HAL Id: tel-01877331
https://tel.archives-ouvertes.fr/tel-01877331
Submitted on 19 Sep 2018HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Impact d’un polluant environnemental, le
benzo[a]pyrène, sur le microbiote intestinal en modèle
murin
Celine Ribière
To cite this version:
Celine Ribière. Impact d’un polluant environnemental, le benzo[a]pyrène, sur le microbiote intestinal en modèle murin. Médecine humaine et pathologie. Université d’Auvergne - Clermont-Ferrand I, 2015. Français. �NNT : 2015CLF1MM18�. �tel-01877331�
Année 2015 N° d’ordre
E
COLED
OCTORALEDES
S
CIENCES DE LAV
IE ET DE LAS
ANTEN° d’ordre :
Thèse
Présentée à l’Université d’Auvergne pour l’obtention du grade de Docteur d’Université Spécialité : Biologie Moléculaire et Microbiologie
Soutenue le 10 Décembre 2015
Céline RIBIERE
I
MPACT D
’
UN
P
OLLUANT
E
NVIRONNEMENTAL
,
LE
B
ENZO
[a]P
YRENE SUR LE
M
ICROBIOTE
I
NTESTINAL
EN
M
ODELE
M
URIN
Jury
Rapporteurs : Hervé BLOTTIERE, Directeur de recherche, UMR 1319 MICALIS, INRA Jouy-en-Josas Philippe N. BERTIN, Professeur des Universités, UMR 7156, CNRS Université de Strasbourg Examinateurs : Annick BERNALIER-DONADILLE, Directrice de recherche, UR 454 INRA Clermont-Ferrand
Benoît FOLIGNE, Chargé de recherche, U1019 UMR 8204, Institut Pasteur Université de Lille
Directeur : Éric PEYRETAILLADE, Maître de conférences, EA 4678 CIDAM, Université d’Auvergne
Co-directrice : Delphine BOUCHER, Maître de conférences, EA 4678 CIDAM, Université d’Auvergne
Invité : Pierre PEYRET, Professeur des Universités, EA 4678 CIDAM, Université d’Auvergne
Année 2015 N° d’ordre
E
COLED
OCTORALEDES
S
CIENCES DE LAV
IE ET DE LAS
ANTEN° d’ordre :
Thèse
Présentée à l’Université d’Auvergne pour l’obtention du grade de Docteur d’Université Spécialité : Biologie Moléculaire et Microbiologie
Soutenue le 10 Décembre 2015
Céline RIBIERE
I
MPACT D
’
UN
P
OLLUANT
E
NVIRONNEMENTAL
,
LE
B
ENZO
[a]P
YRENE SUR LE
M
ICROBIOTE
I
NTESTINAL
EN
M
ODELE
M
URIN
Jury
Rapporteurs : Hervé BLOTTIERE, Directeur de recherche, UMR 1319 MICALIS, INRA Jouy-en-Josas Philippe N. BERTIN, Professeur des Universités, UMR 7156, CNRS Université de Strasbourg Examinateurs : Annick BERNALIER-DONADILLE, Directrice de recherche, UR 454 INRA Clermont-Ferrand
Benoît FOLIGNE, Chargé de recherche, U1019 UMR 8204, Institut Pasteur Université de Lille
Directeur : Éric PEYRETAILLADE, Maître de conférences, EA 4678 CIDAM, Université d’Auvergne
Co-directrice : Delphine BOUCHER, Maître de conférences, EA 4678 CIDAM, Université d’Auvergne
Invité : Pierre PEYRET, Professeur des Universités, EA 4678 CIDAM, Université d’Auvergne
de par la seule force de son obstination. » Robin S. Sharma
Je souhaite exprimer mes remerciements à Hervé Blottière et Philippe Bertin pour m’avoir fait l’honneur d’être rapporteurs de cette thèse, ainsi qu’à Annick Bernalier-Donadille et Benoît Foligné d’avoir accepté d’examiner ce travail et de faire partie de mon jury de thèse.
Je tiens ensuite à remercier Monique Alric, directrice de l’EA 4678 CIDAM, pour m’avoir accueillie au sein de son unité au cours de ces trois années de thèse et pour prêter une oreille attentive aux étudiants lorsqu’ils en ont besoin. Je voudrais également adresser mes sincères remerciements à Pierre Peyret, qui anime le groupe de recherche dans lequel j’ai mené mes travaux de thèse. Merci Pierre de donner aux étudiants qui travaillent avec vous les moyens de réaliser leurs projets et pour votre référence cinématographique inoubliable à propos des westerns.
Une thèse ne serait pas une thèse sans un encadrant. Moi j’ai eu la chance d’en avoir deux. Mes remerciements s’adressent donc bien évidemment à Eric Peyretaillade et à Delphine Boucher, qui ont co-encadré ces travaux de thèse. Je vous remercie du temps que vous m’avez consacré au cours de ces trois années mais également de la liberté que vous avez su me laisser pour mener à bien mes différents projets. J’espère avoir su tirer profit des compétences et des qualités de chacun de vous deux. Merci Delphine pour ton implication et ton soutien notamment au cours de cette troisième année, ainsi que pour ton investissement lors de la rédaction de la publication sur le microbiote intestinal. Merci à toi Éric, en particulier pour m’avoir guidé lors de mes premiers pas à l’IUT.
Je ne peux évoquer l’IUT sans parler des personnes avec qui j’ai eu la chance de réaliser mes enseignements. Mes remerciements vont donc à l’équipe pédagogique du département de génie biologique, et en particulier à Emilie Brasset et à Anne Cayre, aux côtés de qui réaliser mes enseignements aura été un réel plaisir.
Je voudrais également remercier Sylvie Rabot, Annick Bernalier-Donadille et Nicolas Barnich qui ont accepté de participer aux comités de suivi de cette thèse. Merci pour vos discussions enrichissantes sur ces travaux et leurs orientations.
le montage du protocole d’expérimentation animale. Je remercie aussi les personnes de l’animalerie de la Faculté de Médecine pour m’avoir accueillie dans leurs locaux. De la même manière, j’adresse mes sincères remerciements à Annick Bernalier-Donadille qui j’ai également collaboré pour la mise en place d’un protocole d’expérimentation animale en modèle axénique. Je remercie donc aussi Christophe Del’Homme, ainsi que Christophe et Valérie de la plateforme axénique de l’INRA de Theix, pour leur aide et leur grande disponibilité lors de la mise en place de ce protocole.
Au cours de ces trois années de thèse, j’ai aussi eu la chance de travailler avec le CICS des Facultés de Médecine et de Pharmacie, en particulier avec Lorraine Gameiro, que je souhaite vivement remercier pour sa disponibilité, sa réactivité et ses précieux conseils. Mes sincères remerciements vont également aux anatomo-pathologistes Claude Darcha et Pierre Déchelotte qui ont accepté d’examiner les lames histologiques réalisées au cours de ces travaux de thèse. Je souhaite les remercier pour le temps qu’ils ont consacré à mes projets et pour les connaissances et l’expertise qu’ils ont partagées avec moi.
Je remercie l’équipe MASS de l’UR370 de l’INRA de Theix, en particulier Erwan Engel, Jérémy Ratel et Frédéric Mercier, pour leur accueil et leur formation en chimie analytique, même si ces travaux n’ont pas abouti dans le cadre de cette thèse.
Au cours de ces trois années de thèse, j’ai également eu la chance de collaborer avec l’équipe d’écologie microbienne de l’UMR CNRS 5557 de Lyon sur des projets de capture de gènes. Je souhaite notamment remercier Laurence Fraissinet-Tachet, Laurent Vallon et bien sûr Claudia Bragalini qui m’a associé à ses travaux. Au travers des projets de capture de gènes, j’ai également pu travailler avec Sylvain Charlat et Jean-Christophe Simon.
J’ai commencé ma thèse dans un bureau très majoritairement masculin. Merci à Jérémie Denonfoux et à Nicolas Parisot pour cet accueil au cœur de la musique des années 80. Un clin d’œil particulier à mon binôme de vaisselle, tu vois je finis par la soutenir cette thèse, certes avec un an de retard !
Depuis, le bureau s’est radicalement féminisé avec les arrivées successives de Réjane Beugnot, Clémence Defois et Bérénice Batut. Merci les filles pour cette fin de thèse. Merci à toi Clémence, le jukebox ambulant des chansons de Disney et dont les trous de mémoire nous font toujours beaucoup rire (tu te rappelles c’est clé 1-2-3-4 !). Réjane, merci pour toutes nos discussions, pour ton soutien quand je finalisais mon papier cet été, et pour tes conseils en domptage de félins.
avec ma voisine arrivée dans l’équipe peu de temps après moi. Je veux bien sûr parler de Cyrielle Gasc, celle que je considère comme ma « petite sœur de thèse ». Eh oui, Cyrielle l’heure est venue pour toi de « recevoir le retour du bâton de la cuillère ». J’espère que mes remerciements pour toi « ne tomberont pas comme une nouille sur le feu » et qu’ils « enverront des paillettes dans les yeux » autant que ces années passées à côté de toi. Ensemble, on aura beaucoup ri et pleuré, voire parfois les deux en même temps. Merci pour ton aide, ton soutien, les McDo de crise et tout le reste... Je n’oublie pas nos supers aventures parisiennes, je crois que je n’avais jamais eu aussi mal aux pieds de ma vie ! Même si je ne serai plus assise au bureau d’à côté, tu pourras toujours compter sur moi et où que j’atterrisse tu seras toujours la bienvenue, prochaine étape en Irlande !
Je souhaite également remercier le troisième membre du « gang des techniciens », Nicolas Gallois. J’ai eu la chance de te suivre au cours de mes trois années de thèse au travers de ton contrat et de tes différents stages réalisés dans l’équipe. Travailler avec toi aura été un réel plaisir et je te souhaite une bonne continuation dans tes nouveaux projets à Aix.
Je remercie tous les membres de l’EA CIDAM pour leur accueil chaleureux, qu’ils soient au cinquième étage ou au rez-de-chaussée, bien qu’avec les péripéties de ces derniers temps on ne sache plus vraiment où est qui. Merci à Jean-François Jarrige (qui restera toujours « notre Monsieur Jarrige »), Manon Martinet, Sylvain Denis, Stéphanie Blanquet-Diot, Jean-François Brugère, Eric Beyssac, Ghislain Garrait, Valérie Hoffart, Jean-Michel Cardot, Pascale Gauthier et Pierre-Charles Romond. Un clin d’œil spécial à notre « mamie » Sandrine Chalancon dont la bonne humeur et le rire sont communicatifs. Cette équipe a beaucoup de chance de t’avoir, tout comme tes « petits nains de labo ».
Je souhaite également remercier tous ceux qui contribuent à la bonne ambiance générale de cette équipe. Je veux bien sûr parler de tous les étudiants comme Charlotte Cordonnier et Nadia Gaci, qui ont commencé leur thèse en même temps que moi ; les stagiaires devenus des doctorants : Hassana Hsein, Charlène Roussel et Ophélie Uriot ; les doctorants devenus post-doc : Mickaël Fleury (eh oui « poussin » ça c’est toi ! Capitaine Ribière te salue !) ; et pour finir les post-docs : Olivier Le Goff et Prem Prashan Chaudary. Je n’oublie pas non plus tous ceux qui sont déjà partis pour de nouvelles contrées plus ou moins lointaines : Lucie Etienne-Mesmin (mon écrivain mystère), William Tottey (ta bonne humeur me manque beaucoup), Sophie Comtet, Jonathan Thévenot, Faouzi Jaziri, Thierry Allario, Xie Xiaoyu et Suenia De Paiva Lacerda.
de janvier à juin au cours ces trois dernières années. Je remercie en particulier Fanny Matner que j’ai eu la chance d’encadrer lors de son stage de DUT et avec qui j’ai beaucoup apprécié travailler.
Je souhaite clôturer ces remerciements à m’en adressant à mes proches et à ma famille, sans qui tout ceci aurait était bien plus difficile.
Je remercie mes deux meilleurs amis, Julie et Nicolas, pour leur soutien sans faille. Merci Julie pour ces 27 belles années d’amitié dont les souvenirs sont gravés dans ma mémoire (le jeu des senteurs, les chocos vanille-chocolat, les pépitos, l’expédition au bureau de tabac et bien plus encore). Je suis sûre que les 27 prochaines années seront tout aussi réussies. Merci Nicolas, notre amitié n’est pas aussi ancienne mais elle en n’est pas moins précieuse à mes yeux. Je n’oublierai jamais « les courses de raviolis » de notre première année de fac.
Je voudrais remercier des membres de ma famille que malheureusement je ne vois pas assez souvent. Ninou, merci d’avoir été une deuxième maman pour moi en particulier dans les moments difficiles de ma vie. Merci à toi mon frère, Maxime, je te souhaite de trouver une profession dans laquelle tu t’épanouiras même si pour moi tu as déjà trouvé ta voie en devenant pompier volontaire, ce qui montre bien la personne généreuse que tu es.
J’ai aussi une pensée pour mes grands-parents, pour ma mamie Simone partie avant que je puisse apprendre àla connaitre et pour mes grands-parents paternels Anne-Marie et Robert avec qui j’ai eu la chance de passer du temps lors de mes vacances à la ferme mais qui sont eux aussi partis bien trop tôt. Je n’oublie bien sûr pas mon papy, Georges. Merci à toi papy d’avoir aussi bien pris soin de moi quand j’étais petite.
Mes remerciements vont maintenant à ma famille d’adoption. Merci à ma tata Agnès et mon tonton Didier. Merci à vous deux de m’accueillir aussi souvent chez vous, ces séjours sont toujours une grande bouffée d’oxygène (et pas seulement à cause du grand air !!!). Merci aussi à mes petites cousines, Noémie et Camille qui rendent ces séjours encore plus formidables.
Merci à toi, Pauline, ma sœur, ma « petite mimi ». Merci de partager avec moi tous les moments importants et précieux de ma vie, et de rester à mes côtés même dans les moments difficiles. Je suis si fière et impressionnée de la personne que tu es devenue et qui a grandi sous mes yeux.
Je ne peux pas terminer ces remerciements sans parler de mes parents, de ma maman Catherine et de mon papa de cœur Pascal. Merci à toi Pascal de m’élever comme ta fille depuis que j’ai 8 ans et d’avoir donné tout son sens au mot « père ». Merci à toi ma petite maman,
aujourd’hui être là où j’en suis ? Ensemble on a fait face aux difficultés de la vie et ces quelques mots ne pourront jamais exprimer tout l’amour que je te porte. Maman, Papa, merci de m’avoir tant donné et surtout de continuer…
Le microbiote intestinal joue un rôle primordial dans l’homéostasie du tractus gastro-intestinal, et plus généralement dans celle de son hôte. A ce titre, de nombreuses pathologies humaines sont associées à une dysbiose de ce microbiote intestinal, tels que les cancers colorectaux, les maladies inflammatoires chroniques de l’intestin (MICI), les troubles du métabolisme ou encore les maladies auto-immunes. Ces pathologies ont une étiologie mal connue et multifactorielle dans laquelle l’environnement semble jouer un rôle clé. Des études récentes ont ainsi mis en évidence un lien entre la pollution atmosphérique et des pathologies humaines telles que les MICI. Parmi les différentes substances polluantes répertoriées, le benzo[a]pyrène (BaP), qui fait partie de la famille des hydrocarbures aromatiques polycycliques, est soumis à une surveillance accrue en raison de ses effets toxiques sur la santé humaine. De par ses propriétés pro-inflammatoires et mutagènes, le BaP pourrait modifier la composition du microbiote intestinal, induisant alors à une réponse inflammatoire et à une altération des fonctions intestinales. Dans le cadre de ce travail de thèse, une surexposition orale et chronique au BaP en modèle murin a conduit à une inflammation modérée principalement au niveau de la muqueuse iléale. L’analyse des amplicons du gène codant l’ARNr 16S a mis en évidence des modifications de la composition et de l’abondance relative des communautés bactériennes fécales et associées à la muqueuse intestinale avec notamment une augmentation et une diminution des taxa pro et anti-inflammatoires respectivement. Ainsi, dans des conditions de susceptibilité génétique et/ou en association avec d’autres facteurs environnementaux, l’exposition à ce polluant pourrait déclencher et/ou accélérer le développement de pathologies inflammatoires. L’identification des potentialités métaboliques des différentes populations bactériennes caractérisées précédemment et impactées par le polluant revêt donc un caractère primordial. La reconstruction de génomes directement à partir de l’écosystème microbien peut permettre d’établir ce lien entre structure et fonction. C’est également dans ce contexte, qu’une approche innovante de capture de gènes en solution a été développée. En effet, cette technique d’enrichissement permet de reconstruire de larges portions génomiques pouvant relier un biomarqueur phylogénétique à des gènes fonctionnels, y compris pour des populations bactériennes présentes en très faible abondance dans l’écosystème. Mots-clés : microbiote intestinal, benzo[a]pyrène, métagénomique, capture de gènes en solution, inflammation intestinale.
Abstract
Gut microbiota plays a primordial role in gastro-intestinal tract and host homeostasis. Numerous pathologies are associated with a gut microbiota dysbioses, such as colorectal cancer, inflammatory bowel diseases (IBD), metabolism disorders or autoimmune diseases. The physiopathology of these diseases has multifactorial aetiology in which environmental factors seem to play a crucial role. Recent evidences have highlighted a link between air pollution and human diseases such as IBD. Among the different pollutant listed, benzo[a]pyrene (BaP), which belong to the family of polycyclic aromatic hydrocarbons, is subject to an increase surveillance due to its toxic effects on human health. By its pro-inflammatory and mutagenic proprieties, BaP could lead to modifications of gut microbiota composition, then inducing an inflammatory response and an alteration of intestinal functions.
As part of this thesis, BaP subchronic oral exposure in murine model has led to a moderate inflammation mostly in ileal mucosa. The analysis of ARNr 16S amplicons has highlighted composition and abundance alterations of faecal and mucosa-associated microbiota, especially with increase and decrease of pro and anti-inflammatory taxa respectively. Thus, under conditions of genetic susceptibility and/or in association with other environmental factors, exposure to this pollutant could trigger and/or accelerate the development of inflammatory pathologies. Metabolic potential identification of different bacterial populations previously characterized and affected by the pollutant appears therefore primordial. Genome reconstruction directly from microbial ecosystem could allow to establish this link between structure and function. Also in this context, an innovative approach of gene capture in solution was developed. Indeed, this enrichment technique allows to reconstruct large genomic portions that could link phylogenetic biomarker and functional genes, including for bacterial populations present at very low abundance in the ecosystem.
ADN Acide désoxyribonucléique
AFSSA Agence Française de Sécurité Sanitaire des Aliments AGCC Acide Gras à Chaîne Courte
AhR Aryl hydrocarbon Receptor ARN Acide ribonucléique BaP Benzo[a]pyrène
BPDE BaP-7,8-diol-9,10-époxyde CAZy Carbohydrate Active enZymes CCR Cancer colorectal
CEE-NU Commission Européenne des Nations Unies pour l’Europe CL50 Concentration létale 50
CMR Cancérogène Mutagène Reprotoxique CYP450 Cytochrome P450
DGGE Denaturing Gradient Gel Electrophoresis DL50 Dose Létale 50
EFSA European Food Safety Authority FISH Fluorescence In Situ Hybridization HAP Hydrocarbure Aromatique Polycyclique HGC High Gene Content
HITChip Human Intestinal Tract Chip HMP Human Microbiome Project HuGChip Human Gut Chip
IARC International Agency for Research on Cancer IgA Immunoglobuline A
INERIS Institut National de l’Environnement Industriel et des Risques INRS Institut National de Recherche et de Sécurité
KEGG Kyoto Encyclopedia of Genes and Genomes LGC Low Gene Content
MAMP Microbe-Associated Molecular Pattern MC Maladie de Crohn
mcrA méthyl-coenzyme M réductase
MetaHIT The european union project on Metagenomics of the Human Intestinal Tract MICI Maladies Inflammatoires Chroniques de l’Intestin
NGS Next-Generation Sequencing NIH National Institut of Health NOD Nucleotide Oligomerization Domain OH-BaP Hydroxybenzo[a]pyrène
OMS Organisation Mondiale de la Santé OTU Operational Taxonomic Unit pb paire de base
PCB Polychlorobiphényles PCR Polymerase Chain Reaction POP Polluant Organique Persistant qPCR PCR quantitative ou en temps réel RCH Rectocolite hémorragique SCG Single-Cell Genomics SII Syndrome de l’Intestin Irritable SNP Single Nucleotide Polymorphism
SOLiD Sequencing by Oligonucleotide Ligation and Detection TGGE Temperature Gel Gradient Electrophoresis
TLR Toll-Like Receptor Treg Lymphocyte T régulateur
T-RFLP Terminal Restriction Fragment Length Polymorphism UDP Uridine diphosphate
UE Union Européenne
US-EPA United States-Environmental Protection Agency VIH Virus de l’Immunodéficience Humaine XRE Xenobiotic Response Element
Figure 1 : Colonisation et développement du microbiote intestinal. (Modifiée d’après Clemente
et al. 2012)
Figure 2 : Vue d'ensemble des techniques utilisées pour caractériser le microbiote intestinal.
(Modifiée d’après Fraher, O’Toole and Quigley 2012)
Figure 3 : Réparation du microbiote au sein du tractus digestif. (Modifiée d’après Koboziev et
al. 2014 et Jandhyala et al. 2015)
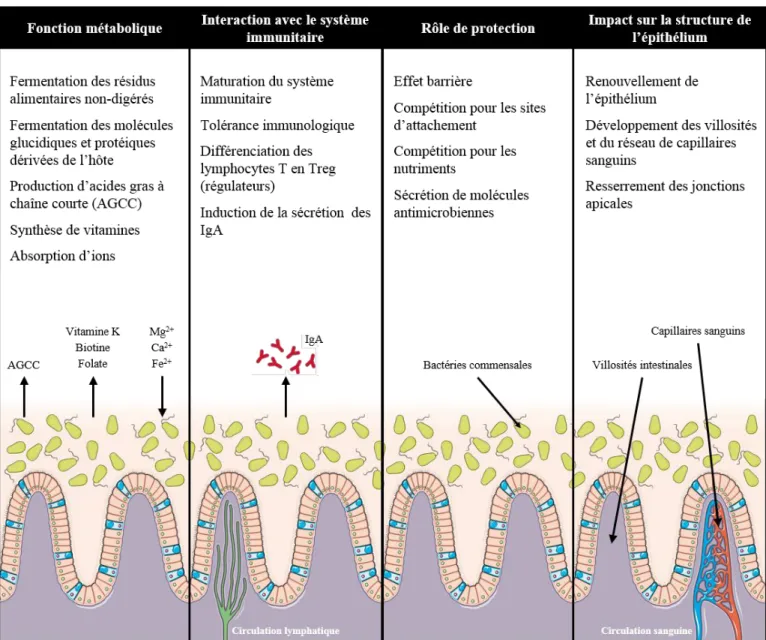
Figure 4 : Les fonctions du microbiote intestinal. (Modifiée d’après O’Hara and Shanahan
2006)
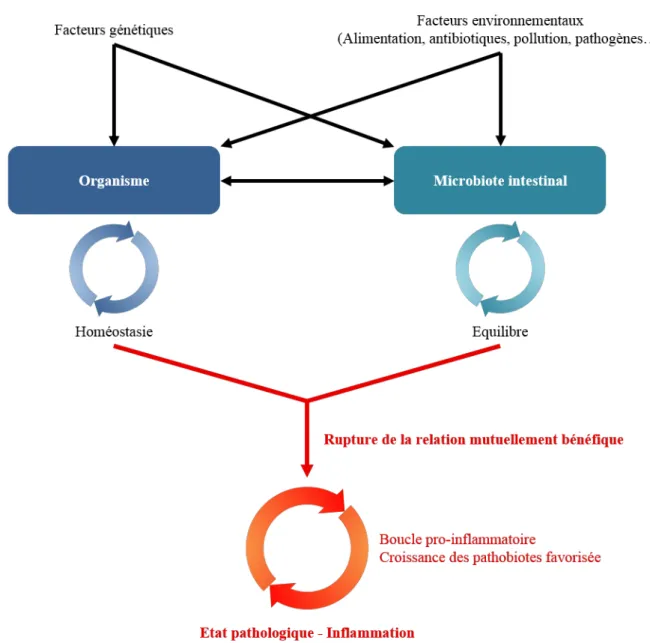
Figure 5 : Des facteurs génétiques et/ou environnementaux peuvent déstabiliser l'équilibre
entre l'hôte et le microbiote intestinal conduisant à un état pathologique.
Figure 6 : Les HAP prioritaires selon l'US-EPA et l'UE. (Adaptée d’après Purcaro, Moret and
Conte 2013)
Figure 7 : Devenir du benzo[a]pyrène absorbé par voie orale dans l'organisme. (Modifiée
d’après Ramesh et al. 2004)
Figure 8 : Les principales voies de contamination des aliments par le BaP. (Modifiée d’après
Bansal and Kim 2015)
Figure 9 : Formation du BaP au cours de la pyrolyse des aliments. (Modifiée d’après (Trafialek
& Kolanowski, 2014)
Figure 10 : Activation métabolique du BaP. (Modifiée d’après Alexandrov, Rojas and Satarug
2010)
Figure 11 : Modulation du métabolisme des xénobiotiques par le microbiote intestinal.
(Modifiée d’après Carmody and Turnbaugh 2014)
Figure 12 : Courbe de suivi du poids des souris traitées avec le BaP. Figure 13 : Scores histologiques d'inflammation au niveau iléal et colique.
Tableau 1 : Techniques utilisées pour caractériser le microbiote intestinal. (Adapté d’après
Fraher, O’Toole and Quigley, 2012)
Tableau 2 : Métabolites produits par le microbiote intestinal. (Adapté d’après Nicholson et al.
2012)
Tableau 3 : Les pathologies humaines associées à une altération du microbiote intestinal. Tableau 4 : Changements entre un style de vie traditionnel et moderne pouvant altérer la
composition et/ou les fonctions du microbiote intestinal. (Adapté d’après Koboziev et al., 2014 et Cortot et al., 2009)
Tableau 5 : Caractéristiques physico-chimique du benzo[a]pyrène (BaP). (Adapté de la fiche
de sécurité de Sigma-Aldrich)
Tableau 6 : Concentration en BaP dans les aliments consommés en France. (Adapté d’après
Veyrand et al. 2013)
Tableau 7 : Estimation de l'exposition au BaP suite à l'ingestion d'aliments contaminés en
fonction des différentes zones géographiques (Adapté d’après AFSSA 2004 et Ma and Harrad 2015)
Tableau 8 : Valeurs limites de concentration en BaP applicables en France depuis décembre
INTRODUCTION GENERALE ... 1 PARTIE I : SYNTHESE BIBLIOGRAPHIQUE ... 5 1. Le microbiote intestinal ... 7
1.1. L’acquisition des communautés bactériennes ... 7 1.2. Richesse et la diversité du microbiote intestinal ... 9 1.2.1. Méthodes d’analyse de la composition du microbiote intestinal ... 9 1.2.2. Gradient longitudinal de richesse et de diversité ... 12 1.2.3. Le microbiote fécal ... 13 1.2.4. Le microbiote associé à la muqueuse ... 14 1.2.5. Les facteurs influençant la composition du microbiote intestinal ... 15 1.2.6. La catégorisation du microbiote intestinal ... 16 1.3. Fonctions du microbiote intestinal ... 17 1.3.1. Fonctions métaboliques ... 18 1.3.2. Développement et maturation du système immunitaire ... 20 1.3.3. Rôle de protection ... 22 1.3.4. Impact sur la structure de l’épithélium intestinal ... 23
2. Pathologies humaines et dysbiose du microbiote intestinal ... 24
2.1. Les différents types de pathologies ... 25 2.1.1. Les pathologies digestives ... 25 2.1.2. Les pathologies extra-digestives ... 27 Pathologies inflammatoires et/ou auto-immunes ... 27 Les maladies métaboliques ... 28 Les pathologies cardio-vasculaires ... 28 Les troubles neurologiques ou comportementaux ... 28 2.2. Les facteurs pouvant induire une dysbiose ... 29
3. Les hydrocarbures aromatiques polycycliques ... 31
3.1. Généralités ... 31 3.2. Le benzo[a]pyrène... 32 3.2.1. Caractéristiques générales ... 32 3.2.2. Toxicité ... 33
Ingestion d’aliments contaminés ... 34 Inhalation de l’air pollué ... 35 Exposition professionnelle ... 36 3.2.4. Cadre législatif et réglementations ... 37 3.2.5. Le métabolisme du BaP chez les mammifères ... 38 3.2.6. Implication des communautés bactériennes dans la dégradation du BaP... 40
PARTIE 2 : TRAVAUX EXPERIMENTAUX ... 41 1. Impact du BaP sur le microbiote intestinal en modèle murin ... 42
1.1. Contexte de l’étude ... 42 1.2. Objectifs de l’étude ... 43 1.3. Principaux résultats obtenus ... 43 1.4. Article n°1 : « Oral exposure to environmental pollutant benzo[a]pyrene impacts the intestinal epithelium and induces gut microbial shifts in murine model. » ... 44 1.5. Conclusions et perspectives ... 77
2. Etude des écosystèmes microbiens complexes ... 78
2.1. Reconstruire des génomes pour étudier les fonctions des microorganismes ... 78 2.1.1. Introduction ... 78 2.1.2. Article n°2 : « Capturing prokaryotic dark matter genomes » ... 80 2.1.3. Conclusions et perspectives ... 98 2.2. La capture de gènes en solution ... 100 2.2.1. Introduction ... 100 2.2.2. Article n°3 : « Targeted gene capture by hybridization to illuminate ecosystem functioning » ... 101 2.2.3. Conclusions et perspectives ... 118
DISCUSSION GENERALE ET PERSPECTIVES ... 119 REFERENCES BIBLIOGRAPHIQUES ... 134
1
2 Le corps humain est habité par un très grand nombre de microorganismes et le tractus digestif, en particulier l’intestin, est le compartiment le plus densément peuplé. Le microbiote
intestinal comporte ainsi 1014 cellules, soit 10 fois plus de cellules que le corps humain. Cette
communauté microbienne intestinale, composée majoritairement par des bactéries, peut être considérée comme un organe à part entière ayant coévolué avec son hôte pour établir une relation symbiotique généralement mutuellement bénéfique. Le microbiote intestinal joue un rôle crucial dans l’homéostasie du tractus digestif, et plus globalement dans celle de son hôte, en remplissant des fonctions métaboliques, immunologiques, et structurales, ainsi qu’en jouant un rôle essentiel dans la protection de l’intestin contre la colonisation par des bactéries pathogènes.
Les premières descriptions du microbiote intestinal ont été réalisées dès le début du
XXème siècle grâce aux approches culturales à partir d’échantillons de selles. L’intérêt était alors
d’identifier les agents pathogènes responsables de diarrhées infectieuses (Campylobacter, Salmonella, Shigella, Escherichia coli enterotoxinogène…). Rapidement, il est apparu que les communautés bactériennes intestinales possédaient d’autres rôles que celui d’agent infectieux et qu’elles étaient indispensables au maintien de l’homéostasie de leur hôte. Au cours de ces dernières décennies, notre compréhension du microbiote intestinal, de sa composition, de son abondance et de ses fonctions, a été révolutionnée par l’avènement des outils moléculaires. Ces approches moléculaires ont en effet permis de donner une vision plus représentative de cet écosystème complexe. De nombreuses études ont montré qu’un grand nombre de pathologies humaines digestives et extradigestives sont associées à une dysbiose du microbiote intestinal c’est-à-dire à une altération de la composition et de l’abondance des populations bactériennes. Le microbiote intestinal pourrait donc être impliqué dans l’apparition et/ou l’évolution de ces maladies. Différents facteurs sont susceptibles d’entrainer une modification de la structure du microbiote intestinal, comme l’alimentation ou les antibiothérapies. Parmi ces facteurs, la qualité de l’environnement semble jouer un rôle important. Des études épidémiologiques ont en effet établi un lien entre pathologies et pollution tel qu’une prévalence plus élevée des maladies inflammatoires chroniques de l’intestin (MICI) en milieu urbain (Kaplan, 2011). Une élévation du niveau de la pollution atmosphérique a également été associée aux poussées inflammatoires chez les personnes atteintes de MICI (Ananthakrishnan et al., 2011). L’industrialisation des sociétés conduit en effet à une production de plus en plus importante d’une grande diversité de molécules chimiques potentiellement toxiques, qui peuvent être déversées accidentellement ou non dans l’environnement. Parmi les différentes substances
3 polluantes répertoriées, les Hydrocarbures Aromatiques Polycycliques (HAP) sont soumis à une surveillance accrue en raison de leurs effets mutagènes, cancérigènes et reprotoxiques sur l’Homme. Au sein de cette vaste famille de molécules polluantes, le benzo[a]pyrène (BaP) est considéré comme le composé le plus toxique et il est par conséquent le plus caractérisé. Or, le tractus digestif est fortement exposé au BaP par la consommation d’aliments contaminés, en particulier ceux qui ont été grillés (barbecue) ou fumés, par l’ingestion de sols pollués (phénomène important chez les enfants), et également par l’inhalation de polluants via le phénomène de clairance mucociliaire. Le BaP pourrait donc perturber le microbiote intestinal, entrainant un dysfonctionnement du système digestif, et participer au développement et/ou à l’aggravation des pathologies associées à une dysbiose de ce microbiote.
C’est dans ce contexte que s’inscrit la première partie des travaux de cette thèse, dont l’objectif est d’étudier l’impact du BaP sur la composition et l’abondance des communautés bactériennes intestinales, ainsi que ses conséquences sur la muqueuse digestive. Le microbiote intestinal est un écosystème microbien très complexe. Or, l’étude de la diversité du rôle des microorganismes dans les environnements complexes reste un défi majeur en écologie microbienne. La reconstruction de génomes à partir de l’environnement peut permettre de relier les populations microbiennes identifiées et leurs capacités métaboliques. Cependant, malgré l’essor des techniques de séquençage haut-débit, les approches de métagénomique ne permettent d’accéder essentiellement qu’aux organismes dominants de l’écosystème. Il apparait donc nécessaire de réduire la complexité des échantillons métagénomiques pour en explorer toute la diversité. C’est avec cet objectif qu’une technique innovante de capture de gènes en solution a été développée au sein de notre équipe.
La suite de ce manuscrit va donc s’articuler autour de deux parties. La première partie aura pour but de replacer le contexte scientifique de ces travaux de thèse au travers d’une synthèse bibliographique. Cette première partie se découpera en trois chapitres consacrés au microbiote intestinal, aux pathologies humaines associées à une dysbiose du microbiote intestinal et aux HAP. La seconde partie de ce manuscrit exposera et commentera les travaux effectués au cours de cette thèse. Les travaux sur l’impact du BaP sur le microbiote intestinal en modèle murin conventionnel feront l’objet du premier chapitre. Le second chapitre portera sur l’étude des écosystèmes microbiens complexes au travers de la reconstruction de génomes, pour lier la structure et la fonction des communautés microbiennes, et de l’exploration de la biosphère rare grâce à une approche innovante de capture de gènes en solution. Enfin, les
4 différents points abordés au long de ce manuscrit seront synthétisés dans une discussion générale, et des perspectives à l’ensemble de ces travaux seront proposées.
5
PARTIE I :
6 Apparus il y a 3,8 milliards d’années, les microorganismes ont été la première forme de vie à peupler la Terre, et durant les trois premiers milliards d’années, ils en ont été les seuls habitants. Aujourd’hui encore, ils représentent la forme de vie la plus abondante, la plus diversifiée et la plus répandue sur Terre. En outre, l’activité de la biosphère dans son ensemble est totalement dépendante de l’action de ces microorganismes. La rapidité de leur cycle cellulaire, favorisant l’apparition de mutations, et une grande versatilité métabolique, leur permettant d’utiliser les différentes sources de carbone et d’énergie présentes dans leur environnement, expliquent la capacité des microorganismes à s’adapter à des conditions de vie variées, y compris les plus extrêmes telles qu’une température et/ou une pression très élevées ou très basses, un pH très acide ou très basique, ou encore de fortes radiations. L’implication des microorganismes dans tous les processus biologiques, tels que les cycles biogéochimiques, le fonctionnement des réseaux trophiques, la régulation des populations en tant qu’agents pathogènes, ou encore le maintien des ressources génétiques globales, est aujourd’hui largement reconnue. Les microorganismes jouent par conséquent un rôle crucial dans l’organisation, l’évolution et le fonctionnement des divers écosystèmes qu’ils colonisent. La nature de ces écosystèmes est très variée, les microorganismes pouvant se développer au sein d’environnements terrestres ou aquatiques, mais également dans et/ou sur des êtres vivants tels que les plantes, les insectes ou encore les animaux.
Ainsi, depuis des millions d’années, l’Homme, et les animaux avant eux, ont co-évolué avec les microorganismes, conduisant ainsi à l’établissement d’une relation mutuelle, le plus souvent bénéfique. Le nombre de cellules microbiennes hébergées par un individu est bien supérieur au nombre de cellules qui composent le corps humain. L’Homme est donc considéré comme un « super-organisme », c’est-à-dire qu’il est constitué à la fois par les composants du corps humain et par les différents microorganismes qui le colonisent. En effet, toutes les surfaces du corps humain en contact avec le milieu extérieur, comme la peau et les tractus respiratoire supérieur, uro-génital et digestif, constituent des niches écologiques abritant des microorganismes. Le tractus digestif, en particulier l’intestin, est le compartiment le plus densément peuplé, et parmi les différents microbiotes, le microbiote intestinal est celui dont la composition et les fonctions sont les plus étudiées.
Figure 1 : Colonisation et développement du microbiote intestinal. (Modifiée d’après Clemente et al.
2012)
Le fœtus n’est pas stérile mais son microbiote intestinal est très peu abondant et très peu diversifié. La colonisation du tractus digestif débute réellement au moment de la naissance. Selon le type d’accouchement, le microbiote intestinal du nourrisson va ressembler au microbiote vaginal (voie basse) ou au microbiote cutané (césarienne) de sa mère. la diversité et l’abondance du microbiote de l’enfant vont augmenter jusqu’à l’âge de 3 ans où ce dernier ressemble alors à celui d’un adulte. Durant cette période de colonisation et de développement, le microbiote intestinal est particulièrement instable et est donc très sensible aux facteurs environnementaux.
7
1. Le microbiote intestinal
Si le corps humain est colonisé par un très grand nombre de microorganismes, le tractus gastro-intestinal reste cependant le système qui présente la plus forte abondance microbienne
avec 1014 microorganismes, soit plus de 10 fois le nombre de cellules humaines (Sommer &
Bäckhed, 2013). Ce microbiote intestinal est très majoritairement composé de bactéries, les archées, les eucaryotes et les virus n’en représentant qu’une part minoritaire (Lozupone et al., 2012).
1.1. L’acquisition des communautés bactériennes
Le fœtus a longtemps été considéré comme stérile, il était ainsi admis que la colonisation microbienne du tractus digestif ne débutait qu’à la naissance. Cependant, certaines études ont montré que le liquide amniotique n’est pas stérile, y compris en dehors d’un état pathologique (Steel et al., 2005). Des bactéries ont également été détectées au niveau du cordon ombilical (Jiménez et al., 2005) et dans le méconium (premières selles du nouveau-né) (Jiménez et al., 2008; Hu et al., 2013). Ceci soutient donc l’hypothèse que des bactéries d’origine maternelle ont accès au fœtus in utero. Cependant aucun mécanisme n’a encore pu être mis en évidence (Arrieta et al., 2014). Le microbiote hébergé par le tractus digestif du fœtus reste très faible en
termes d’abondance, entre 100 et 101 bactéries/g de méconium, et est essentiellement constitué
d’entérocoques et de staphylocoques (Jiménez et al., 2008). L’acquisition des communautés bactériennes a par conséquent principalement lieu au cours de la naissance et durant l’enfance, en particulier durant les trois premières années de la vie. Cette colonisation est influencée par plusieurs facteurs tels que le mode de délivrance, l’allaitement, l’alimentation ou encore l’administration d’antibiotiques (Koenig et al., 2011) (Figure 1).
L’intestin du nouveau-né est un environnement aérobie au sein duquel seuls des microorganismes anaérobies aérotolérants peuvent se développer, comme certains membres de la famille des Enterobacteriaceae. Quelques jours après la naissance, la lumière intestinale s’appauvrit en oxygène laissant alors place à un environnement anaérobie et permettant la colonisation par des microorganismes anaérobies stricts tels que les Bifidobacterium, les
Clostridium et les Bacteroides (Arrieta et al., 2014). Durant les premières semaines de la vie,
le microbiote intestinal du nouveau-né ressemble au microbiote vaginal ou cutané de sa mère, c’est-à-dire au premier environnement auquel l’enfant est exposé au moment de la naissance.
8 En effet, le premier facteur influençant la composition du microbiote intestinal chez le nourrisson est le type d’accouchement pratiqué (Figure 1). Ainsi, lorsque la naissance a lieu par voie basse, le microbiote intestinal du nouveau-né ressemble au microbiote vaginal de sa mère et est dominé par les genres bactériens Lactobacillus et Prevotella, tandis que lors d’un accouchement par césarienne le microbiote va s’apparenter au microbiote cutané de sa mère et être dominé par les genres Staphylococcus, Corynebacterium et Propionibacterium (Dominguez-Bello et al., 2010). De la même manière, le système digestif des enfants nés par césarienne hébergent moins d’espèces de Bifidobacterium et de Bacteroidetes (Adlerberth et al., 2006; Penders et al., 2006; Biasucci et al., 2008). Cette différence semble perdurer dans le temps, puisqu’à deux ans la diversité du microbiote intestinal est toujours plus faible chez les enfants nés par césarienne par rapport aux enfants nés par voie naturelle (Jakobsson et al., 2013).
Le type d’alimentation est un autre facteur qui influence fortement la colonisation, l’abondance et la composition du microbiote intestinal chez l’enfant. Durant les premiers mois de sa vie, un nourrisson consomme exclusivement du lait, qui peut être d’origine maternelle et/ou artificielle. Le microbiote intestinal va être différent selon le type d’allaitement (Figure 1) notamment car les oligosaccharides présents dans le lait maternel vont favoriser la croissance de certains taxas bactériens tels que les Lactobacillus ou les Bifidobacterium (Harmsen et al., 2000; Conlon & Bird, 2015) (Figure 1). De plus, le lait maternel est une source de microorganismes et il est estimé qu’un nourrisson qui consomme environ 800 mL de lait
maternel par jour ingère entre 105 et 107 bactéries commensales (Heikkila & Saris, 2003; Martín
et al., 2003). L’origine de ces bactéries reste cependant incertaine. Lors de la tétée, le nourrisson est exposé au microbiote maternel cutané, mais des études ont montré que les bactéries retrouvées dans le lait maternel sont en grande partie différentes de celles du microbiote cutané présent au niveau du sein (Martín et al., 2003). Il a alors été suggéré que certaines bactéries retrouvées dans les selles de l’enfant proviendraient de l’intestin de la mère. Elles rejoindraient les glandes mammaires grâce à une voie entéro-mammaire qui impliquerait les cellules dendritiques et les macrophages maternels (Perez et al., 2007; Jost et al., 2014). Le type d’allaitement n’est pas le seul facteur alimentaire influençant la composition du microbiote intestinal. La période de sevrage avec l’introduction d’aliments solides est un événement majeur qui structure le microbiote intestinal chez le jeune enfant (Figure 1). Cette période de sevrage induit des changements importants au sein du microbiote intestinal de l’enfant car il est exposé à de nouveaux substrats, notamment à une gamme plus diversifiée d’hydrates de carbone, qui
Tableau 1 : Techniques utilisées pour caractériser le microbiote intestinal. (Adapté d’après Fraher,
O’Toole and Quigley, 2012)
Technique Description Avantages Inconvénients
Culture Isolement des bactéries sur milieux sélectifs. Coût peu élevé, semi-quantitatif. Laborieux, actuellement moins de 30% des bactéries intestinales sont cultivées.
qPCR Amplification et quantification du gène codant l’ARNr 16S. Détection de la fluorescence émise.
Identification phylogénétique, quantitatif, rapide.
Biais PCR, impossible d’identifier de nouvelles espèces.
DGGE/TGGE Amplicons du gène codant l’ARNr 16S séparés sur gel en utilisant un gradient de dénaturation ou de température.
Rapide, quantitatif, les bandes peuvent être excisées pour une analyse ultérieure.
Pas d’identification phylogénétique directe, Biais PCR.
T-RFLP Utilisation d’amorces marquées par fluorescence et d’enzymes de restriction pour digérer les amplicons du gène codant l’ARNr 16S générés. Les fragments digérés sont séparés grâce à un séquenceur capillaire.
Rapide, semi-quantitatif, coût peu élevé.
Biais PCR, faible résolution.
FISH Sondes oligonucléotiques marquées par un fluorochrome s’hybridant sur des séquences cibles complémentaires du gène codant l’ARNr 16S. Lorsque l’hybridation se produit, la fluorescence peut être dénombrée en utilisant la cytométrie en flux.
Identification phylogénétique, quantitatif, pas de biais PCR.
Dépendant des séquences des sondes, impossible d’identifier de nouvelles espèces.
Biopuce phylogénétique
Sondes oligonucléotiques marquées par un fluorochrome s’hybridant avec des séquences nucléotidiques complémentaires. Fluorescence détectée avec un laser.
Identification phylogénétique, quantitatif, rapide.
Hybridations croisées, les espèces présentes en très faible abondance peuvent être difficiles à détecter.
Clonage-séquençage du gène codant l’ARNr 16S
Clonage du gène complet codant l’ARNr 16S, séquençage Sanger suivi d’une électrophorèse capillaire.
Identification phylogénétique, quantitatif.
Biais PCR, laborieux, coûteux, biais de clonage.
Séquençage direct des amplicons du gène codant l’ARNr 16S
Séquençage massif parallèle d’amplicons partiels du gène codant l’ARNr 16S. Technologies de séquençage haut-débit (pyrosequençage 454, Illumina, SOLiD)
Identification phylogénétique, quantitatif, rapide, identification de nouvelles espèces possible.
Biais PCR, coûteux.
Séquençage massif du microbiome
Séquençage massif parallèle de l’ensemble des gènes du microbiome.
Identification phylogénétique, quantitatif, pas de biais PCR, identification de nouvelles espèces possible.
Coûteux, l’analyse des données générées est complexe et nécessite une puissance de calcul importante.
Abréviations : DGGE, denaturing gradient gel electrophoresis ; FISH, fluorescence in situ hybridization ; qPCR, quantative polymerase chain reaction, TGGE, temperature gradient gel electrophoresis, T-RFLP, terminal restriction fragment length polymorphism.
9 vont favoriser la croissance d’espèces bactériennes diversifiées (Fallani et al., 2011). L’introduction d’aliments solides et variés dans le régime alimentaire de l’enfant initierait la maturation du microbiote intestinal vers celui de l’adulte (Arrieta et al., 2014).
La densité et la diversité des populations bactériennes intestinales augmentent durant les premières années de la vie jusqu’à l’âge de trois ans où elles s’apparentent alors à celles de l’adulte (Koenig et al., 2011). Une fois arrivé à maturité et en dehors de tout facteur environnemental, le microbiote intestinal reste relativement stable à l’âge adulte excepté au cours du vieillissement, où une diminution de la diversité bactérienne est observée chez certaines personnes âgées et est à corréler avec un état de fragilité (Biagi et al., 2010; Claesson
et al., 2012).
1.2. Richesse et la diversité du microbiote intestinal
1.2.1. Méthodes d’analyse de la composition du microbiote intestinal
Les premières descriptions du microbiote intestinal ont été réalisées par approches
culturales à partir d’échantillons de selles dès le début du XXème siècle. Même si ces techniques
culturales ont été largement améliorées depuis les années 1960 (ex. cultures anaérobies, milieux sélectifs) (Moore & Holdeman, 1974), elles ne permettent qu’une vision limitée de la composition du microbiote intestinal puisque moins de 30% des bactéries intestinales sont à ce jour cultivées (Eckburg et al., 2005; Fraher et al., 2012).
Dans les années 1990, l’avènement des outils moléculaires a révolutionné notre compréhension du microbiote intestinal. Ces techniques moléculaires se sont tout d’abord basées sur l’analyse du gène codant l’ARN ribosomique (ARNr) 16S. Ce gène est utilisé comme marqueur phylogénétique car il possède à la fois des domaines conservés permettant son amplification par PCR, et des régions variables représentant des horloges moléculaires de l’histoire évolutive des organismes (Woese, 1987). Dans les techniques dites d’empreintes génétiques, les amplicons du gène codant l’ARNr 16S sont soumis soit à une séparation directe sur gel d’électrophorèse avec un gradient de dénaturation (DGGE, Denaturing Gradient Gel Electrophoresis) ou de température (TGGE, Temperature Gradient Gel Electrophoresis), soit digérés par des enzymes de restriction préalablement à la migration électrophorétique, comme dans la T-RFLP (Terminal Restriction Fragment Length Polymorphisme) permettant l’analyse des fragments terminaux (Tableau 1). Bien que quantitatives, ces approches d’empreinte génétique ne permettent pas une identification taxonomique directe des communautés
10 bactériennes. En revanche, les profils électrophorétiques obtenus permettent de comparer la structure du microbiote intestinal entre des individus sains (Zoetendal et al., 1998; Marsh, 1999) ou entre des patients atteints de pathologies différentes (Noor et al., 2010).
D’autres outils moléculaires utilisent la complémentarité entre une sonde spécifique et une région cible du gène codant l’ARNr 16S pour étudier la composition du microbiote intestinal (Tableau 1). Ces outils reposent sur la détection d’un signal fluorescent. Dans la technique FISH (Fluorescence In Situ Hybridization) ce sont les sondes qui sont marquées avec un composé fluorescent, tandis que dans l’approche biopuce ce sont les cibles qui sont marquées. Ces cibles peuvent être soit de l’ADN génomique soit des amplicons du gène codant l’ARNr 16S. Différentes biopuces phylogénétiques ont été mises au point pour étudier le microbiote intestinal comme la HITChip (Human Intestinal Tract Chip) (Rajilić-Stojanović et al., 2009) ou la HuGChip (Human Gut Chip) (Tottey et al., 2013). Ces approches basées sur l’hybridation entre une sonde spécifique et une région cible du gène codant l’ARNr 16S sont quantitatives et permettent une identification taxonomique des populations composant le microbiote intestinal mais uniquement pour les taxa déjà décrits dans les bases de données. En effet, la détermination des sondes nécessite de connaître les séquences à cibler. Comme ces techniques permettent de comparer facilement des échantillons entre eux, elles sont principalement utilisées pour mettre en évidence les différences dans la structure du microbiote. Par exemple, Bezirtzoglou et al. (2011) ont mis en évidence grâce à la technique FISH, que chez les nourrissons allaités avec du lait maternel, l’abondance des Bifidobacterium est deux fois supérieure à celle des enfants nourris avec du lait synthétique. Grâce à l’utilisation de la HITChip, de Goffau et al. (2014) ont montré que l’abondance des Bacteroidetes était augmentée chez les enfants diabétiques par rapport à des enfants sains.
Une quantification fiable des communautés microbiennes est possible grâce à l’approche de PCR en temps réel ou PCR quantitative (qPCR). Cette technique utilise des amorces spécifiques, ciblant des populations bactériennes à différents niveaux taxonomiques, et détecte la fluorescence émise soit par un composé qui fluoresce lorsqu’il se lie à l’ADN double brin soit par un fluorochrome. La fluorescence émise est proportionnelle à la quantité d’amplicons produits et permet ainsi la quantification des taxa ciblés. De nombreuses études ont appliqué cette approche pour décrire les variations dans la composition et l’abondance des communautés bactériennes intestinales, par exemple chez des individus d’âge (Mariat et al., 2009) ou atteints d’une pathologie telle que la maladie de Crohn (Sokol et al., 2008). Cet outil
11 ne permet pas de détecter de nouveaux taxa microbiens, puisque la détermination des amorces de qPCR nécessite de connaitre la séquence du gène codant l’ARNr 16S des taxa ciblés.
L’étude d’un écosystème aussi complexe que le microbiote intestinal nécessite une description plus précise des populations microbiennes qui le composent. Le séquençage du gène codant l’ARNr 16S permet d’identifier les microorganismes déjà décrits mais également de mettre en évidence de nouveaux taxa (Tableau 1). Jusque dans les années 1990, il était nécessaire de cloner les amplicons du gène codant l’ARNr 16S pour pouvoir individualiser chaque molécule et assurer son séquençage par la méthode Sanger. Cette approche dite de clonage-séquençage étant laborieuse, il a fallu attendre le développement des techniques de séquençage de nouvelle génération (NGS) pour que l’exploration du microbiote intestinal grâce au séquençage du gène codant l’ARNr 16S se généralise. Le séquençage massif direct des amplicons utilise des billes (pyroséquençage 454) ou des surfaces solides (Illumina et SOLiD, Sequencing by Oligonucleotide Ligation and Detection). Le pyroséquençage génère des séquences dont la longueur permet de couvrir, de manière individuelle, la plupart des régions variables de l’ARNr 16S (Claesson et al., 2009). Il a donc été largement utilisé pour décrire ou comparer la composition et l’abondance des communautés bactériennes intestinales (e.g. Brim et al., 2013; Durbán et al., 2012; Jeffery et al., 2012). Les progrès constants et le développement de nouvelles techniques de séquençage haut-débit ont permis d’augmenter la profondeur de séquençage. C’est notamment le cas de la technologie Illumina qui grâce au séquençage « paired-end », génère des séquences dont la longueur est comparable au pyroséquençage mais avec une meilleur qualité et un taux d’erreur plus faible (Rodrigue et al., 2010). Ce séquençage Illumina est de plus en plus utilisé pour caractériser le microbiote intestinal et pour comparer par exemple le microbiote des individus sains et atteints par le VIH (virus de l’immunodéficience humaine) (Lozupone et al., 2013), des personnes saines et obèses (Yasir et al., 2015) ou encore décrire les communautés bactériennes chez des patients atteints de rectocolite hémorragique (Walujkar et al., 2014). La technologie de séquençage SOLiD pour le séquençage des amplicons du gène codant l’ARNr 16S, bien que moins répandu, peut être utilisée pour l’étude du microbiote intestinal, car elle permet notamment d’obtenir une résolution plus importante au niveau de l’espèce (Mitra et al., 2013).
Le séquençage du gène codant l’ARNr 16S a considérable étendu la vision de la structure du microbiote intestinal, mais il ne permet qu’une identification taxonomique des communautés microbiennes. Le développement de la métagénomique, approche dans laquelle l’ensemble des gènes est séquencé, permet d’obtenir une vision globale et sans a priori du
Figure 2 : Vue d'ensemble des techniques utilisées pour caractériser le microbiote intestinal. (Modifiée
d’après Fraher, O’Toole and Quigley 2012)
Représentation schématique des outils disponibles pour étudier la composition et/ou l’abondance des communautés bactériennes du microbiote intestinal.
Abréviations : DGGE, denaturing gradient gel electrophoresis ; FISH, fluorescence in situ hybridization ; NGS, next-generation sequencing ; PCR, polymerase chain reaction ; TGGE, temperature gradient gel electrophoresis ; T-RFLP, terminal restriction fragment length polymorphism.
12 microbiote intestinal. En effet, elle permet d’identifier les taxa présents mais également d’explorer les potentialités métaboliques du microbiote intestinal et donc d’étudier son rôle dans la physiologie de l’hôte (Gill et al., 2006). Cependant, le microbiote intestinal est un écosystème très complexe et variable, ainsi pour en obtenir une description exhaustive il est nécessaire de conduire des approches de métagénomique avec une grande profondeur de séquençage et couvrant un échantillonnage important d’individus. C’est avec cet objectif que le consortium américain « the Human Microbiome Project » (HMP) et européen « MetaHIT » (the european union project on Metagenomics of the Human Intestinal Tract) ont été constitués. Le consortium américain HMP a été fondé en 2008 par le NIH (National Institut of Health). Ce projet vise à décrire de manière exhaustive les microbiomes humains intestinaux, oraux, respiratoires, cutanés et vaginaux de 242 donneurs sains, au travers de l’échantillonnage de 15 (pour les hommes) et 18 (pour les femmes) sites anatomiques (The Human Microbiome Project, 2012a). Le consortium européen MetaHIT a lui été constitué afin d’explorer uniquement le microbiote intestinal et en particulier son association avec l’état de santé des individus. Majoritairement financé par l’Union Européenne (UE), ce consortium a été fondé en 2008 et regroupe huit pays différents (la France, l’Allemagne, l’Espagne, l’Italie, le Danemark, les Pays-Bas, le Royaume-Uni et la Chine). Son objectif est de réaliser un catalogue des gènes portés par le microbiote intestinal grâce à des approches de métagénomique conduites sur une cohorte de 370 individus (sains, obèses ou atteints de maladies inflammatoires chroniques de l’intestin) (Ehrlich & The MetaHIT Consortium, 2011).
L’ensemble des outils moléculaires appliqués au microbiote intestinal a permis d’améliorer notre connaissance de cet écosystème complexe (Figure 2). Si les approches de métagénomique permettent une exploration taxonomique et fonctionnelle du microbiote intestinal, la profondeur de séquençage qu’elles nécessitent engendre des coûts encore très élevés. Les études menées sur le microbiote intestinal se limitent donc généralement à une description phylogénétique basée sur l’amplification du gène codant l’ARNr 16S (séquençage NGS, qPCR…).
1.2.2. Gradient longitudinal de richesse et de diversité
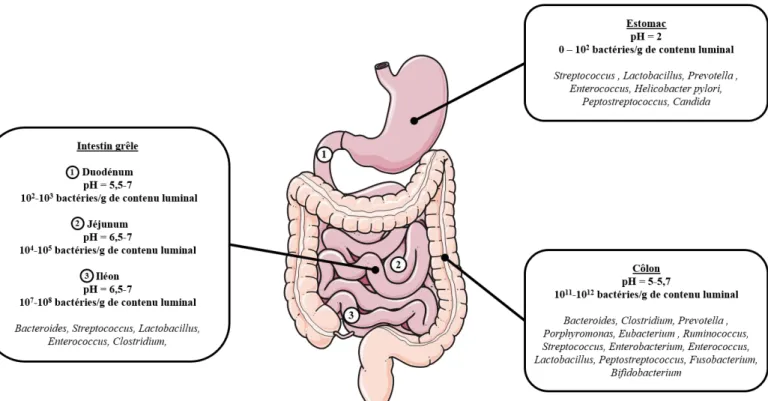
Le tractus digestif est un tube musculeux continu qui assure la digestion des aliments. Pour assurer ce processus, le tube digestif est divisé en plusieurs compartiments : la bouche, l’œsophage, l’estomac, l’intestin grêle, le côlon et le rectum. Chaque segment du tube digestif exerce une fonction différente dans la digestion, comme l’estomac qui est entre autre
Figure 3 : Réparation du microbiote au sein du tractus digestif. (Modifiée d’après Koboziev et al. 2014
et Jandhyala et al. 2015)
La concentration bactérienne augmente de l’estomac jusqu’au côlon, pour atteindre 1012 bactéries/g de
contenu luminal. Les principaux genres bactériens dominant le microbiote de chaque organe sont également indiqués.
13 responsable du brassage des aliments, l’intestin grêle de l’absorption des nutriments ou encore le côlon dans lequel l’eau est réabsorbée et les fèces formées. Les différents compartiments du tractus digestif ont donc des caractéristiques différentes ce qui crée des niches écologiques variées permettant la colonisation par les microorganismes. Ainsi, un gradient croissant longitudinal (oro-anal) est observé aussi bien en terme de densité que de diversité des populations bactériennes (O’Hara & Shanahan, 2006) (Figure 3). Le microbiote stomacal ne
comprend que 0 à 102 bactéries/g de contenu luminal, car le pH très acide de l’estomac et un
temps de transit rapide du bol alimentaire tendent à limiter la croissance des microorganismes (Walter and Ley, 2011). La densité bactérienne augmente progressivement de
102-103 bactéries/g de contenu luminal dans le duodénum (partie proximale de l’intestin grêle)
à 107-108 bactéries/g de contenu luminal dans l’iléon distal et atteint un maximum de
1012 bactéries/g de contenu luminal au niveau colique (Koboziev et al., 2014). Le côlon
représente ainsi le compartiment intestinal le plus densément peuplé mais également le plus diversifié avec de nombreux genres bactériens détectés tels que Bacteroides, Clostridium, Prevotella, Porphyromonas, Ruminococcus, Lactobacillus, ou encore Bifidobacterium (Figure 3).
1.2.3. Le microbiote fécal
La très grande majorité des études sur la diversité et la richesse du microbiote intestinal est menée à partir d’échantillons de selles, ces derniers étant les plus faciles à collecter. Il est généralement admis que la composition du microbiote fécal reflète celle du microbiote luminal au niveau du côlon distal.
Parmi les populations bactériennes retrouvées au sein du microbiote fécal, 90% appartiennent à deux phyla : les Firmicutes et les Bacteroidetes (Jeffery et al., 2012). Le phylum des Firmicutes est toujours fortement représenté, notamment par des espèces appartenant aux genres Eubacterium, Clostridium, Ruminococcus, et Butyvibrio. Les membres du genre Bacteroides, appartenant au phylum des Bacteroidetes, sont également abondamment retrouvés (Doré & Corthier, 2010). Les Actinobacteria constituent généralement le troisième phylum dominant du microbiote fécal, et sont principalement représentés par des membres du genre Bifidobacterium (Doré & Corthier, 2010). D’autres phyla bactériens minoritaires sont fréquemment détectés tels que les Proteobacteria, les Fusobacteria et les Verrucomicrobia (Silva et al., 2015). Cette forte conservation du microbiote intestinal au niveau du phylum, avec moins d’une dizaine de phyla représentés sur les 55 actuellement décris (Ley et al., 2006;
14 Geuking et al., 2014), indique l’existence d’une forte pression de sélection au sein de l’environnement intestinal (Schippa & Conte, 2014). Le tractus intestinal apparait donc comme une niche écologique hautement spécialisée dont les membres ont été sélectionnés au cours de l’évolution (Geuking et al., 2014). Cependant, si les bactéries intestinales appartiennent à un nombre restreint de phyla, elles constituent en revanche une richesse importante en espèces. Un seul individu abrite en effet au moins 160 espèces bactériennes différentes, ce qui représente environ 1 150 espèces à l’échelle de la population humaine (Qin et al., 2010).
1.2.4. Le microbiote associé à la muqueuse
De la même manière que le microbiote hébergé par le tractus digestif présente un gradient longitudinal croissant de diversité et de densité bactérienne de l’œsophage à l’anus, le microbiote intestinal montre un gradient axial. Bien que de nombreuses espèces bactériennes diversifiées peuplent la lumière de l’intestin, très peu adhèrent et résident dans la couche de mucus. Cette dernière, produite par les cellules caliciformes, comprend deux parties : la plus interne fortement réticulée qui adhère intimement à l’épithélium et est imperméable aux bactéries, tandis que la plus externe moins réticulée abrite des bactéries commensales (Natividad & Verdu, 2013). La couche de mucus forme une niche écologique particulière colonisée par des espèces bactériennes spécialisées dont certaines protéobactéries (Sommer & Bäckhed, 2013) et des taxa mucolytiques comme Akkermansia muciniphila (Derrien et al., 2004) et Mucispirillum (Wang et al., 2010).
Un gradient d’oxygène est également présent au niveau de la muqueuse intestinale. Effectivement, la concentration en oxygène diminue fortement et rapidement depuis l’épithélium jusqu’à la lumière intestinale, où seuls des taxa anaérobies stricts sont retrouvés (Espey, 2013). La présence de cette zone micro-oxygénée proche de l’épithélium permet le développement de bactéries micro-aérophiles et anaérobies aérotolérants (Espey, 2013), appartenant notamment aux genres Clostridium, Lactobacillus et Enterococcus (Jandhyala et
al., 2015).
Les communautés bactériennes présentes dans la lumière intestinale diffèrent donc nettement de celles associées à la muqueuse. Or, ces dernières, de par leur proximité avec l’épithélium sous-jacent, pourraient jouer un rôle complètement différent de celui des bactéries luminales, notamment dans les échanges d’éléments nutritifs et l’induction du système immunitaire inné de l’hôte (Eckburg et al., 2005). L’analyse du microbiote associé à la
15 muqueuse serait donc plus pertinente que celle du microbiote fécal. Cependant, l’échantillonnage des communautés bactériennes associées à la muqueuse, par biopsie du tissu intestinal sous endoscopie, est un acte invasif et donc plus difficile à mettre en œuvre qu’une simple collecte de fèces, en particulier chez des donneurs sains. La richesse et la diversité du microbiote associé à la muqueuse sont donc moins explorées que celles du microbiote fécal.
1.2.5. Les facteurs influençant la composition du microbiote intestinal
Les différents travaux visant à étudier la composition du microbiote intestinal ont cependant mis en exergue que ce dernier n’est pas identique d’une personne à l’autre. Chaque individu possède un microbiote intestinal unique et héberge une combinaison d’espèces bactériennes qui lui est propre (Holmes et al., 2012). Une grande variabilité dans la composition mais également dans l’abondance des taxas bactériens est observée entre les individus (Bäckhed
et al., 2012). Ainsi, l’abondance des Firmicutes et Bacteroidetes varie au sein de la population
humaine, le microbiote intestinal d’un individu pouvant être dominé par l’un ou l’autre de ces deux phyla (Arumugam et al., 2011 ; The Human Microbiome Project consortium, 2012). De nombreux éléments sont évoqués pour expliquer cette variabilité tels que des facteurs génétiques ou environnementaux. Parmi ces derniers l’alimentation ainsi que l’antibiothérapie sont les plus documentés (Koboziev et al., 2014; Jandhyala et al., 2015).
L’alimentation est un paramètre qui influence de manière radicale la composition du microbiote intestinal avec des structures différentes observées en fonction du type de régime alimentaire. Ainsi, une prévalence plus importante des phyla Actinobacteria et Bacteroidetes est observée chez des personnes ayant un régime alimentaire riche en fibres et pauvre en graisses, tandis que les phyla Firmicutes et Proteobacteria prédominent dans un régime riche en graisses et pauvre en fibres (Silva et al., 2015). De plus, le passage d’un régime riche en graisses et en sucres à une régime riche en fibres induit des changements notables et rapides au sein du microbiote intestinal (Jumpertz et al., 2011; Wu et al., 2011). Les taxa affectés par ce changement de régime alimentaire diffèrent selon les individus (Wu et al., 2011).
L’administration d’antibiotiques est un autre facteur largement reconnu pour influencer la composition du microbiote intestinal. Les antibiotiques sont employés afin d’éliminer les bactéries pathogènes responsables de maladies infectieuses. Cependant, ils provoquent également un fort dérèglement du microbiote intestinal en créant une pression de sélection qui favorise le développement de certains microorganismes au détriment de la croissance d’autres






![Figure 7 : Devenir du benzo[a]pyrène absorbé par voie orale dans l'organisme. (Modifiée d’après Ramesh et al](https://thumb-eu.123doks.com/thumbv2/123doknet/14587443.541722/93.892.217.675.145.931/figure-devenir-benzo-pyrène-absorbé-organisme-modifiée-ramesh.webp)
