Speciation of naturally-accumulated uranium in an organic-rich soil of an alpine region (Switzerland)
17
0
0
Texte intégral
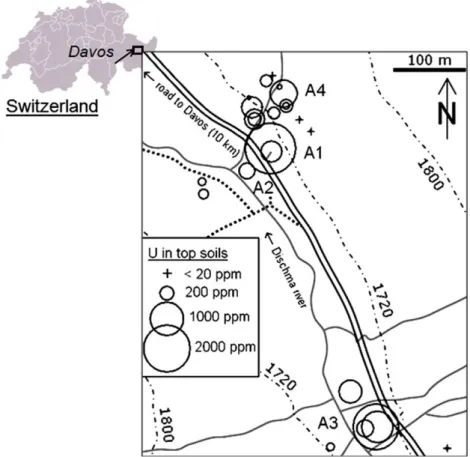
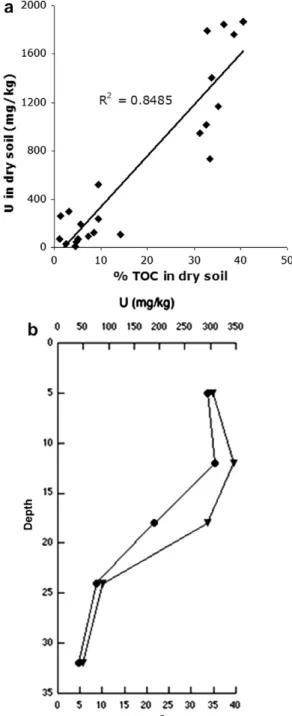
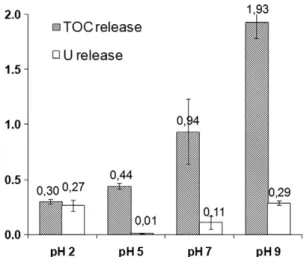
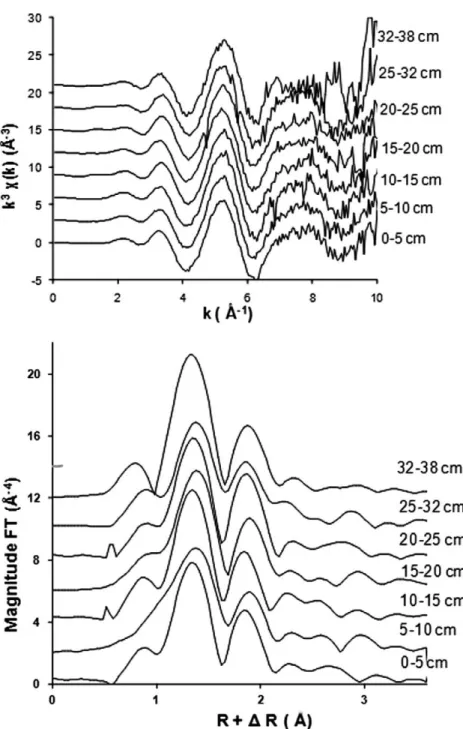
Figure


+5


Documents relatifs