about genetic variants with small effect sizes. In their meta-analysis of 752 GWAS studies, Ioannidis et al. find that, in the median case, a given study would require a 13-fold increase in the number of individual subjects in order to identify meaningful gene-disease associations [13]. Second, the presence of biases such as population heterogeneity and environmental factors further increase the required
sample sizes [19]. In particular, many existing datasets suffer from highly limited genetic diversity due to bias towards European ancestry in study populations [12]. This limits the applicability of specific GWAS findings to biomedical research settings across the world unless datasets with better coverage of the broader population are incorporated into the analysis.
1.2
Genomic Privacy Protections
In order for researchers to reliably and accurately answer questions about the most complex diseases, modern GWAS studies require datasets ranging from 500,000 to several million individual genomes [13]. Since the vast majority of research institu-tions and biobanks cannot provide the sheer amount of data required for analyzing complex diseases by themselves, there is a growing consensus that collaborative stud-ies are critical for modern genomics research. By pooling together their datasets, researchers from distinct labs and institutions can generate more meaningful insights than they otherwise could alone.
Unfortunately, data sharing in the genomic space is easier said than done, since such datasets contain highly sensitive information. Violations of genomic privacy are especially concerning because they leave individuals vulnerable to discrimination by employers or insurance companies on the basis of their genetic profile, which may reveal risk factors for certain inherited disorders [24]. Additionally, typical approaches for secure data sharing such as de-identification have been proven to be insufficient in the genomic setting. In one instance, researchers found that querying publicly accessible genetic genealogy databases alongside de-identified genomic sequence data could reveal the surnames of subjects; this information, along with metadata like age and location, could then be used to accurately re-identify the subject [10]. Similar studies have demonstrated successful re-identification attacks on beacons that answer queries about allele frequency within a database [23], electronic health records [8], and genomic databases containing familial records (such as pedigrees) [16].
research institutions are often subject to strict standards of protection (such as the NIH’s HIPAA guidelines) or access control policies that limit or explicitly prevent data-sharing [25]. Therefore, even in instances when two or more labs may wish to collaborate on a GWAS study, regulatory guidelines often prevent them from doing so.
1.3
Our Approach
In this thesis, we present a new approach for genomics research that realizes the bene-fits of collaborative GWAS efforts while maintaining compatibility with international standards on genomic data privacy. We accomplish this by building a secure, cus-tomizable, high-performance cloud platform that allows independent, geographically separated entities to analyze crowdsourced genomic datasets at scale, while keeping each participating entity’s data hidden from all other collaborators. By doing so, we address three major gaps within the existing infrastructure for launching collaborative genomic studies.
First, we implement collaborative GWAS in a manner that fully respects the privacy constraints of all participating institutions by leveraging a technique known as secure computation. Secure multi-party computation (MPC) is a cryptographic framework that allows distinct entities to jointly compute functions on a set of inputs while keeping each entity’s individual input hidden from all others [7]. MPC effec-tively allows parties to compute on encrypted data, which offers an opportunity for achieving the dual goals of collaboration and privacy. By using the MPC framework to build out a secure GWAS protocol, we give researchers the ability to secret-share their individual datasets with each other and accurately analyze the joined input without revealing the raw data at any point in the process. While prior research has demonstrated practical applicability of MPC techniques to genomics research [4], we expand on such research by building, to our knowledge, the first ever fully collaborative, end-to-end GWAS pipeline based on MPC techniques.
collab-orative studies by implementing our secure GWAS pipeline in the public cloud. To launch a collaborative GWAS study from the ground up, researchers must agree on protocols for communicating between and orchestrating computation across multiple different compute environments that may be legally and geographically separated. This presents an incredibly challenging distributed systems problem that researchers may not have the time, budget, or technical know-how to solve. Some researchers may not even have access to the computational resources needed for real-world GWAS studies in the first place. We address these practical issues by building a single stream-lined cloud platform that abstracts away the technical details involved in setting up a GWAS study. We expose our platform as a RESTful web service that turns complex cloud operations - such as launching a GWAS compute instance, securely uploading and preprocessing a dataset, establishing networked connections with compute in-stances belonging to partner institutions, and asynchronously initiating the GWAS protocol - into simple, one-click actions for users. This dramatically simplifies the time and computational literacy needed for genomics researchers to participate in joint studies.
Finally, we overcome computational hurdles to our goal of a successful real-world GWAS protocol by focusing on scalability. Since large-scale GWAS studies must in-clude data from millions of individual genomes and hundreds of thousands of candi-date SNPs to identify statistically significant correlations between specific genetic vari-ants and disease, the scale of the necessary input size may easily lead to prohibitively high computation runtimes (and, therefore, monetary costs). Our MPC-based GWAS protocol is particularly vulnerable to this issue, since cryptographic operations are intrinsically more computationally expensive than corresponding plaintext ones. We address these computational concerns by providing a parallel implementation of our GWAS protocol that greatly improves on the performance of the basic, single-threaded one. Through extensive profiling, we demonstrate that our final secure GWAS pro-tocol is practically feasible on real-world inputs, even on the large, pooled datasets needed to accurately analyze the most complex phenotypes.
1.4
Overview
The remainder of this thesis is organized as follows.
In Chapter 2, we present a detailed overview of secure computation, along with a survey of related work in the field of secure genomic analysis.
In Chapter 3, we describe our efforts towards building a secure GWAS pipeline that is customizable and scalable enough to handle genomic studies in complex, real-world research settings.
In Chapter 4, we provide a detailed design of our cloud platform and describe the end-to-end workflow of setting up and initiating a secure GWAS study.
In Chapter 5, we provide profiling results that show how the performance of our final GWAS platform scales with a variety of factors, such as the input dataset size and the network configuration between participants.
In Chapter 6, we wrap up our analysis with some concluding remarks, includ-ing discussion of ongoinclud-ing efforts to improve the overall functionality of our GWAS platform.
Chapter 2
Background
In this chapter, we provide a survey of prior research that forms the foundation of our work. First, we give a more detailed treatment of multi-party computation and its previous applications to genomics research. Next, we take a look at an existing codebase that provides an MPC-based implementation of secure GWAS and the lim-itations within it that motivate our work. Finally, we discuss other approaches to building a genomics research platform in the public cloud.
2.1
Secure Multi-Party Computation
The core cryptographic technique at the heart of most popular secure genomic data analysis pipelines is secure multi-party computation (MPC) [7]. To illustrate the workings of MPC, let us consider a simplified setting: we have two entities named 𝑆1
and 𝑆2, each of which owns a (secret) dataset, represented here as 2 numbers: 𝑥 and 𝑦.
We also have 3 computing parties named 𝐶𝑃0, 𝐶𝑃1, and 𝐶𝑃2. None of the computing
parties are allowed to learn the values of 𝑥 or 𝑦; similarly 𝑆1 cannot learn the value of
𝑦, nor can 𝑆2 learn the value of 𝑥. However, the three computing parties must work
together to compute some function of the secret data. For sake of demonstration, let’s assume a simple yet instructive function of interest: 𝑓 (𝑥, 𝑦) = 𝑥𝑦.
The first stage of the pipeline is secret sharing. In this step, the data-owning entities decompose the secret dataset into shares such that the original data cannot be
recovered from any one share alone. In particular, 𝑆1 decomposes 𝑥 into the two secret
shares [𝑥]1 and [𝑥]2 by choosing a random number 𝑟 and setting [𝑥]1 = 𝑟, [𝑥]2 = 𝑥 − 𝑟.
𝑆1 can then send the share [𝑥]1 to 𝐶𝑃1 and [𝑥]2 to 𝐶𝑃2; since 𝐶𝑃2 doesn’t know the
value of 𝑟, and since neither party receives both shares, neither 𝐶𝑃1 nor 𝐶𝑃2 can
reconstruct the secret data 𝑥. 𝑆2 decomposes 𝑦 in the same way, sending [𝑦]1 to 𝐶𝑃1
and [𝑦]2 to 𝐶𝑃2.
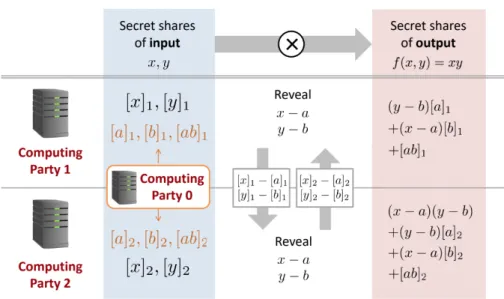
Figure 2-1: Secure Computation of 𝑥𝑦 from secret shares of 𝑥 and 𝑦. Figure taken from [5].
The next stage, or evaluation, is illustrated in Figure 2-1. First, 𝐶𝑃0 generates
two random numbers 𝑎 and 𝑏, then secret-shares them as well as their product 𝑎𝑏 with 𝐶𝑃1 and 𝐶𝑃2. Next, 𝐶𝑃1 computes the value [𝑥]1 − [𝑎]1 and sends it to 𝐶𝑃2.
Note that now, 𝐶𝑃2 can compute the value 𝑥 − 𝑎 as ([𝑥1] − [𝑎]1) + [𝑥]2 − [𝑎]2 since
𝑥 = [𝑥]1+ [𝑥]2 and 𝑎 = [𝑎]1+ [𝑎]2. However, 𝐶𝑃2 still hasn’t learned the values of 𝑥
or 𝑎; in other words, this computation has not exposed the value of the raw input. Through a similar process, both 𝐶𝑃1 and 𝐶𝑃2 can calculate the values 𝑥 − 𝑎 and
𝑦 − 𝑏. Finally, 𝐶𝑃1 computes the final value (𝑦 − 𝑏)[𝑎]1+ (𝑥 − 𝑎)[𝑏]1− [𝑎𝑏]1, while 𝐶𝑃2
computes the final value (𝑥 − 𝑎)(𝑦 − 𝑏) + (𝑦 − 𝑏)[𝑎]2+ (𝑥 − 𝑎)[𝑏]2− [𝑎𝑏]2. Since these two
share of 𝑥𝑦 to be fed into the next downstream MPC subroutine, or they can publish their calculated values allowing both parties to reconstruct the final output. In some cases, the latter approach might be insecure, since a party (such as 𝑆1) could infer the
value of 𝑦 from 𝑥 and 𝑥𝑦 or vice versa; however, the real functions of interest in typical MPC routines are far more sophisticated and therefore avoid such vulnerabilities.
In the above process, the secretly shared triple of (correlated) random numbers (𝑎, 𝑏, 𝑎𝑏) is known as a Beaver multiplication triple. Although the process above only demonstrates how to implement secure multiplication, this specific subroutine can be composed with other similar protocols to perform more complex computations, ulti-mately resulting in a broader framework for evaluating arbitrary arithmetic circuits [4]. Note, however, that a naive approach that requires a new Beaver triple to be generated for every arithmetic operation will scale very poorly on large datasets, due to the performance bottleneck incurred in the communication channel between 𝐶𝑃0
and the other computing parties.
2.2
MPC for Genomics Research
Multiple researchers have documented successful efforts at applying the MPC frame-work to the GWAS task in an effort to create secure pipelines for analyzing genomic data. However, most existing approaches suffer from one of two problems: either they greatly oversimplify the GWAS task thereby reducing their own usefulness, or they suffer from prohibitive computational slowdowns. For instance, Constable et al. use a garbled-circuit approach to implementing GWAS in an MPC framework, but their implementation is based on an MPC engine that assumes there are only 2 participants [6]. As a result, their secure analysis pipeline cannot be used in studies involving more than 2 participating labs, institutions, hospitals, or biobanks. Jagadeesh et al. also use a similar garbled-circuit approach more recently but for a slightly different use case than pure GWAS, placing a greater emphasis on finding rare mutations. As a result, they remark that their implementation cannot practically handle computa-tions involving tens of thousands of individuals without hardware advances, the scale
necessary for any meaningful GWAS workflow [14].
On the other hand, Bonte et al. use a different MPC implementation to achieve extremely good performance, completing analysis of 1 million subjects in just 2 ms [2]. However, their methodology greatly simplifies the GWAS task, outputting a single yes-no answer for each queried SNP that represents whether or not the SNP is a possible marker; as they note in their work, they do not reveal the actual sig-nificance value of the correlation, nor do they calculate the statistics necessary to draw causal inferences from the data. Perhaps most importantly, Bonte et al. also omit quality control data filters and population structure correction from their secure GWAS workflow. Since identifying and correcting for population biases ensures that interpopulation differences (i.e., differences in allele frequency distributions between two or more racial subpopulations) are not incorrectly identified as false associations with disease, this omission limits the applicability of their approach to actual GWAS studies.
Cho et al. recently published an MPC implementation of GWAS that achieves good performance (namely, a practical runtime of 3 days to analyze 20,000 individuals’ genomes) without sacrificing any complexity in the GWAS problem formulation [4]. They generalize the Beaver multiplication triples approach outlined above into a framework for efficiently evaluating arbitrary polynomial functions while maximizing data reuse, reducing the total network cost associated with repeatedly communicating new Beaver triples. This allows them to implement efficient subroutines for matrix multiplication, exponentiation, and iterative algorithms that are used extensively in a typical GWAS workflow. By leveraging this technique alongside a tool known as random projection, Cho et al. also implement an extremely efficient routine for PCA, allowing them to support population structure correction without adversely affecting runtime performance. The resulting MPC implementation is able to achieve a total inter-CP network communication complexity that scales linearly with the number of individuals and genetic variants, compared to the quadratic cost of naively implementing GWAS in the MPC framework outlined above. Finally, in theory, their implementation can be readily extended to support an arbitrary number of
data-contributing study participants while maintaining robust security guarantees.
2.3
Secure GWAS Codebase
Along with the description of their secure GWAS protocol, Cho et al. also pro-vide a proof-of-concept, open-source C++ implementation of the protocol (which can be viewed at this Github repository: https://github.com/hhcho/secure-gwas). This codebase provides client software for any user to run a secure GWAS study using their own compute environment and datasets. Their specific implementation sup-ports GWAS studies that consist of four different participants:
∙ CP0 - A computing party who plays a complementary role in the protocol by precomputing and sharing necessary random values (such as Beaver multiplica-tion triples) used by CP1 and CP2.
∙ CP1 and CP2 - The two computing parties who receive secretly shared genomic data and carry out the actual analysis of it. At the end of the protocol, the final output (the calculated SNP-phenotype association statistics) is calculated and saved by CP2.
∙ SP - The data-contributing study participant who actually owns the private genomic data. The protocol guarantees that no CP ever sees the data that SP provides for analysis in plaintext form. Note that SP does not actually participate in the genomic analysis itself.
Compiling the secure GWAS codebase produces 3 different C++ executables: the GenerateKey program, which is used by GWAS participants to generate and save random 128-bit keys for encrypted AES communication; and the two client programs DataSharingClient and GwasClient, which implement protocols for secret-sharing genomic data and carrying out GWAS analysis, respectively. To run a secure GWAS study, each participating entity must join the protocol by running an instance of the corresponding client program on their own machine. Once all participants have
initiated the protocol in this way, the programs begin working jointly to carry out the protocol to completion.
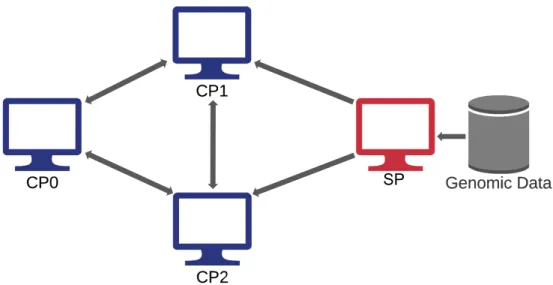
However, in order for these client programs to successfully orchestrate compu-tation, the GWAS protocol assumes the existence of a preconfigured network setup between the client machines for each party, as shown in Figure 2-2. In other words, all users of the secure GWAS software are responsible for creating the necessary networked connections that allow participants connected in the below graph to com-municate with each other.
Figure 2-2: Required network setup for secure GWAS protocol. Arrows represent secure communication channels.
This reveals the first major limitation of Cho et al.’s secure GWAS codebase: it is implemented as an entirely client-side package (i.e. all code runs in the user’s own environment). This means that for any researcher to run their program, they must first set up their own machine with adequate compute and storage resources, install all necessary packages, and create a network over which they can successfully connect with the machines of other collaborating parties. In the best case, this overhead poses a logistical or technical challenge for researchers; in the worst case, it actively prevents them from using the secure GWAS implementation at all. For instance, government entities like the NIH have strict regulations for network traffic that would prevent them from using their own computing resources for collaborative studies, like this
secure GWAS codebase requires.
Additionally, although the theoretical MPC framework that Cho et al. describe in their paper supports an arbitrary number of datasets sourced by an arbitrary number of entities, their actual implementation only provides support for analysis of a single dataset, provided by a single entity playing the role of SP, per run. Therefore, the existing secure GWAS codebase cannot be used for GWAS studies where more than one lab, biobank, or institution wishes to contribute data, making it ill-suited in its current form for the type of large, collaborative studies we describe in section 1.2.
Finally, one more major limitation of this secure GWAS implementation is that it presents users with a well-defined, relatively low upper bound on computational performance. Running the protocol on their own machines means that users are constrained by the compute and storage resources they are able to obtain and maintain themselves. Even when users have access to sophisticated computational resources, Cho et al.’s secure GWAS implementation suffers from a number of performance bottlenecks due to extremely limited use of multi-threading (particularly for expensive MPC operations), making secure GWAS studies impractical in many cases. And although the codebase is open-source, researchers must read through and understand thousands of lines of code if they wish to improve performance by, for instance, writing their own custom multi-threaded routines for core functionalities.
Our solution is twofold. First, we implement an improved and more customizable secure GWAS pipeline by forking and building directly on top of Cho et al.’s exist-ing secure GWAS codebase. By utilizexist-ing the existexist-ing codebase for our own GWAS pipeline, we are able to automatically support secure GWAS functionalities out-of-the-box without having to write our own MPC or GWAS routines. At the same time, we also add valuable functionalities to the codebase, including support for multiple in-put datasets as well as extensive parallelization of the entire data sharing and GWAS programs that significantly improve their runtime performance. We expose our cus-tom optimizations to users as easily tunable parameters, giving them fine-grained control over the end-to-end performance of the entire pipeline.
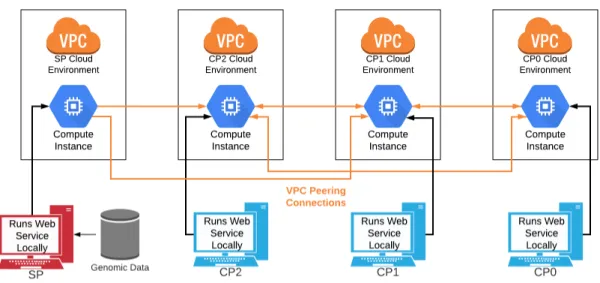
com-pute and storage services for their GWAS studies. By deploying our updated secure GWAS implementation on a public cloud service and adding additional features (such as the ability to configure study parameters), our project creates a platform that ab-stracts away all the engineering and infrastructure considerations into an easy-to-use interface, enabling researchers to launch a GWAS study with just a few clicks. This has the added benefit of creating a single, centralized pipeline for launching secure GWAS studies, eliminating the need for researchers in distinct labs to repeat the work of implementing the MPC protocol and setting up the necessary cloud com-puting infrastructure themselves. Finally, a customizable public cloud interface also allows users to configure compute instances with user-defined specs such as number of CPUs/cores, amount of RAM, and type and amount of persistent storage. These tunable specifications give further flexibility to researchers in controlling the cost and runtime performance of their GWAS compute environment without being constrained by their on-premise compute capabilities.
2.4
Existing Public Cloud Approaches
While no one has, to our knowledge, initiated a similar task of deploying a secure MPC-based GWAS framework on the public cloud to date, we note that similar ef-forts at providing cloud-based toolkits for genomic analysis do exist. For instance, the Neale lab maintains the open-source data analysis library Hail, which supports common genomic workflows such as GWAS in the public cloud [9]. Additionally, the NIH’s National Human Genome Research Institute has recently pushed an ambitious effort to improve genomic data access, sharing, and analysis capabilities using a com-putational resource called AnVIL, which is built on the Broad Institute’s Terra cloud platform [20]. However, as of yet, both Hail and AnVIL do not include any modules for secure data analysis, nor do either of them support the cryptographic primitives used by our implementation of MPC-based GWAS. Therefore, building our cloud platform for secure GWAS on top of these platforms would require substantial en-gineering work, including re-implementing the existing cryptographic protocols. For
this reason, we consider Hail and AnVIL in their current forms as unsuited towards the task of deploying secure GWAS on the cloud and have worked to build an in-dependent cloud platform to achieve this goal, one that nevertheless might be easily integrated into these broader efforts in the future.
Chapter 3
Implementing a More Scalable Secure
GWAS Pipeline
In this chapter, we describe our efforts towards building a more flexible and scalable pipeline for users to conduct MPC-based secure GWAS studies. First, we give a detailed description of the structure and usage of the existing secure GWAS code-base that our own implementation is forked from. Next, we discuss how we added support for multiple datasets and data-sourcing participants to this GWAS pipeline. Finally, we describe how we improved the overall computational performance of the pipeline by multi-threading the most expensive steps of the protocol. Our final im-plementation of the secure GWAS pipeline is open-source and can be viewed here: https://github.com/shreyanj/secure-gwas.
3.1
Overview of Codebase
As mentioned in section 2.3, Cho et al.’s secure GWAS pipeline requires four par-ticipants to each independently run the protocol code on their own machine. The protocol identifies the role of each these four participants through a unique party ID that users pass in as an argument to the client programs: 0 for CP0, 1 for CP1, 2 for CP2, and 3 for SP. This argument is used to specify the exact behavior of the executable.
3.1.1
MPC Library
In addition to the client programs themselves that provide an entryway for users to initiate the GWAS protocol, the secure GWAS codebase contains an extensive collection of MPC-based implementations of common mathematical operations and subroutines which are used by the protocol to accurately compute on secret-shared data. These MPC library methods are packaged together in a single C++ class called MPCEnv. Each time a user runs a client program, the program first initializes a MPCEnv object which will be used to perform computations on secret shares for its corresponding party (which we henceforth refer to as the caller ). Upon initialization, a newly created MPCEnv object immediately calls the following two functions:
∙ SetupChannels: This function establishes a separate encrypted communication channel with each party connected to the caller. The channel itself is imple-mented as web socket, and a distinct AES key is used for encrypting messages on each channel. The code uses the following convention for passing messages between 2 parties: the party with lower ID listens on one of its own ports, while the other party connects to that port.
∙ SetupPRGs: This function sets up different pseudo-random number generators (PRGs) to be used for sampling random numerical values throughout the pro-tocol. In addition to initializing a private PRG for the caller, this function also generates one global PRG (using a global AES key file that should be shared by all users prior to running the client program) and one shared PRG for each communicating party connected to the caller. To ensure that both members of a connected pair generate the "same" shared PRG (i.e., a PRG that will pro-duce an identical stream of random values), the party with lower ID generates a random key used to create the PRG and then sends this key to the other party through their shared communication channel. The additional PRGs are used by the protocol so that different parties can generate consistent random values without having to communicate these values across the network, thereby reducing the overall necessary amount of network communication. This can
lead to significant performance improvements when the machines for each party are located geographically far from each other [4].
Each MPCEnv object also contains the following data structures: ∙ pid: The caller’s party ID.
∙ buf: A data buffer (implemented as a char*) used for reading/writing data from the filesystem, as well as for sending/receiving messages to and from connected parties.
∙ sockets: A C++ std::map where each key is a party ID, and each value is the web socket used to communicate with that party.
∙ prg: A C++ std::map where each key is a party ID, and each value is the corresponding private or shared PRG. The special key -1 is used for the global PRG.
∙ cur_prg_pid: An int used to keep track of the key corresponding to the PRG being currently used at any moment.
The MPCEnv class leverages the C++ package called NTL (Number Theory Li-brary) to efficiently store and represent secret-shared genomic data. In particular, the secure GWAS framework designed by Cho et al. utilizes a cryptographic scheme known as additive secret sharing [4]. In this scheme, each piece of data used by the protocol is represented by some value in the finite field Z𝑝, which consists of the
in-tegers mod 𝑝 for some large prime number 𝑝. To secret-share some value 𝑥 ∈ Z𝑝, SP
samples a element 𝑟 uniformly at random from the field and splits the value 𝑥 into the two shares 𝑟 (called the mask ) and 𝑥 − 𝑟 (called the masked value), sending these shares to the computing parties CP1 and CP2, respectively. The MPC library in the secure GWAS codebase uses NTL classes such as ZZ_p to store and operate on the finite field representations of secretly shared genomic data. All collaborating client programs use the same base prime 𝑝 throughout the protocol, choosing a sufficiently
large prime that allows for high-precision storage of fixed point values as finite field elements.
Each MPC subroutine in the MPCEnv class is implemented as a single C++ func-tion that implicitly takes the caller’s party ID as an argument. In order to execute some mathematical operation on secret-shared data, all of the involved computing parties call the corresponding MPCEnv function, passing in their own secret share as the argument. For operations requiring orchestration, the code within the MPC function body sends or receives the necessary data using the communication channels, with the computation blocking until both parties have entered into the same function call. Therefore, in order to successfully execute a long, complex MPC protocol such as GWAS, the client program for each party must call the same sequence of MPC functions in the same exact order. The secure GWAS codebase ensures this by pro-viding a single executable to be used by all parties and minimizing the amount of party-specific behavior.
Finally, we note that Cho et al.’s MPC implementation does not use any explicit parallelization to speed up computationally expensive MPC operations, as has been shown to be feasible in the literature [1] [15]. Instead, the existing secure GWAS codebase relies on a feature of NTL called thread boosting to improve performance. This feature spawns a certain number of threads for NTL operations at the beginning of the program, allowing the NTL environment to automatically determine at runtime when to use multi-threading to speed up expensive NTL vector and matrix operations.
3.1.2
Client Programs
In order for SP to begin secret-sharing its genomic dataset with CP1 and CP2, SP must first save the genomic data (in plaintext) on the machine it will use to run the DataSharingClient program. For a dataset consisting of 𝑛 individuals and 𝑚 candidate SNPs, SP must save the genomic data using the following format:
∙ geno.txt: A 𝑛-by-𝑚 matrix containing the minor allele dosage for each SNP for every individual (0 for homozygous dominant, 1 for heterozygous, 2 for
homozygous recessive, and -1 for missing data).
∙ pheno.txt: A 𝑛-by-1 binary vector containing the phenotype for every individ-ual.
∙ cov.txt: A 𝑛-by-𝑘 matrix containing covariate values for every individual, where 𝑘 is the number of covariates. The protocol assumes every covariate is binary. ∙ pos.txt: A file containing the genomic position of all 𝑚 SNPs, one per line, in
the same order as that of the columns of geno.txt. Additionally, before running the DataSharingClient, SP should share this file with all of the CPs.
All of the above files must be saved in the same directory; this directory is provided to the DataSharingClient program run by the SP as a command line argument.
Once all parties have begun running the DataSharingClient, SP’s machine be-gins reading each of the above data files, splitting each value into secret shares by generating random masks, and streaming the masked values to CP2. To reduce net-work communication, SP uses the PRG it shares with CP1 to generate the random masks; this allows CP1 to simply re-generate the same random masks using the same shared PRG and thereby compute the exact shares it would have received from SP.
Upon receiving all streamed data, the computing parties then use a Beaver pre-computation trick to speed up future MPC operations. In particular, for each value 𝑥 in the genotype matrix, CP1 and CP2 both turn the secret shares [𝑥]1 = 𝑟 and
[𝑥]2 = 𝑥 − 𝑟 that they receive, respectively, from SP into the masked data value 𝑥 − 𝑎,
where 𝑎 is a random number generated by CP0. To accomplish this while minimizing network communication, CP0 and CP1 generate the same random value 𝑎1 from their
shared PRG, and similarly CP0 and CP2 generate the same random value 𝑎2 from
their shared PRG. Then CP0 sets the random value 𝑎 as simply 𝑎1 + 𝑎2. Now CP1
can calculate [𝑥]1− 𝑎1 and send this value to CP2, who can then calculate the desired
value 𝑥 − 𝑎 as [𝑥]2 − 𝑎2 + [𝑥]1 − 𝑎1 = (𝑥 − 𝑟 + 𝑟) − (𝑎1 + 𝑎2) = 𝑥 − 𝑎; through a
similar process, CP1 too can calculate the same value. This precomputation step will allow CP1 and CP2 to perform complex operations on the genotype matrix during the
GWAS stage without having to generate new Beaver multiplication triples each time, providing a significant performance boost. Note that this step keeps each genomic value 𝑥 hidden from all CPs, since CP1 never receives 𝑎2 and CP2 never receives 𝑎1;
this prevents both of them from reconstructing 𝑎 and therefore from reconstructing 𝑥.
After this Beaver precomputation step, CP1 and CP2 save the secret-shared phe-notype and covariate matrices along with the masked gephe-notype matrix to their local filesystem. Additionally, all three CPs export the CP0-CP1 and the CP0-CP2 shared PRGs to their local filesystem as well; this allows CP0, CP1, and CP2 to reconstruct the masks 𝑎, 𝑎1, and 𝑎2, respectively, during the GWAS stage of the protocol.
Once the DataSharingClient has been successfully run by all parties to comple-tion, CPs 0, 1, and 2 can then run the GwasClient program to actually carry out a secure GWAS study on the secret-shared data. The GWAS protocol code reads in the data files from the local filesystem, filters out individuals and SNPs with too much missing data, performs population structure correction, and calculates association values for each SNP and the target phenotype. Once each party has successfully run the GWAS client program to completion, the resulting association values are saved in plaintext form on CP2’s machine. CP2 can then share these values with all other parties.
Similar to the MPC routines, the protocol code within these client programs is also not explicitly multi-threaded, relying on automatic NTL thread boosting instead.
3.1.3
Usage
After compiling the secure GWAS codebase, users can run the client programs directly from the command line of their machine. In order to do this, they must pass in two required command line arguments: the party ID and the location of a parameter file containing information on all the necessary settings for the entire protocol. Each line of the parameter file contains the setting for a single parameter, formatted as a key-value pair separated by whitespace. The client program begins by reading the parameter file from the local filesystem and initializing the corresponding values in a
container class called Param, which is accessible by the entire codebase.
In order for the protocol to execute correctly, all users must come to a consensus on the values of all shared parameters before running the client program. These shared settings include:
∙ Network information. For example, IP_ADDR_P0 is used to set the IP address of the machine used by CP0, and PORT_P2_P3 is used to set the port number that CP2 and SP will use to communicate.
∙ Secret sharing parameters. For example, BASE_P is used to set the base prime 𝑝 for NTL operations, NBIT_K is used to set the bit length of an NTL data value, and NBIT_F is used to set the bit length of the fractional part of an NTL fixed point data value.
∙ GWAS parameters. For example, NUM_INDS, NUM_SNPS, and NUM_COVS are used to set the dimensions of the input dataset, IMISS_UB and GMISS_UB are used to set the acceptable missing rate levels for filtering out individuals and genotypes, and NUM_POWER_ITER is used to set the number of iterations for the PCA step during population structure correction.
∙ Software parameters. For example, NTL_NUM_THREADS is used to set the number of threads for NTL thread boosting, and MPC_BUF_SIZE is used to set the size of the data buffer buf.
Additionally, each party’s parameter file also includes non-shared settings spe-cific to that user, most notably directory and file names for reading and writing the necessary data. The relevant settings include:
∙ SNP_POS_FILE: the location of the pos.txt file, which is shared by all parties. ∙ CACHE_FILE_PREFIX: the filename prefix for saving files containing secret-shared
data as well as cached intermediate GWAS results. For example, the secret-shared genotype matrix is saved as CACHE_FILE_PREFIX + "_input_geno.bin".
∙ KEY_PATH: the name of the directory where the AES key files used to create shared communication channels and the global PRG are located.
3.2
Supporting Multiple Data-Sharing Parties
The first goal in modifying the forked secure GWAS codebase was adding support for multiple input datasets. In particular, we wanted to redesign the secure GWAS pipeline so that during the data sharing stage, the computing parties would be able to receive and save secret-shared genomic data sourced from more than one party playing the role of SP; then, during GWAS, the computing parties would analyze a single, joined dataset by pooling together the contributions of each SP. We made one key assumption: although the datasets contributed by each SP could contain data for a different number of individuals (i.e. have different values for NUM_INDS), each secret-shared dataset would use the same set of SNPs and the same set of covariates, ordered in a universal way. This assumption simplifies the secure GWAS logic by treating the secure GWAS codebase as a blackbox that receives perfectly formatted data, with datasets to be pooled together differing only in their number of rows. This makes logical sense since two data-contributing participants in the same GWAS study should be interested in the same phenotype and SNPs, but provide data for different individuals. Additionally, we found such an assumption reasonable for the time being since we could most effectively relax it by adding a preprocessing step to the public cloud interface layer between the user and the secure GWAS code, which we describe in Chapter 4.
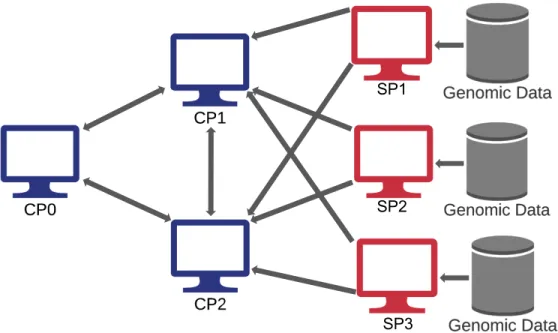
In order to add support for multiple SPs, we had to make a few key design choices. The first was choosing a new network setup for our modified GWAS task. We observed that 2 different SPs had no reason to collaborate with each other, since neither was involved in any MPC computations. Furthermore, sending data between SPs would naturally lead to privacy concerns and additional security considerations. Therefore, we easily settled on the network setup shown in Figure 3-1.
Figure 3-1: Network setup for modified secure GWAS protocol. Arrows represent secure communication channels. All SPs communicate with CP1 and CP2, but no two SPs communicate with each other.
for a single GWAS study during a single execution of the DataSharingClient pro-gram, or whether to have the CPs run a separate instance of the DataSharingClient program for each dataset/SP. These two approaches yield two different corresponding mechanisms for pooling datasets. With the former approach, all datasets would be joined during the data sharing stage, and each CP would save a single cache file con-taining secret shares for all individuals; with the latter approach, secret shares for each dataset would be saved in separate cache files, and the pooling of the datasets would occur during the GWAS stage. At first, the former approach seemed ideal since it compartmentalizes the logic of crowdsourcing datasets within the data sharing stage, leaving the GWAS stage of the pipeline completely untouched. However, the second approach proved superior for a variety of reasons. First, since each MPCEnv object only creates communication channels with a single SP, receiving data from more than one SP within a single execution of the DataSharingClient would require signifi-cant changes to the core MPC logic. On the other hand, with the latter approach, each DataSharingClient execution would initialize a separate MPCEnv object with
a separate secure channel to communicate with the corresponding SP. Additionally, this approach allows data sharing of two different datasets to occur asynchronously: rather than having to wait for all SPs to join the protocol, the CPs can begin reading and saving streamed data the second any SP joins. Asynchronicity is also useful in the case that an additional SP joins the GWAS study after the study has already begun; in this case, the data for SPs who have already run the DataSharingClient program need not be re-shared. Finally, the multiple dataset-multiple execution model offers a final benefit of parallelism; rather than have the SPs stream the datasets in some sequential order, the CPs can instead receive secret-shared datasets from different SPs in parallel, with each corresponding execution of the DataSharingClient pro-gram running in a separate process. This leads to a significant performance boost, especially when the datasets are quite large.
Finally, once we decided on having each CP run the DataSharingClient once for each input dataset, we had to decide whether to use a separate parameter file for each such execution. Looking at the various parameters, we realized only three parameters would need to differ between DataSharingClient executions for separate SPs: the port numbers that CPs receive messages on, the number of individuals in the dataset, and the cache file prefix used for saving secret-shared data. Since the vast majority of the parameters remained the same, and since the GwasClient would also use essentially the same parameter file, we decided to keep one parameter file per user per GWAS study.
3.2.1
Parameter File Changes
In order to support analysis of multiple datasets with a single parameter file, we made several modifications to the Param class. Specifically, we changed the type of three parameters all of the port numbers, NUM_INDS, and CACHE_FILE_PREFIX -to be std::vec-tor’s instead. This modification allows CPs -to s-tore information for many different input datasets using a single parameter file/Param class. After making this change, the updated parameters can be understood as follows: PORT_P1_P3[i] is the port number that CP1 and SP𝑖 will use to communicate, PORT_P0_P2[i] is
the port number that CP0 and CP2 will use to send messages about SP𝑖’s dataset, and so on for the other ports; NUM_INDS[i] is the number of individuals (rows) in the dataset sourced by SP𝑖; and CACHE_FILE_PREFIX[i] is the filename prefix used for saving secret shares for the dataset sourced by SP𝑖. Note that the actual ordering of the SPs does not matter, as long as all three CPs maintain a consistent ordering. Additionally, the SPs themselves need to only store a single element in each of their vector-type parameters: the information for their own dataset. Once again, we require consistency, i.e. that the values that SP𝑖 stores in these parameters are consistent with each CP’s values for the 𝑖th element of these parameters. Ensuring this consistency is easily taken care of by the cloud interface layer, which we describe in Chapter 4.
In order to distinguish between different datasets, we also added a parameter called CUR_ROUND to the parameter file and Param class. This integer valued parameter takes a single, fixed value between 0 and 𝑁 − 1 (representing the corresponding SP) for the entirety of one DataSharingClient execution and therefore can be used by the program as the correct index into the modified, vector-type parameters.
Finally, since the secret shares for each dataset will be saved in the locations CACHE_FILE_PREFIX[i] + "_input_geno.bin" and so on, we require that the values at any two indices of the CACHE_FILE_PREFIX vector be distinct from one another. Similarly, since using the same port for two different communication channels would make it impossible to distinguish between messages from two different parties, we also require that no two port numbers (across all values stored by all five port parameter vectors) overlap with each other.
3.2.2
Updated Client Programs
After updating the logic for parsing parameter files based on the modifications de-scribed above, we updated the DataSharingClient program to take the CUR_ROUND parameter as a command line argument. This one update immediately made the DataSharingClient code compatible with the multiple SP/dataset case. In par-ticular, in order to secret-share data from three different SPs once all parameter
files have been correctly configured as described above, each CP must simply run the DataSharingClient executable three times, passing in the values 0, 1, and 2 as the CUR_ROUND argument once each. Meanwhile, each SP must simply run the DataSharingClient executable once, passing in the value 0 for the CUR_ROUND ar-gument. Once all executions of DataSharingClient have completed, the three CPs will have secret-shared data for every dataset available in a separate cache file in their local filesystem.
To make the GWAS pipeline compatible with multiple input datasets, we did not need to change the usage of the GwasClient executable. In other words, after data sharing is successfully completed, each CP must simply run the GwasClient executable once as before to analyze the pooled dataset. However, we did have to change the body of the GWAS protocol code to ensure that each party uses all secret-shared data across cache files. To accomplish this, in each place where the GWAS protocol code reads in secret-shared data from the filesystem, we added a loop over all values in the CACHE_FILE_PREFIX vector to make sure the program reads data from every single cache file. Similarly, any place where the code used the parameter NUM_INDS, we replaced it with the appropriate updated value (either NUM_INDS[i], or the total number of individuals found by summing up the values in the NUM_INDS vector, depending on the usage).
3.3
Synchronized Multi-Threading to Improve
Scal-ability
Once we successfully added support for multiple input datasets, we turned our at-tention towards improving the overall performance of the end-to-end secure GWAS pipeline, in order to ensure the code would achieve reasonable runtime with respect to the size of the input dataset (both the total number of individuals, and the number of SNPs). As mentioned earlier, the existing codebase used an NTL feature called thread boosting, in which the NTL runtime environment automatically multi-threads
expensive operations. We thought such an approach would be effective in practice, given that the most common subroutines in the data sharing and GWAS protocols consisted of embarrassingly parallel computations on NTL objects: either iterating over some large, matrix-structured dataset and sequentially processing every row, or MPC batch operations on long vectors (i.e. operations where the value at any in-dex of the output vector depends only on the values at the same inin-dex of the input vectors). However, when we profiled the performance of the existing secure GWAS pipeline, we were surprised to notice that these very same operations that should have been easily parallelized were the biggest bottleneck sections in the code, and that increasing the number of threads spawned for NTL thread boosting had little to no effect whatsoever on the runtimes of these sections and of the pipeline as a whole. These observations made us realize that NTL’s automatic thread boosting func-tionality was a highly ineffective means of parallelizing the GWAS pipeline. Although NTL was spawning threads, these threads were incredibly underutilized because of the frequent network communication steps in MPC routines, which introduced a large number of synchronization points throughout the protocol. As a result, the actual ex-ecution of the program was essentially proceeding serially in practice. To address this issue, we decided to replace the NTL thread boosting approach with a synchronized multi-threading approach to speed up the execution of these bottleneck sections. By writing our own routines for explicitly splitting input data into chunks and spawning a new thread to compute on each chunk, and by ensuring the parallel execution of these routines would be identical across machines in the network, we could leverage the benefits of parallelization much more effectively. We knew such manual multi-threading would require adding a lot of code to the codebase, but we also knew that it would lead to significantly better parallelism behavior as we increased the number of cores/threads, thereby making our improved pipeline much more scalable than the existing one.
For our custom multi-threaded routines, we decided to use OpenMP, a simple yet high-performance C++ compiler extension for shared-memory parallel programming. OpenMP was perfect for our purposes since it allows one to parallelize existing code
without having to significantly rewrite it. For example, to parallelize a C++ for loop using OpenMP, we only needed to add the following line above the loop: #pragma omp parallel for num_threads(N). Since we already had an extensive secure GWAS codebase, this simple, low-overhead usage was ideal for our project. Additionally, as seen in the previous example, OpenMP allows users to easily specify the number of threads used to execute parallel sections of code as a variable; this would allow us to ultimately expose the desired parallelism of our code as a parameter setting to be specified by end users, improving the overall customizability of our protocol. In order to achieve this, we added a parameter called NUM_THREADS to the Param class which we then used to specify the number of threads to be used for any parallel OpenMP section. Note that this parameter is distinct from the NTL_MUM_THREADS parameter used for NTL thread boosting; we kept both parameters to allow us to easily compare the performance of NTL thread boosting against that of our custom multi-threading approach using the same number of spawned threads.
3.3.1
Support for Parallel Inter-Party Communication
Before we could proceed with parallelizing the codebase, we had to make a number of changes to the MPEnv class to enable parallel computation. In particular, we ob-served that parallelizing any MPC functions that required orchestrating computation across parties would be impossible without significantly changing the logic for inter-party network communication. As an illustrative example, consider the case where we wanted to parallelize some MPC vector operation that requires CP1 to send a different value to CP2 for each element of the input vector. Since there’s no guar-antee that different threads will execute the communication code in the same order across machines, we could easily run into the case where the thread responsible for performing the computation on the first element of the vector on CP1’s machine sends a value to CP2, only for it to be received by the thread on CP2’s machine that is responsible for performing the computation on some totally different element of the vector. As a result, naively parallelized MPC protocols would be unable to accurately communicate between machines and therefore return incorrect values.
In order to prevent such race conditions, we realized that with parallelization, we now needed one separate secure communication channel for each pair of corresponding threads on each pair of connected machines. As long as each party spawned threads to compute on the same exact sections of inputs (easily accomplished since we only implement each MPC function once, and since OpenMP allows one to specify fixed input ranges for each spawned thread), and as long as these pairs of corresponding threads had a dedicated communication channel, the threads spawned by two con-nected machines could execute in any arbitrary order and still output the correct result. In a similar fashion, we realized we had to create a separate dedicated shared PRG for each pair of corresponding threads on each pair of connected machines. Oth-erwise, in any parallel section that requires two connected parties to generate the same random value, differences in thread order execution due to context switching could cause parties to generate inconsistent random values and calculate incorrect results.
To formalize the notion of "corresponding threads," we took advantage of OpenMP’s method for assigning IDs to threads spawned in a parallel section. In particular, in any OpenMP parallel section, the thread corresponding to the 𝑖th input range is iden-tified by the thread number 𝑖. Additionally, at any point within an OpenMP parallel section, one can access the thread number of the currently executing thread using the special function omp_get_thread_num(). Thus OpenMP thread numbers provide a consistent and programmatic way of identifying the specific thread responsible for computation on each section of the input. These thread numbers can therefore allow the protocol to automatically keep track of which desired communication channel or PRG to use at any point within a parallel section.
With this in mind, we made the following changes to the MPCEnv class. First, we updated the SetupChannels functions to initialize Param::NUM_THREADS differ-ent secure communication channels for each pair of connected parties, while ensur-ing no two channels use the same port on a given machine. Similarly, we updated the SetupPRGs functions to initialize Param::NUM_THREADS different sets of private, shared, and global PRGs for each party. In order to effectively store all of these
differ-ent channels and PRGs while making sure that all accesses to shared data structures were thread safe, we made the following changes to the core MPCEnv data structures: ∙ buf: now implemented as a map such that buf[i] is the data buffer used by
the thread numbered i
∙ sockets: now implemented as a two-level map such that sockets[i][j] is the web socket used by the thread numbered i on this machine to communicate with the thread numbered i on the machine for the party whose ID is j ∙ prg: now implemented as a two-level map such that prg[i][j] is a PRG for
the thread numbered i (private if j = pid, global if j = -1, and shared with the thread numbered i on party j’s machine otherwise)
∙ cur_prg_pid: now implemented as a map such that cur_prg_pid[i] represents the current party ID index into prg for the thread numbered i
Finally, for all MPCEnv functions that send or receive data via a communication chan-nel, sample a random element from a PRG, or read or write data from the filesystem, we added code that calls omp_get_thread_num() and uses the result as the key into the corresponding data structure. This ensures that within any parallel section, threads may read data into the buffer or message another party via the network in parallel, while ensuring all such operations operate consistently across machines for different parties. Additionally, since omp_get_thread_num() always returns 0 when called outside of an OMP parallel section, this ensures that code outside of parallel blocks also executes consistently across all parties.
Note that with these changes, all reads and writes of MPCEnv shared data structures from a given thread only mutate the values of a map stored at a specific key unique to that thread. This guarantees that no two threads will ever access the same socket, PRG, or buffer, ensuring that all data accesses within the MPCEnv class are in fact thread safe.
3.3.2
Parallel MPC Routines
With the infrastructure for parellel communication in place, we decided to begin parallelizing the most expensive MPC operations. In particular, extensive profiling with real GWAS datasets revealed that three specific MPC functions proved to be significant bottlenecks in our protocol: FPSqrt (element-wise square root of a fixed point ZZ_p vector), FPDiv (element-wise division of two fixed point ZZ_p vectors), and IsPositive (element-wise sign test of a fixed point ZZ_p vector). These results were not surprising, given that all three of these functions shared the following features: 1) they are used repeatedly by the GWAS protocol code on input vectors of length NUM_SNPS (which ranges in the hundreds of thousands for real-world datasets); and 2) they all operate on NTL finite field representations of fixed point (i.e. decimal) values, which means they are implemented using complex approximation algorithms rather than straightforward, one-step mathematical operations. Additionally, all three functions also operate element-wise on their input vectors, so they offer ripe targets for parallelization.
We therefore wrote parallel implementations of these routines using the same template. First, the parallel long vector routine splits the input vector into a number of nearly equal-sized chunks that is a perfect multiple of Param::NUM_THREADS. Next, the parallel routine spawns a parallel OpenMP for loop over these chunks, assigning the chunks (in contiguous pieces) to Param::NUM_THREADS number of threads. Finally, for each iteration of the for loop, the function copies the corresponding chunk of the input vector(s) to temporary variables, calls the corresponding non-parallel base routine on the temporary variables, and copies the result to the single, shared output vector. After implementing parallel routines for all three of these MPC functions, we extensively profiled them on vectors of varying lengths and using a varying number of threads on Google Cloud instances with many cores. The results of these experiments are detailed in section 5.1. Once we verified reproducible runtime improvements using parallelization, we replaced all calls to these long vector routines in the GWAS protocol with the corresponding parallel routines.
After successfully optimizing these long vector routines, we then turned our at-tention to a more complex MPC function called OrthonormalBasis. This function implements a linear algebra routine known as QR factorization for rectangular ma-trices, and it posed a bottleneck in our protocol code because of its repeated use on matrices of dimension 𝑘-by-NUM_SNPS during the iterative PCA step of GWAS (here 𝑘 is a small integer, usually 10 or 15). Since the OrthonormalBasis function was a similar routine to the above long vector operations (computes on "long" matrices, i.e. those with a very large number of columns; implemented as a complex approximation algorithm), we thought parallelization would be effective in improving the runtime of this function as well.
Profiling of the OrthonormalBasis function revealed that it spent the majority of its runtime in calls to two other MPC operations: a matrix multiplication rou-tine, and a function called Trunc (element-wise bit truncation of a fixed point ZZ_p matrix, usually performed after a multiplication). We therefore wrote parallel ver-sions of both of these routines using a similar approach as before; this time, since the data was structured as a matrix, we used parallel for loops over the rows of the input matrix (this leads to ideal cache behavior for matrix multiplication). We then wrote a new version of OrthonormalBasis in which all calls to MultMat and Trunc were replaced with their corresponding parallel versions, and used this new optimized implementation of OrthonormalBasis in the GWAS protocol code instead.
3.3.3
Chunked Data Streaming and Analysis
After successfully optimizing all bottleneck MPC routines, we turned our attention to the full GWAS protocol code instead. Here, we realized the main computational bottlenecks were sections that read the genotype matrix row by row, iterating over each individual to calculate some statistics or result. Parallelizing these for loops, however, proved trickier than expected; since the genotype matrix values were masked by random numbers computed during Beaver precomputation, and since these random values could only be re-generated using the cached PRGs from the data sharing stage, we couldn’t parallelize these loops without finding some way to non-sequentially draw