RESEARCH OUTPUTS / RÉSULTATS DE RECHERCHE
Author(s) - Auteur(s) :
Publication date - Date de publication :
Permanent link - Permalien :
Rights / License - Licence de droit d’auteur :
Bibliothèque Universitaire Moretus Plantin
Institutional Repository - Research Portal
Dépôt Institutionnel - Portail de la Recherche
researchportal.unamur.be
University of Namur
Taxol-induced unfolded protein response activation in breast cancer cells exposed to
hypoxia. ATF4 activation regulates autophagy and inhibits apoptosis
Notte, Annick; Rebucci, Magali; Fransolet, Maude; Roegiers, Edith; Genin, Marie; Tellier,
Celine; Watillon, Kassandra; Fattaccioli, Antoine; Arnould, Thierry; Michiels, Carine
Published in:
The international journal of Biochemistry & Cell biology
DOI:
10.1016/j.biocel.2015.02.010
Publication date:
2015
Document Version
Publisher's PDF, also known as Version of record
Link to publication
Citation for pulished version (HARVARD):
Notte, A, Rebucci, M, Fransolet, M, Roegiers, E, Genin, M, Tellier, C, Watillon, K, Fattaccioli, A, Arnould, T &
Michiels, C 2015, 'Taxol-induced unfolded protein response activation in breast cancer cells exposed to hypoxia.
ATF4 activation regulates autophagy and inhibits apoptosis', The international journal of Biochemistry & Cell
biology, vol. 62, pp. 1-14. https://doi.org/10.1016/j.biocel.2015.02.010
General rights
Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain
• You may freely distribute the URL identifying the publication in the public portal ? Take down policy
If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim.
ContentslistsavailableatScienceDirect
The
International
Journal
of
Biochemistry
&
Cell
Biology
j ou rn a l h o m ep a g e :w w w . e l s e v i e r . c o m / l o c a t e / b i o c e l
Taxol-induced
unfolded
protein
response
activation
in
breast
cancer
cells
exposed
to
hypoxia:
ATF4
activation
regulates
autophagy
and
inhibits
apoptosis
Annick
Notte
1,
Magali
Rebucci
1,
Maude
Fransolet,
Edith
Roegiers,
Marie
Genin,
Celine
Tellier,
Kassandra
Watillon,
Antoine
Fattaccioli,
Thierry
Arnould,
Carine
Michiels
∗LaboratoryofBiochemistryandCellularBiology(URBC),NAmurResearchInstituteforLifeScience(NARILIS),UniversityofNamur,61ruedeBruxelles, 5000Namur,Belgium
a
r
t
i
c
l
e
i
n
f
o
Articlehistory: Received26August2014
Receivedinrevisedform4February2015 Accepted17February2015
Availableonline24February2015 Keywords: Breastcancer Hypoxia UPR ATF4 Autophagy Apoptosis
a
b
s
t
r
a
c
t
Understandingthemechanismsresponsiblefortheresistanceagainstchemotherapy-inducedcelldeath isstillofgreatinterestsincethenumberofpatientswithcancerincreasesandrelapseiscommonly observed.Indeed,thedevelopmentofhypoxicregionsaswellasUPR(unfoldedproteinresponse) acti-vationisknowntopromotecancercelladaptiveresponsestothestressfultumormicroenvironmentand resistanceagainstanticancertherapies.Therefore,theimpactofUPRcombinedtohypoxiaonautophagy andapoptosisactivationduringtaxolexposurewasinvestigatedinMDA–MB-231andT47Dbreastcancer cells.TheresultsshowedthattaxolrapidlyinducedUPRactivationandthathypoxiamodulated taxol-inducedUPRactivationdifferentlyaccordingtothedifferentUPRpathways(PERK,ATF6,andIRE1␣).The putativeinvolvementofthesesignalingpathwaysinautophagyorinapoptosisregulationinresponseto taxolexposurewasinvestigated.However,whilenolinkbetweentheactivationofthesethreeERstress sensorsandautophagyorapoptosisregulationcouldbeevidenced,resultsshowedthatATF4activation, whichoccursindependentlyofUPRactivation,wasinvolvedintaxol-inducedautophagycompletion.In addition,anATF4-dependentmechanismleadingtocancercelladaptationandresistanceagainst taxol-inducedcelldeathwasevidenced.Finally,ourresultsdemonstratethatexpressionofATF4,inassociation withhypoxia-inducedgenes,canbeusedasabiomarkerofapoorprognosisforhumanbreastcancer patientssupportingtheconclusionthatATF4mightplayanimportantroleinadaptationandresistance ofbreastcancercellstochemotherapyinhypoxictumors.
©2015ElsevierLtd.Allrightsreserved.
1. Introduction
Resistance to chemotherapy is a major current issue in the outcome ofcancertreatment. Themechanisms leadingtodrug resistancearecomplexandmultifactorial.Althoughtheinadequate drugconcentrationatthetumorsitecancontributetoclinical resis-tance,intrinsic cellularmechanismsora reprogrammingofthe intracellularsignaling pathwayscausedby themodifications of thetumormicroenvironmentcontribute,foramajorpart,todrug
Abbreviations: ATF4,activatingtranscriptionfactor4;ATF6,activating tran-scriptionfactor6;ER,endoplasmicreticulum;IRE1␣,inositol-requiringenzyme-1␣; PERK,proteinkinasedouble-strandedRNA-dependent(PRK)-likeERkinase;UPR, unfoldedproteinresponse.
∗ Correspondingauthor.Tel.:+3281724131;fax:+3281724135. E-mailaddress:carine.michiels@unamur.be(C.Michiels).
1 Theseauthorscontributedequallytothiswork.
resistance(Gattiand Zunino,2005).Oneof thesemodifications isthepresenceofhypoxicareasinthetumormicroenvironment ofsolidtumors.Theresponsesinducedbyhypoxiaresultincell adaptationandresistancetothechemotherapy-inducedcelldeath. One ofthe mostcharacterizedtranscription factorsresponsible forthehypoxic responseisthehypoxiainduciblefactors(HIFs) (Semenza,2010;RohwerandCramer,2011).However,anumberof HIFindependentmechanismsarealsoabletopromotecancercell adaptationandsurvivalandareofgreatinteresttofurtherimprove cancertreatmentsandoutcomes.(RouschopandWouters,2009)In thiswork,twobiologicalprocessesconnectedtocelldeathand/or stressadaptiveresponseshavebeeninvestigated:autophagyand unfoldedproteinresponse(UPR).
Autophagy is a degradation process in which cells form double-membranevesicles,calledautophagosomes,sequestering a portionof cytoplasm. These autophagosomes ultimately fuse with lysosomes, resulting in the degradation of their content allowingtherecycling of smallmolecules. Autophagy initiation http://dx.doi.org/10.1016/j.biocel.2015.02.010
andcompletionrequiresmTOR(mammaliantargetofrapamycin) inhibition,therecruitmentandactivationofVps34(class-III phos-phoinositide3-kinase)withinaproteincomplexcontainingbeclin 1,severalproteinsencodedbyAtg(Autophagyrelatedgenes)and twoconjugationsystemsleadingtoAtg12–Atg5aswellasLC3-PE (PhosphatidylEthanolamine)conjugation(Mehrpouretal.,2010). Inbasal and activatedconditions,autophagycontributes tothe eliminationofdamagedorganellesaswellastotheturnoverof long-livedproteins.Inaddition,autophagycanalsobestimulated inresponse todifferent conditionsof stress suchasstarvation, hypoxia,oxidativestress,drugtreatmentorDNAdamage(Heand Klionsky,2009).Inbothcases,autophagyrepresentsapro-survival mechanismallowingqualitycontrol,metabolicreprogrammingor cellfitnessafterastress(Semenza,2010;Mehrpouretal.,2010; Codognoand Meijer,2005; Shannonet al.,2003;Brahimi-Horn andPouyssegur,2006).Moreover,evidencethatautophagyplays aroleinmaintainingcancercellsurvivalintheir microenviron-mentwherenutrientsandoxygenarereducedorinresponseto anticancerchemotherapieshavebeenreported(Notteetal.,2013; Checinskaand Soengas,2011;Notteetal.,2011;Morsellietal., 2009;Shenetal.,2012;White,2012;Songetal.,2009;Zhangetal., 2008).
Thesecondsignalingmechanismthatcouldplayarolein can-cercell resistanceis UPR that is activatedby ER(endoplasmic reticulum)stresses. UPRis induced byseveral perturbations in ERhomeostasis (change in cellular energy, redoxstate or Ca2+
homeostasis) leading to the accumulation of mis- or unfolded proteins in the ER compartment. In mammalian cells, UPR is transducedbythreeERtransmembraneproteinsensorsnamely PERK(proteinkinaseRNA(PKR)-likeERkinase),IRE1␣ (inositol-requiringenzyme-1)andATF6(activatingtranscriptionfactor-6) (Ron and Walter, 2007). The ER chaperone GRP78/BIP (78kDa glucose-regulatedprotein/immunoglobulin heavy chain-binding protein)bindstotheluminaldomainsofPERK,IRE1␣,andATF6in unstressedcellstomaintaintheminaninactivestate(Shenetal., 2002;Bertolottietal.,2000).Unfoldedproteinloadtriggersthe dissociationofGRP78/BIPfromthesesensorstriggeringtheUPR activation.PERKthenformshomodimersandisactivatedby trans-autophosphorylation.PERKphosphorylatesthealpha-subunit of eIF2(eukaryotic translationinitiationfactor-2)leadingto trans-lationalattenuationofcappedmRNAandthustoproteinsynthesis inhibition topreventadditionalaccumulation ofnewly synthe-sizedproteinsintotheER.However,PERKsignalingalsoselectively inducestheexpressionofATF4(activatingtranscriptionfactor-4) throughthebypassofamicro-ORFupstreamofATF4open read-ingframe(Hardingetal.,2000).IRE1containsaserine/threonine proteinkinasedomainandan endoribonucleasedomainwithin theC-terminalcytosolic region.Activationof IRE1is driven by itsoligomerizationandtrans-autophosphorylation.Onceactivated, IRE1splicesanunconventionalintronfromtheXBP1(X-box bind-ingprotein-1)mRNAtogenerateanactivesplicedformofXBP1 transcriptionfactorthatcontrolstheexpressionofUPRtargetgenes leadingtoanadaptive response.UponERstress,ATF6is trans-portedtotheGolgiapparatuswhereitiscleaved generatingan activefragmentcontainingaDNAbindingdomain.CleavedATF6 translocatetothenucleusandactivatesthetranscriptionofUPR targetgenes(Bravoetal.,2013;Chakrabartietal.,2011;Walter andRon,2011;Uranoetal.,2000).TheprimaryfunctionofUPR isthustoreestablishERhomeostasisbyloweringprotein synthe-sisand translocation into theER,by increasingthe capacity of theERtohandleunfolded proteinsand bytheremovalof mis-foldedproteinsviatheERAD(ER-associatedproteindegradation) system(Traversetal.,2000).WhentheERADsystemissaturated andunabletodegrademisfoldedoraggregatedproteins,UPRcan alsoinduceautophagyasanalternative mechanismfor protein degradation(YorimitsuandKlionsky,2007)PERK,IRE1␣,andATF6
signalingpathwaysarenowknowntocontrolautophagy(Deegan et al., 2012; Hoyer-Hansen and Jaattela, 2007; Kouroku et al., 2007;Garcia-Navasetal.,2012).Ifactivationofthesepro-survival mechanismsisinsufficienttoovercometheERstress,prolonged activationofUPRtriggersapoptosis(Vannuveletal.,2013;Szegezdi etal.,2006,2009;Heath-Engeletal.,2008).Interestingly,thehigh proliferativerateofcancercellsgeneratesastressfulhypoxic envi-ronmentinwhichUPRisoftenchronicallyactivatedandactsasa pro-survivalmechanism(Rouschopetal.,2010;Koumenis,2006; Bietal.,2005;Romero-Ramirezetal.,2004;Shajahanetal.,2009). While the role of UPR in chemotherapy resistancehas already beendemonstratedinseveralworks(Jiangetal.,2008;Fengetal., 2011;Rzymskietal.,2009;Milanietal.,2009;LiandLee,2006; Wangetal.,2009;Kumandanetal.,2013;Chenetal.,2011;Mann andHendershot,2006),ithasneverbeenstudiedincombination withahypoxicenvironmentintheresistanceagainsttaxol-induced apoptosis.SinceUPRisinvolved intheregulationof autophagy andapoptosis(Kiviluotoetal.,2013),a betterunderstandingof themechanismsbywhichUPRcontrolscellfatedecisioninthese conditionsrepresentsanecessarystepinthefieldofanti-cancer therapy(Healyetal.,2009;Schonthal,2013).
Wepreviouslyshowedthatapoptosisisactivatedin MDA–MB-231breastcancercellsincubatedwithtaxol(Notteetal.,2013).In theseconditions,autophagyisactivatedmuchearlierthan apopto-sisandisaprotectivemechanism.MDA–MB-231cellsincubated withtaxolunderhypoxiaaremoreresistantthancellsincubated withtheanticanceragentalone,becausehypoxiastimulatesamore effectiveautophagicflow(Notteetal.,2013).Inthepresentwork, we studiedwhethertaxol-inducedUPR activationwould bean upstreameventleadingtotheactivationofautophagyand/or apo-ptosis.TheroleofUPRinthehypoxia-inducedresistanceagainst taxol-inducedcelldeathanditslinkwithautophagyand apopto-sisactivationwerethusinvestigated.Resultsshowaspecificrole forATF4intaxol-inducedautophagyasamechanismpromoting cancercelladaptationandsurvivalunderhypoxia.
2. Materialandmethods
2.1. Cellcultureandhypoxiaincubation
HumanbreastcancercellsMDA–MB-231weremaintainedin culturein75cm2polystyreneflasks(Costar,Lowell,MA,USA)with
15ml of Roswell Park Memorial Institutemedium (RPMI1640 Invitrogen,Carlsbad,CA,USA)containing10%offetalcalfserum (Invitrogen).HumanbreastcancercellsT47Dweremaintainedin culturein75cm2polystyreneflasks(Costar,Lowell,MA,USA)with
15mlofDulbecco’smodifiedEaglemedium(DMEM,Invitrogen) containing10%offetalcalfserum(Invitrogen)and1%glutamine (Sigma,St.Louis,USA).Bothcelltypeswereincubatedunderan atmospherecontaining5%CO2.
For hypoxia experiments (1% O2), cells were incubated in
serum-freeCO2-independentmedium(Invitrogen)supplemented
with1mML-glutamine(Sigma)withorwithout50Mpaclitaxel (MolecularProbes,Carlsbad,CA,USA)at50M.Normoxiccontrol cellswereincubatedinthesameconditionsbutinnormal atmo-sphere(20%O2).
2.2. Plasmidtransfectionanddual-luciferasereporterassay Cellswereseededin12-wellcultureplates(100,000cells/well) 24hbeforebeingtransfectedwith0.9g/welloftheplasmid con-tainingthefireflyluciferasegeneand0.1g/welloftheplasmid containingtherenillaluciferasegene(fornormalizationof transfec-tionefficiency)usingthelipofectaminetransfectionreagent(from Invitrogen).ATF6activitywasmeasuredusingaluciferasereporter
geneconstructinwhichthefireflyluciferasegeneisdrivenbya promotercontainingfivecopiesoftheATF6/XBP1bindingelement (Zengetal.,2004).ATF6reporterplasmidwasalsousedtoconfirm ATF6invalidation.Intheseexperimentalconditions,cellswere co-transfectedwith50nMofATF6siRNA.Transfectionmediumwas removedandreplacedbycompletemediumfor24h.Cellswere thenincubatedaspreviouslydescribed(Notteetal.,2013).After incubation,theluciferasereporterassaywasperformedusingthe dual-luciferasereporterassaysystemkit(E1910,fromPromega) accordingtothemanufacturer’sinstructions.
2.3. Caspase3activityassay
ThefluorogenicsubstrateAc-DEVD-AFCwasusedtomeasure caspase3activityaccordingtoLozanoetal.(2001)Cellextracts werepreparedasdescribedbyWellingtonetal.(1998).Cellswere seededin25cm2 polystyreneflasks(Costar) (800,000cells/well)
24hbeforetheincubation.Aftertheincubation,themediumwas recoveredandcentrifugedat1200rpmfor5min.Cellsthatwere stillattachedtothewellwerescrappedin 500lcold PBSand recoveredintoamicrotube. Pelleteddetachedcells were resus-pendedin100lPBSat4◦Candpooledwithrespectiveattached cellsample tothe microtube.The sampleswere centrifugedat 1200rpmfor5min.at4◦Candthepelletresuspendedin50lof lysisbuffer(10mMHepes/KOH,pH7.0,10%sucrose,2mMEDTA, 0.1%CHAPS,5mMdithiothreitol,and10g/mlaprotinin).After incubationonarotatingwheelfor15minat4◦C,thelysateswere centrifugedat13,000rpmfor5minat4◦Candthesupernatants wererecoveredfortheassay.
Theproteinconcentrationwasdetermined(Pierce660,Thermo Scientific)and10gofproteinscompletedtoavolumeof75l withlysisbufferweremixedwith1lAc-DEVD-AFC(BD Pharmin-gen, 556574) and 75l reaction buffer (40mM PIPES, pH 7.2, 200mMNaCl,2mMEDTA,0.2%CHAPS,0.10%sucrose,and10mM dithiothreitol).Thereactionwasallowedtotakeplacefor1hat 37◦C andthefluorescencegenerated bythereleaseofthe fluo-rogenicgroupAFCaftercleavageby caspase3/7wasmeasured ina spectrophotometrer (excitation: 400nm andemission : 505nm).
2.4. LDHreleaseassay
LDHreleaseassayhasbeendescribedinNotteetal.(2013). 2.5. Westernblotting
ProcedureforWesternblottinganalyseshasbeendescribedin
Notteetal.(2013).Primaryantibodiesusedarereferencedin sup-plementaryTable1.
2.6. RT-qPCR
ProcedureforRT-qPCRhasbeendescribedinNotteetal.(2013). ForwardandreverseprimersforRPL13A,PERK,IRE1␣,andATF6 were designed using the Primer Express 1.5 software (Applied Biosystem).
2.7. siRNAtransfection
Silencing of PERK, IRE1␣, ATF6, and ATF4 expression was achieved using ON-TARGET plus SMARTpool human PERK (L004883,fromDharmacon),ON-TARGETplusSMARTpoolhuman ERN1 (IRE1␣) (L004951, from Dharmacon), ON-TARGET plus SMARTpoolhumanATF6(L009917,fromDharmacon),ON-TARGET plusSMARTpool humanCREB2(ATF4)(L005225,from Dharma-con),ON-TARGETplusSMARTpoolhumanGCN2(L005314,from
Dharmacon),ON-TARGETplusSMARTpoolhumanHRI(L005007, fromDharmacon),and ON-TARGETplusSMARTpool humanPKR (L003527, from Dharmacon). Risc-ree control siRNA purchased fromDharmaconwasusedasanegativecontrol.ForsiRNA exper-iments, 8×105 cells were seeded in 25cm2 polystyrene flasks
(Costar)with4mlofRPMI1640medium(RPMI)containing10%of fetalcalfserumandincubatedfor24hunderanatmosphere con-taining5%CO2.Cellswerethentransfectedfor24hunderstandard
cultureconditionswith50nMsiRNAwhentransfectedalone,or 25nMofeachsiRNAwhenco-transfectedusingtheDharmaFECT1 (Dharmacon)transfectionreagentaccordingtothemanufacturer’s instructions.Thetransfectionmediawereremovedandreplaced byculturemediafor24h.Cellswerethenincubatedaspreviously described(Notteetal.,2013).
2.8. Kaplan–Meiercurves
AnAffymetrixdatasetcontaininggene expressiondata from 249breastcancerpatientsforwhomcompletedemographic, histo-logicalgrade,stage,differentiationinformationareavailable,was usedforsurvivalstudies(downloadedfromGEO,GSE4922, Upp-salacohort).ProceduresofRNAextractionandhybridizationare describedinIvshinaetal.(2006).RawdatainCELfilesarepublicly availableandwereprocessedaccordingtothemethoddefinedby
Pierreetal.(2010),byusingalternativeCDFsfromAffyProbeMiner andGCRMAfornormalizationandsummarizationofdownloaded dataintheRstatisticalenvironment.Kaplan–Meiercurveswere drawn in theR statistical environment for each subgroup. 249 patientswereseparatedintwoequalsubgroups(n=124/n=125) accordingtotheexpressionofone gene.Thesesubgroupswere thensubdividedwhencombinationsofseveralgeneexpressions werestudied.
2.9. Statisticalanalysis
SigmaStatsoftware(JandleScientific,Germany)wasusedforthe statisticalanalysis.Dataarepresentedasmeans±standard devia-tion(SD)andwereevaluatedbyone-wayortwo-wayANOVA,using theHolm–Sidakmethod.
3. Results
3.1. PERK,IRE1˛,andATF6pathwaysareactivatedinbreast cancercelllinesduringtaxolexposureundernormoxiaand hypoxia
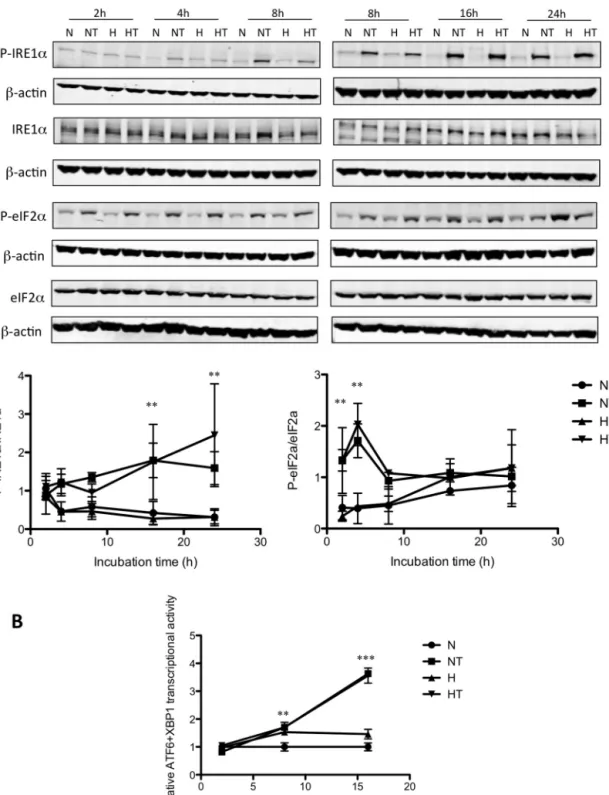
To examine whether taxol induces an ER stress in breast cancer cells, activation of the three branches of the UPR was studied. MDA–MB-231cells wereincubated under normoxia or hypoxiawithor without50Mtaxol.Phosphorylation ofeIF2␣ and IRE1␣wasassessed, byWestern analysis,after2, 4,8, 16, and 24h ofincubation (Fig.1A). Theresultsshowed that taxol induced IRE1␣ phosphorylation already after 4h of incubation undernormoxiaorhypoxia.Thelevelofphosphorylationincreased withtimeandbecamestatisticallysignificantat16h.Moreover, taxol also induced the phosphorylation of eIF2␣: a significant increase of respectively 3.5 and 5 fold was observed after 2 and4hofincubation.HypoxiaaloneinducedeIF2␣ phosphory-lation asobserved after24hof incubation. For longincubation times,taxol-induced eIF2␣phosphorylationdecreased. Hypoxia didnot influenceeIF2␣and IRE1␣phosphorylationinducedby taxol.
Inordertoconfirmtheseresults,weusedanotherbreastcancer cellline,T47Dcells.Thiscelllineisalsoprotectedagainst taxol-inducedapoptosisand celldeathbyhypoxiaasevidenced bya reduced activationofcaspase3and PARPcleavageaswellasa
Fig.1. PERK,IRE1␣,andATF6pathwaysareactivatedaftertaxolexposureundernormoxiaandhypoxiainMDA-MB-2331cells.MDA–MB-231cellswereincubatedunder normoxia(N)orhypoxia(H)withoutorwithtaxol(T)at50M.A.After2,4,8,16,and24hofincubation,theabundanceofphospho-eIF2␣,eIF2␣,phospho-IRE1␣,and IRE1␣wasassessedintotalcellextractsbywesternblottinganalysis,usingspecificantibodies.-actinwasusedtoassessthetotalamountofproteinsloadedonthegel. Uncroppedwesternblotsarepresentedinthesupplementaryfigure6.Thegraphsbelowrepresentthequantificationofphospho-IRE1␣abundancenormalizedtototal IRE1␣abundanceandofphospho-eIF2␣abundancenormalizedtoeIF2␣abundanceforthreeindependentexperiments.Theresultsarepresentedasmeans±1S.D.(n=3).** p<0.01vstimematchedcontrolcells,usingANOVA2andSidak–Holmtestasposthoctest.B.Cellswereco-transfectedduring4hwithfirefly-luciferasereporterATF6/XBP1 plasmidandrenillaluciferasereporterplasmid24hbeforeincubation.After2,8,and16hofincubation,ATF6/XBP1activitywasassayedusingthedualluciferasereporter assay.DataareexpressedinrelativeATF6transcriptionalactivitynormalizedtothecontrolcells(N)andpresentedasmeans±1S.D.(n=3).**p<0.01;***p<0.001vstime matchedcontrolcells,usingANOVA2andSidak–Holmtestasposthoctest.
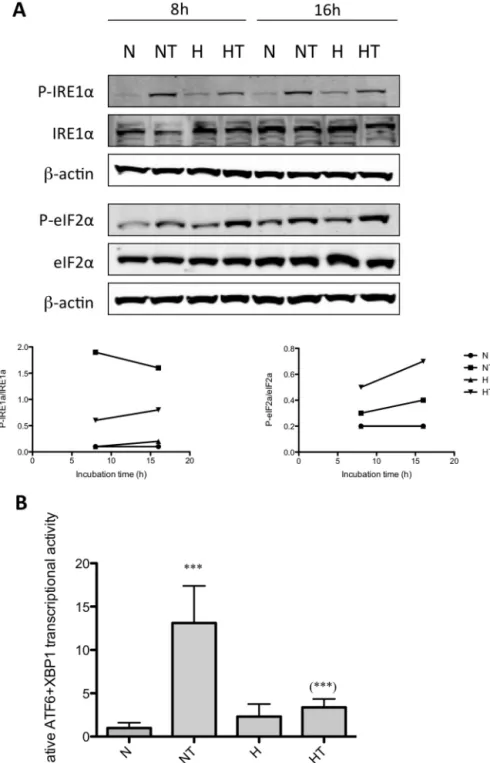
lowerLDH release (supplementary Figure1).Similarresultson thephosphorylationof eIF2␣andIRE1␣inducedbytaxolwere obtainedfor T47D cells as shown for 8 and 16h of incubation (Fig.2A).Next, ATF6activitywasmeasuredafter2,8,and 16h
ofincubationincellstransientlytransfectedwithan ATF6/XBP1-specificluciferasereporterconstructinwhichthefireflyluciferase reporter gene is driven by a promoter containing five copies ofthe ATF6/XBP1bindingelement (Zeng etal., 2004)(Fig.1B).
Fig.2. PERK,IRE1␣,andATF6pathwaysareactivatedaftertaxolexposureundernormoxiaandhypoxiainT47Dcells.
T47Dcellswereincubatedundernormoxia(N)orhypoxia(H)withoutorwithtaxol(T)at50M.A.After8and16hofincubation,theabundanceofphospho-eIF2␣,eIF2␣, phospho-IRE1␣,andIRE1␣wasassessedintotalcellextractsbywesternblottinganalysis,usingspecificantibodies.-actinwasusedtoassessthetotalamountofproteins loadedonthegel.Thegraphsbelowrepresentthequantificationofphospho-IRE1␣abundancenormalizedtototalIRE1␣abundanceandofphospho-eIF2␣abundance normalizedtoeIF2␣abundance.Uncroppedwesternblotsarepresentedinthesupplementaryfigure6.B.Cellswereco-transfectedduring4hwithfirefly-luciferasereporter ATF6plasmidandrenilla-luciferasereporterplasmid24hbeforeincubation.After16hofincubation,ATF6activitywasassayedusingthedualluciferasereporterassay. DataareexpressedinrelativeATF6transcriptionalactivitynormalizedtothecontrolcells(N)andpresentedasmeans±1S.D.(twoindependentexperimentswithn=4).*** p<0.001vsnormoxiccontrolcells;***p<0.001vsNTusingANOVA2andSidak–Holmtestasposthoctest.
ATF6wasactivatedinMDA–MB-231cellsincubatedfor8hwith taxolbutnodifferencecouldbeobservedbetweencellsincubated withtaxolundernormoxiaorhypoxia.Taxolsimilarlyincreased ATF6/XBP1transcriptionalactivityin T47Dcellsbut,in thiscell line, hypoxia inhibited the taxol-induced ATF6/XBP1 activation (Fig.2B).
3.2. ActivationofUPRisneitherinvolvedinapoptosisnorin autophagyinducedbytaxol
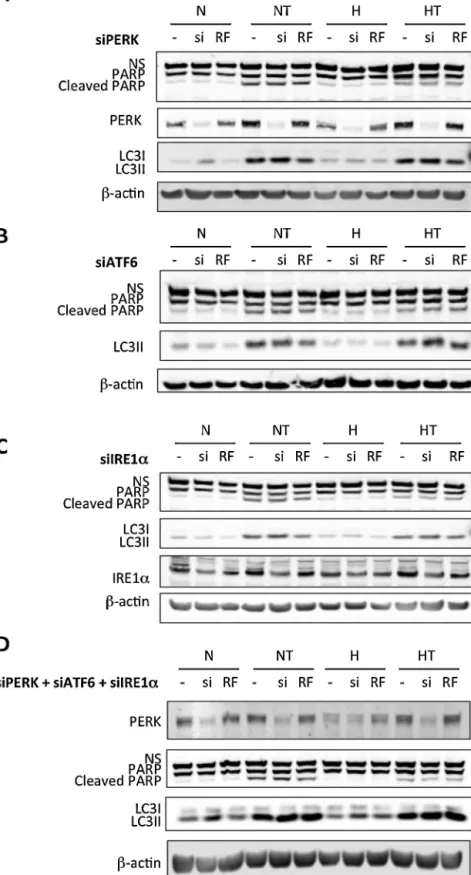
Recentstudies have shown that UPRactivationleadsto the inductionofeitherautophagyorapoptosis(Deeganetal.,2012; RashevaandDomingos,2009).Severalargumentssupporttherole
Fig.3.ActivationofUPRisneitherinvolvedintaxol-inducedapoptosisnorinautophagy.MDA–MB-231cellswereuntransfected(−)ortransfectedwith50nMofPERK(A), ATF6(B),orIRE1␣(C)siRNA(si)ornegativecontrolRiscFreesiRNA(RF)for24h(D).MDA–MB-231cellswereuntransfected(−)orco-transfectedwith25nMofPERK,ATF6, andIRE1␣siRNA(si)or75nMofnegativecontrolRisc-freesiRNA(RF)for24h.Thetransfectionmediawereremovedandreplacedbyculturemediafor24h.Cellswerethen incubatedundernormoxia(N)orhypoxia(H)for16h,withoutorwithtaxol(T)at50M.PARPcleavage,LC3II,PERK,andIRE1␣abundancewasdetectedintotalcellextracts bywesternblottinganalysis,usingspecificantibodies.-actinwasusedtoassessthetotalamountofproteinsloadedonthegel.Uncroppedwesternblotsarepresentedin thesupplementaryfigure6.NS:nonspecificband.
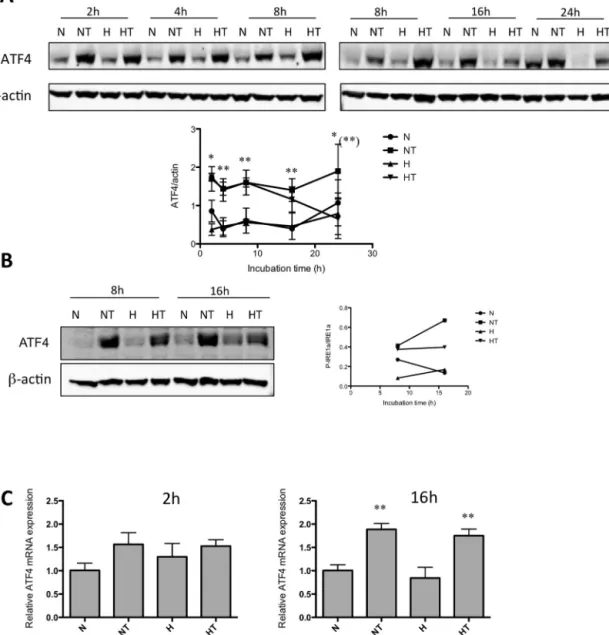
Fig.4.ATF4pathwayisactivatedaftertaxolexposure.MDA–MB-231(A,C)andT47D(B)cellswereincubatedundernormoxia(N)orhypoxia(H)withoutorwithtaxol (T)at50Mfordifferentincubationtimes.A,B.Aftertheincubation,theabundanceofATF4wasassessedintotalcellextractsbyWesternblottinganalysis,usingspecific antibodies.-actinwasusedtoassessthetotalamountofproteinsloadedonthegel.ThegraphsbelowrepresentthequantificationofATF4abundancenormalizedtoß-actin abundance.Uncroppedwesternblotsarepresentedinthesupplementaryfigure6.Thegraphsbelowrepresentthequantificationofphospho-IRE1␣abundancenormalized tototalIRE1␣abundanceforthreeindependentexperiments.Theresultsarepresentedasmeans±1S.D.(n=3).*p<0.05;**p<0.01vstimematchedcontrolcells;**p<0.01 HTvsNT,usingANOVA2andSidak–Holmtestasposthoctest.C.After2or16hofincubation,totalRNAwasextracted,mRNAswereretrotranscribedandATF4mRNAlevels analyzedbyRT-qPCR.Theresultsarepresentedasmeans±1S.D.(n=3).**p<0.01vstimematchedcontrolcellsusingANOVA1andSidak–Holmtestasposthoctest.
ofUPRinthesebiologicalprocesses.PERKinduceseIF2␣ phospho-rylationleadingtoATF4activationthatcaninduceAtg12andLC3 expression(Kourokuetal.,2007;Rouschopetal.,2010).Moreover, ATF4inducesCHOP10expressionandactivation,atranscription factorthatcontrolstheexpressionofTRB3(Ohokaetal.,2005),a proteinfromtheBcl2 family(McCulloughetal.,2001)andAtg5
(Rouschopetal.,2010),andcouldthusactasapro-apoptoticora pro-autophagictranscriptionfactor.Inaddition,ATF6andXBP1are alsoknowntoenhanceCHOP10expression(OyadomariandMori, 2004).IRE1␣mayalsoregulateautophagyandapoptosisby acti-vatingtheJNKsignalingpathway(Uranoetal.,2000;Nishitohetal., 2002;Ogataetal.,2006).
Toinvestigatewhetheroneofthesepathwaysregulates taxol-inducedautophagyorapoptosis,PERK,ATF6,andIRE1␣expression wassilencedinMDA–MB-231cellstransfectedwithPERK(Fig.3A), ATF6 (Fig. 3B), or IRE1␣ (Fig. 3C) siRNAs independently or in
combination(Fig.3D).Theefficiencyofinvalidationofthetarget proteinswasanalyzedbywesternblotting(forPERKandIRE1␣,
Fig. 3) as well as by RT-qPCR (for PERK, IRE1␣, and ATF6) or bymeasuringtranscriptionalactivity(forATF6)(see supplemen-taryfigures 2 and3).Results showedthat neitherindependent nor combined knock down of the three ER-stress sensors did modulatetheabundanceoftaxol-inducedautophagyor apopto-sismarkers. Indeed,nodifferencein theabundanceofLC3IIor in PARP cleavage wasobserved betweencells transfected with the specific siRNAs and cells transfected with control siRNA (Fig.3).
3.3. ATF4isinvolvedintaxol-inducedactivationofautophagy ATF4isanotherplayerofUPR,whichisdownstreamofPERK. It is known tobe induced bytumor microenvironment factors
Fig.5.eiF2␣andATF4pathwaysareactivatedaftertaxolexposureindependentlyofPERK.MDA–MB-231(A,C)orT47D(B)cellswereuntransfected(−)ortransfected with50nMofPERKsiRNA(si)ornegativecontrolRisc-freesiRNA(RF)for24h.Thetransfectionmediawereremovedandreplacedbyculturemediafor24h.Cellswere thenincubatedundernormoxia(N)orhypoxia(H)for16h,withoutorwithtaxol(T)at50M.Phospho-eIF2␣,eIF2␣,ATF4andPERKabundancewasassessedintotalcell extractsbyWesternblottinganalysis,usingspecificantibodies.-actinwasusedtoassessthetotalamountofproteinsloadedonthegel.A,B.Thetablesbelowrepresent thequantificationofphospho-eIF2␣abundancenormalizedtoeIF2␣abundanceandofATF4normalizedtoß-actinabundance.Uncroppedwesternblotsarepresentedin thesupplementaryfigure6.C.ThegraphsbelowrepresentthequantificationofATF4abundancenormalizedtoß-actinabundanceforthreeindependentexperiments.The resultsarepresentedasmeans±1S.D.(n=3).$$p<0.01vscorrespondingRFtransfectedcells,usingANOVA2andSidak–Holmtestasposthoctest.
suchashypoxia(Koumenis, 2006)and toplay a role incancer progression(AmeriandHarris,2008)aswellasinautophagy reg-ulation(Rzymskietal.,2009;Milanietal.,2009).Moreover,other kinasesthanPERKareabletophosphorylateeIF2␣leadingtoATF4 activation(Donnelly etal.,2013).Thesekinases, GCN2(general controlnon-derepressible-2),HRI(heme-regulatedinhibitor),and
PKR(doublestranded RNA-dependentproteinkinase), are acti-vatedafteraminoacidstarvation(Hardingetal.,2000),oxidative stress/proteasomedegradationinhibition(Luetal.,2001;Yerlikaya etal.,2008),andviralinfection(Kiviluotoetal.,2013)respectively. Theybelongtothefamilyofkinasesthatcontroltheintegrated stressresponse(ISR).
Fig.6.ATF4isinvolvedinautophagyactivationaftertaxolexposureinMDA–MB-231cells.(A,B,C)MDA–MB-231cellswereuntransfected(−)ortransfectedwithATF4 siRNA(si)ornegativecontrolRiscFreesiRNA(RF)at50nMfor24h.Thetransfectionmediawereremovedandreplacedbyculturemediafor24h.Cellswerethenincubated undernormoxia(N)orhypoxia(H)for16h,withoutorwithtaxol(T)at50M.A.TheabundanceofPARP,cleavedPARP,cleavedcaspase3,LC3,andp62wasdetectedin totalcellextractsbyWesternblottinganalysis,usingspecificantibodies.-actinwasusedtoassessthetotalamountofproteinsloadedonthegel.Uncroppedwesternblots arepresentedinthesupplementaryfigure6.NS:nonspecificband.B.Caspase3and7activitywasassayedbymeasuringfluorescenceintensityassociatedwithfreeAFC releasedbythecleavageoftheflouorogenicAc-DEVD-AFCsubstratebycaspase3and7.Resultsareexpressedinrelativecaspase3/7activitynormalizedtofluorescence intensityoftheuntransfectedcontrolcellsandpresentedasmeans±1S.D.(n=3).C.After40hofincubation,LDHreleasewasassessed.Resultsareexpressedinpercentages ofcytotoxicityasmeans±1S.D.(n=3).***p<0.001vsnormoxiccontrolcells;(*)p<0.05vsNT;***p<0.001vscorrespondingRF-transfectedcellsusingANOVA2and Sidak–Holmtestasposthoctest.
ATF4activationwasthusinvestigatedinMDA–MB-231(Fig.4A) andin T47D(Fig.4B)cellsexposed totaxolunder normoxiaor hypoxia.Results showedthat ATF4proteinlevel wasincreased in cells incubatedin the presenceof taxol.Hypoxia seemedto decreasethetaxol-inducedincreaseinATF4proteinlevelatlong incubationtime(24h).IthastobenotedthatATF4wasobservedas adoubletonwesternblotanalysis.Thishasalreadybeenobserved inseveralotherstudies.(Panetal.,2013;Yuetal.,2013)Thismay beduetophosphorylation,asdemonstratedinLassotetal.(2001). Sincethereisastrongdecreaseintheintensityofthesebandsin lanescorrespondingtoproteinextractsofcellstransfected with ATF4siRNA(Fig.7),weconcludedthatthedetectionwasspecific forthisprotein.
InordertoanalyzethemechanismbywhichATF4proteinlevel wasincreased,ATF4mRNAlevelhasbeenanalyzed.Theresults
showedthat taxolsignificantlyincreasedby2-foldATF4mRNA levelafter16hofincubation(Fig.4C).Hypoxiaalonehasnoeffect anddidnotaffecttheeffectoftaxol.ItmustbenotedthatATF4 pro-teinlevelwasupregulatedby2foldalreadyafter2hofincubation inthepresenceoftaxol(Fig.4A).Whileitdoesnotexcludethatan increaseinATF4mRNAwasresponsiblefortheupregulationofthe proteinlevelatlongerincubationtimes,theincreaseobservedat 2hismoreprobablyduetopost-translationalregulation.Indeed, themRNAlevelofATF4wasalsomeasuredafter2hincubationin thepresenceoftaxolandtherewasnosignificantchangeinduced bytaxol(Fig.4C).
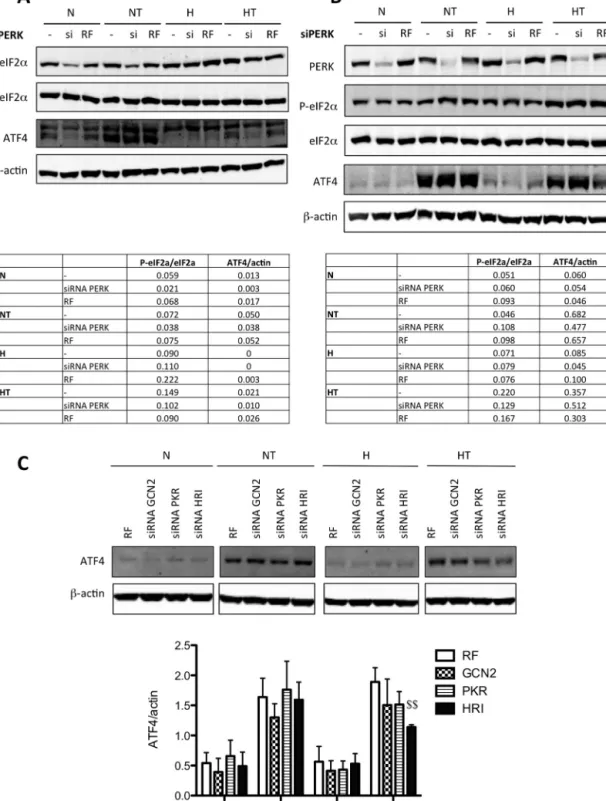
SinceweshowedthatPERKisactivatedbytaxol,we investi-gatedwhetherthiskinaseistheenzymethatcouldberesponsible for ATF4accumulation inboth cell breast cancercelllines. For that, PERKwasthus invalidated usingsiRNA but noeffect was
Fig.7.ATF4isinvolvedinautophagyactivationaftertaxolexposureinT47Dcells.T47Dcellswereuntransfected(−)ortransfectedwithATF4siRNA(si)ornegativecontrol RiscFreesiRNA(RF)at50nMfor24h.Thetransfectionmediawereremovedandreplacedbyculturemediafor24h.Cellswerethenincubatedundernormoxia(N)or hypoxia(H)for16h,withoutorwithtaxol(T)at50M.TheabundanceofATF4,PARP,cleavedPARP,p62,cleavedcaspase3andLC3wasdetectedintotalcellextractsby Westernblottinganalysis,usingspecificantibodies.-actinwasusedtoassessthetotalamountofproteinsloadedonthegel.Uncroppedwesternblotsarepresentedinthe supplementaryFig.6.NS:nonspecificband.
observedoneiF2␣phosphorylationinresponsetotaxol,exceptin MDA–MB-231cellsundernormoxia(Fig.4Cand4D).Furthermore, thetaxol-inducedincreaseinATF4proteinlevelwasnotmodified inMDA–MB-231cellsmaintainedinnormoxia(Fig.4C)norinT47D cellsmaintainedinnormoxiaorexposedtohypoxia(Fig.4D).These resultssuggestthatATF4accumulationresultsfromtheactivation oftheISRwhenbreastcancercellsareexposedtotaxol.
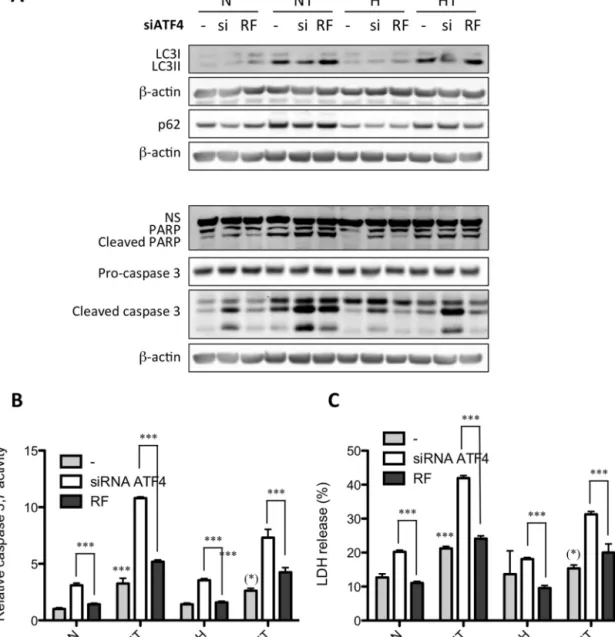
Inordertoverifythishypothesis,eachofthethreekinasesfrom theIRShasbeensilencedbysiRNA.Thecontrolsforsilencingare presentedinthesupplementaryFig.4.Threeindependent experi-mentswereperformed.TheresultssuggestthatHRIisthekinase involvedinthiseffectsincetherewasasignificantdecreaseinthe taxol-inducedincreaseinATF4proteinlevelwhenthiskinasewas silenced,onthecontrarytothetwootherkinases,GCN2andPKR (Fig.5C).TheseresultssuggestthatIRSmayberesponsibleforthe taxol-inducedincreaseinATF4proteinlevel.
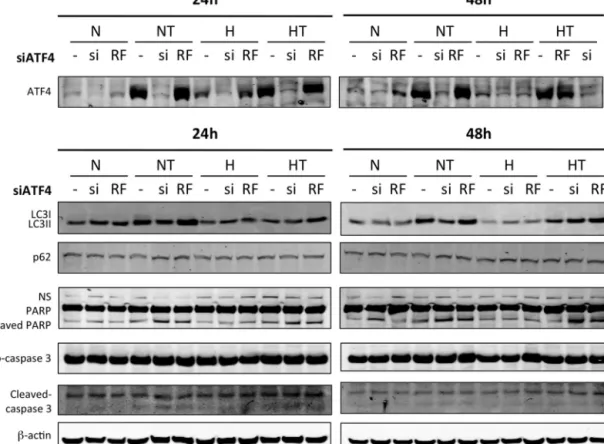
Since ATF4 is activated by an UPR-independent signaling pathway, the role of ATF4 in the adaptation to hypoxia or in taxol-induced autophagy was investigated. MDA–MB-231 cells weretransfectedwithATF4siRNAorRisc-freesiRNAasanegative control.ItresultedinATF4mRNAsilencingtoalevellowerthan5% (supplementaryFig.5A).TheeffectsofATF4silencingonautophagy orapoptosiswerethenstudiedbymeasuringtheabundanceof LC3II,cleaved PARP,and cleaved caspase3by westernblotting (Fig.6A)as wellasbymeasuringcaspase3/7activity(Fig.6B). In addition,overall cytotoxicity wasassessed bya LDH release assay(Fig.6C).InresponsetoATF4silencing,taxol-inducedLC3II conversion was decreased both under normoxia and hypoxia. PARPandcaspase3cleavageaswellascaspase3/7activityand cytotoxicityweresignificantlyincreasedbyATF4silencingincells incubated with or without taxol, under normoxia or hypoxia.
ATF4 invalidation also reduced the upregulation of LC3 mRNA level induced by taxol (data not shown). In order to confirm these results, the effect of ATF4 invalidation was also studied inT47D (Fig.7).Silencingreachedmore that90%at themRNA level(supplementaryFig.5B).Longerincubationtimeswereused sincetaxolat50MislesstoxiconT47Dcellswhencompared with MDA–MB-231 cells. Results showed that ATF4 silencing decreased the taxol-induced LC3I to LC3IIconversion, while it increasedtaxol-inducedPARPandcaspase3cleavage,asobserved for MDA–MB-231 cells. All together,these results suggest that ATF4notonlyplaysarole inthesurvivalof breastcancercells underbasalconditionsbutisalsoinvolvedintheresistanceagainst taxol-inducedcelldeathinpartbypromotingautophagy. 3.4. ATF4expressionisassociatedwithapoorprognosisin humanbreastcancer
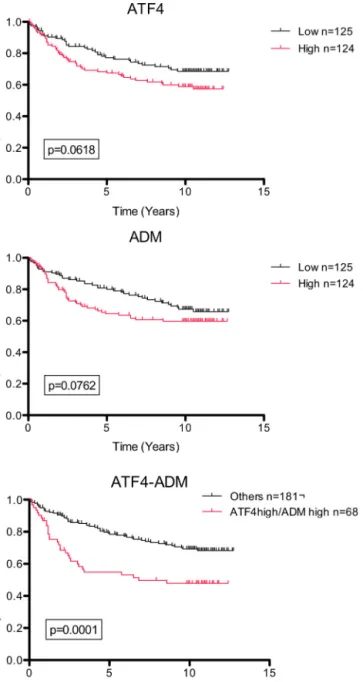
Finally,wewantedtodeterminewhetherATF4expression com-bined tohypoxia couldbeof clinical interest for breast cancer treatmentorprognosisbecause anincrease inATF4expression andthepresenceofhypoxicareasareoftenassociatedwithtumor development(Koumenis, 2006;Ye and Koumenis, 2009; Ameri etal., 2004).We thus analyzedgene expressiondata ofa pub-licAffymetrixdataset(GSE4922)involving249patients(fromthe Uppsalacohort)withinvasivebreasttumors.Inordertodetermine whetherATF4expressioncouldbeusedasaprognosisbiomarker, survivalanalysesusingtheKaplan–Meiermethodwereperformed. Thecohortofpatientswasdividedintotwogroups,withone show-inghighexpressionandthesecondonelowexpressionofATF4, and/orADM(adrenomedullin,chosenasahypoxicmarker(Winter etal.,2007)).Therelapse-freesurvivalprobabilityforpatientswith
Fig.8. ATF4expressioninhypoxicbreastcancerisamarkerofpoorprognostic. PublicAffymetrixarraydataof249breastcancerpatients(GSE4922,Uppsalacohort) forwhomcancerspecificsurvivaltimeisknownwereanalyzed.Patientswere stratifiedintotwosubgroupsaccordingtothemedianvalueandKaplan–Meier plotswere drawn. Graphs represent the relapse-free survival probability for patientswhodisplayhighexpressionversuslowexpressionofATF4and/orADM. Gehan–Breslow–Wilcoxontestswereusedforstatisticalanalysis.
loworhighgeneexpressionwascompared(Fig.8).Resultsshowed thatahighATF4expressionisassociatedwithadecreaseinthe probabilityofsurvivalinpatientswithbreasttumors.ADM,byits ownmRNAexpression,doesseemtoinfluencethesurvival prob-abilityofpatients, butthisdidnotreachstatisticalsignificance. Patientswerethensubdividedasafunctionoftheexpressionof thesetwogenestakensimultaneously.Weparalleledthe probabil-ityofcancersurvivalbetweenpatientsdisplayingasimultaneous highexpressionofATF4/ADMvs.allotherpatients.Patientswith aATF4high/ADMhigh phenotypehadasignificantlylowersurvival
(p<0.0001).ATF4expressionisthusabiomarkerforapoor prog-nosisforbreastcancer.ThisprognosisisevenworsewhenATF4 expressionisassociatedwithahypoxicsignature.
4. Discussion
Theimprovementofchemotherapyis stillabigchallengeas thenumberofpatientswithcancerincreasesandchemotherapy resistanceandrelapsearecommon.Inthis study,wesoughtto unravelsomemechanismsinvolvedintheresistanceofcancercells againstchemotherapywhenproliferatinginahypoxic microenvi-ronment.MDA–MB-231andT47Dbreastcancercellswereused andincubatedwithtaxolundernormoxiaorhypoxia.Ina previ-ouswork,weshowedthatonemechanismpromotingresistance againsttaxol-inducedcelldeathistheactivationofautophagy dur-ingtaxolexposure.Indeed,hypoxiaincreasesautophagyactivation andeffectiveness,whencomparedwithnormoxia,incells incu-batedwithtaxol(Notteetal.,2013).Inthiswork,theroleofUPR andATF4wasinvestigated.TaxolinducedERstressasshownby arapideIF2␣andIRE1␣phosphorylation(respectivelyafter2and 4hofincubation)andalateractivationofATF6(after8hof incu-bation).Hypoxiahadnoeffectonthetaxol-inducedUPR.Hypoxia alonewasalsoabletoinduceeIF2␣phosphorylationafter24hof incubation.
SincetaxolexposureinducedUPR,wedecidedtoinvestigate whetherthisactivationcouldbeinvolvedinautophagyinitiation orinapoptosisactivationtriggeredbytaxolexposure.Liaoetal. showedthattaxolinducesERstress,UPRactivation,andapoptosis inU937cellsbyanearlyreleaseofCa2+andthelateamplificationof
mitochondria-mediatedapoptoticsignals(Liaoetal.,2008). How-ever,theydidnotstudytheroleofPERK,IRE1␣,andATF6signaling inapoptosisactivationbytaxol.ThethreeERstresssensorswere thussilenced,eitherindependentlyorsimultaneouslyusing siR-NAsandtheeffectsofUPRinhibitiononautophagyandapoptosis werestudied.NolinkbetweenPERK,IRE1␣,orATF6activationand autophagyorapoptosisregulationcouldbehighlighted.
Next,theinvolvementofATF4intaxol-inducedautophagyand apoptosiswasinvestigated.Weshowedthatthetranscription fac-torATF4promotescancercellsurvivalunderbasalconditionsand duringtaxolexposure.Indeed,inhibitionofATF4expressionusing ATF4siRNAsresultedinanincreaseinapoptosisactivationandcell death.TheprotectiveroleofATF4aftertaxolexposurecouldbe duetoitscapacitytoregulateautophagy.Indeed,ATF4inhibition alsoledtoadecreaseintaxol-inducedLC3IIaccumulation.Inthis study,thedecreaseinLC3IIaccumulationcanbeassociatedwith adecreaseinautophagyflow.Furthermore,weshowedthatATF4 invalidationdecreaseLC3bmRNAexpression.
Inapreviousstudy(Notteetal.,2013),weshowedthattaxol induced autophagy activation already after 2h of incubation both under normoxia and hypoxia. Autophagy activation after taxolexposurewasshowntobeaprotectivemechanismagainst taxol-inducedcelldeathbothundernormoxiaandhypoxia. How-ever,atlongerincubationtime,theautophagic processreached asaturationpointundernormoxialeadingtocelldeath,whereas underhypoxia,autophagyflowstillcorrectlytookplaceallowing the cells to survive. The conclusionwas reached using several experimentalapproaches aswellasusing detailedtime curves. Whenlookingatonly16hofincubationinthepresenceoftaxol, nodifferenceinLC3IIabundanceunderhypoxiaascomparedto normoxiawasobserved,asshownhere(Figs.3,6and7).Inthis previousstudy,weshowedthattheinhibitionofthetaxol-induced activationofautophagyledtoanincreaseincytotoxicityandin thecurrentstudy,thedecreaseinLC3IIaccumulationobservedin responsetoATF4silencingalsoledtoanincreaseincytotoxicity. Inconclusion,ATF4isnecessaryforaneffectiveautophagic pro-cessduringtaxolexposure.Theseresultsareinaccordancewith previousreportsfromRouschopetal.(Rouschopetal.,2010)and Rzymskietal.(Rzymskietal.,2009;Milanietal.,2009;Rzymski etal.,2010)demonstratingthatATF4transcriptionallyregulates LC3expressionallowingacontinuoussupplyof thisproteinfor
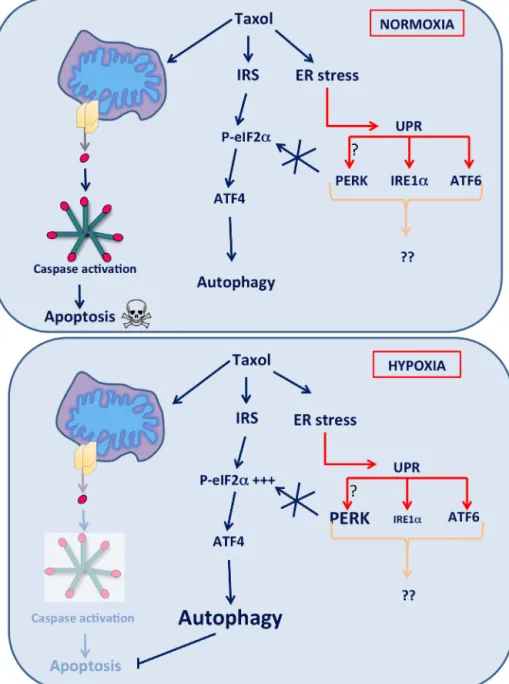
Fig.9.Mechanismspromotingcancercellsurvivalaftertaxoltreatmentunderhypoxia.
Taxolinducescaspaseactivationandsubsequentapoptosisandcelldeathinbreastcancercells.However,differentmechanismsareactivatedtoinhibittaxol-induced apoptosisactivation.WeshowedthattaxolinducesanERstressleadingtoactivationofthethreeER-stresssensorsandUPRactivation.UPRactivationisneitherinvolvedin autophagyactivation,norinapoptosisinhibitionaftertaxolincubation.However,aftertaxolexposure,autophagyisactivatedinanATF4-dependentwaythatpromotescell survivalagainsttaxol-inducedcelldeathbothundernormoxiaandhypoxia.
an effective autophagic process. However, these studies were performed under severe hypoxia or after the inhibition of the 26SproteasomebyBortezomib.Rouschopetal.alsoshowedthat ATF4activation under severehypoxia wasdependent onPERK activationwhereas Rzymski etal. showed that ATF4activation afterBortezomibtreatmentwasPERK-independent.
ATF4isamediatoroftheintegratedstressresponse(ISR)and is activated downstream of eIF2␣ phosphorylation (Ameri and Harris,2008).ThreekinasesotherthanPERKareableto phospho-rylateeIF2␣duringtheISR:GCN2,PKR,andHRI.Yerlikayaetal. showedthatBortezomiborMG-231activatesHRI.(Yerlikayaetal., 2008)ATF4dependent-autophagyactivationinresponseto pro-teasomeinhibitioncouldbeduetoHRIactivation.In ourwork, taxol-induced ATF4activation could also bePERK-independent sinceinvalidationofPERKwithsiRNAdidnotinfluenceautophagy norapoptosisactivation.BysilencingthethreeISRkinases,HRI,
PKR,orGCN2, weshowedthat thekinaseresponsiblefor ATF4 inductionisHRI.
Inconclusion,differentmechanismspromotingbreastcancer celladaptationto hypoxiaand resistanceagainsttaxol-induced apoptosiswerehighlighted(Fig.9).Weshowedthattaxol-induced UPRactivationisneitherinvolvedinautophagynorinapoptosis activation. On the other hand, ATF4 promotes cancer cell sur-vivalunderbasalconditionsandaftertaxolexposure.ATF4also allowsLC3ItoLC3IIconversionandautophagycompletion sug-gesting that ATF4 is important for autophagosome elongation andautophagyexecutionthatpromotesresistanceagainst taxol-induced celldeath. Autophagy regulation byATF4 seemstobe independentofPERKactivationandseemstooccurthroughthe activationoftheISRviatheHRIkinase.Moreover,ATF4isdescribed tobeexpressedathigherlevelincancerwhencomparedtonormal tissue(Amerietal.,2004),thismaybeduetotheinductionofits
expressionbysignalsspecifictothetumormicroenvironmentsuch ashypoxia,nutrientdeprivation,oxidativestress,and/orERstress. Finally,breast cancerpatientscarrying theATF4high/ADMhigh
hadalowersurvivalprobablycomparedtobreastcancerpatients withlowexpressionofthesegenes.ThisresultsuggeststhatATF4 expressionmightbeassociatedwithhypoxiacellresponse.This molecularsignaturecouldthusbeusedasabiomarkerreflectinga poorprognosisinhumanbreastcancer.Thisfurthersupportsthe conclusionthatATF4playsaroleintheadaptationandresistance ofbreastcancercellsagainstchemotherapy.
Conflictofintereststatement
Theauthorsdeclarenoconflictofinterest.
Acknowledgments
We acknowledge Professor J.Y. Shyy (University of Califor-nia,Riverside,CA,USA)forgenerouslyprovidingtheATF6/XBP1 luciferasereporter plasmid.A. Notteand C. Tellierare PhD fel-lowsfromFNRS(NationalFundsforScientificResearch,Brussels, Belgium).M.Geninbenefitsfroma PhDgrantfromFRIA(FNRS, Brussels,Belgium)andM.RebucciandE.Roegiersarerespectively apost-doctoralresearcherandaPhDgranteefromTelevie(FNRS, Brussels,Belgium).
AppendixA. Supplementarydata
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j. biocel.2015.02.010.
References
AmeriK, Harris AL.Activatingtranscriptionfactor 4.Int JBiochem Cell Biol 2008;40(1):14–21.
AmeriK,LewisCE,RaidaM,SowterH,HaiT,HarrisAL.AnoxicinductionofATF-4 throughHIF-1-independentpathwaysofproteinstabilizationinhumancancer cells.Blood2004;103(5):1876–82.
BertolottiA,ZhangY,HendershotLM,HardingHP,RonD.Dynamicinteractionof BiPandERstresstransducersintheunfolded-proteinresponse.NatCellBiol 2000;2(6):326–32.
BiM,NaczkiC,KoritzinskyM,FelsD,BlaisJ,HuN,etal.ERstress-regulated transla-tionincreasestolerancetoextremehypoxiaandpromotestumorgrowth.EMBO J2005;24(19):3470–81.
Brahimi-HornC,PouyssegurJ.Theroleofthehypoxia-induciblefactorintumor metabolismgrowthandinvasion.BCancer2006;93(8):E73–80.
BravoR,ParraV,GaticaD,RodriguezAE,TorrealbaN,ParedesF,etal.Endoplasmic reticulumandtheunfoldedproteinresponse:dynamicsandmetabolic integra-tion.IntReviewCellMolBiol2013;301:215–90.
ChakrabartiA,ChenAW,VarnerJD.Areviewofthemammalianunfoldedprotein response.BiotechnolBioeng2011;108(12):2777–93.
ChecinskaA,Soengas MS.Thegluttonous sideofmalignantmelanoma:basic andclinical implicationsofmacroautophagy. PigmentCell MelanomaRes 2011;24(6):1116–32.
ChenR,DaiRY,DuanCY,LiuYP,ChenSK,YanDM,etal.Unfoldedproteinresponse suppressescisplatin-inducedapoptosisviaautophagyregulationinhuman hep-atocellularcarcinomacells.FoliaBiol2011;57(3):87–95.
Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ 2005;12(Suppl 2): 1509–18.
DeeganS,SaveljevaS,GormanAM,SamaliA.Stress-inducedself-cannibalism:on theregulationofautophagybyendoplasmicreticulumstress.CellMolLifeSci 2012.
DonnellyN,GormanAM,GuptaS,SamaliA.TheeIF2alphakinases:theirstructures andfunctions.CellMolLifeSci2013.
FengR,ZhaiWL,YangHY,JinH,ZhangQX.InductionofERstressprotectsgastric cancercellsagainstapoptosisinducedbycisplatinanddoxorubicinthrough activationofp38MAPK.BiochemBiophResCo2011;406(2):299–304.
Garcia-NavasR,MunderM,MollinedoF.DepletionofL-arginineinducesautophagy asacytoprotectiveresponsetoendoplasmicreticulumstressinhumanT lym-phocytes.Autophagy2012;8(11):1557–76.
GattiL,ZuninoF.Overviewoftumorcellchemoresistancemechanisms.Methods MolMed2005;111:127–48.
HardingHP,NovoaI,ZhangY,ZengH,WekR,SchapiraM,etal.Regulatedtranslation initiationcontrolsstress-inducedgeneexpressioninmammaliancells.MolCell 2000;6(5):1099–108.
HeC,KlionskyDJ.Regulationmechanismsandsignalingpathwaysofautophagy. AnnuRevGenet2009;43:67–93.
HealySJ,GormanAM,Mousavi-ShafaeiP,GuptaS,SamaliA.Targetingthe endo-plasmicreticulum-stressresponseasananticancerstrategy.EurJPharmacol 2009;625(1–3):234–46.
Heath-EngelHM,ChangNC,ShoreGC.Theendoplasmicreticuluminapoptosisand autophagy:roleoftheBCL-2proteinfamily.Oncogene2008;27(50):6419–33.
Hoyer-HansenM,JaattelaM.Connectingendoplasmicreticulumstresstoautophagy by unfolded protein response and calcium. Cell Death Diff 2007;14(9): 1576–82.
IvshinaAV,GeorgeJ,SenkoO,MowB,PuttiTC,SmedsJ,etal.Geneticreclassification ofhistologicgradedelineatesnewclinicalsubtypesofbreastcancer.CancerRes 2006;66(21):10292–301.
JiangCC,LucasK,Avery-KiejdaKA,WadeM,deBockCE,ThorneRF,etal. Up-regulationofMcl-1iscriticalforsurvival ofhumanmelanomacells upon endoplasmicreticulumstress.CancerRes2008;68(16):6708–17.
KiviluotoS,VervlietT,IvanovaH,DecuypereJP,DeSmedtH,MissiaenL,etal. Reg-ulationofinositol1,4,5-trisphosphatereceptorsduringendoplasmicreticulum stress.BiochimBiophysActa2013;1833(7):1612–24.
KoumenisC.ERstress,hypoxiatoleranceandtumorprogression.CurrMolMed 2006;6(1):55–69.
Kouroku Y, Fujita E, TanidaI, Ueno T, Isoai A, Kumagai H, et al. ER stress (PERK/eIF2alphaphosphorylation)mediatesthepolyglutamine-inducedLC3 conversion, an essential step for autophagy formation. Cell Death Differ 2007;14(2):230–9.
KumandanS,MahadevanNR,ChiuK,DeLaneyA,ZanettiM.Activationofthe unfoldedproteinresponsebypassestrastuzumab-mediatedinhibitionofthe PI-3Kpathway.CancerLett2013;329(2):236–42.
LassotI,SegeralE,Berlioz-TorrentC,DurandH,GroussinL,HaiT,etal.ATF4 degrada-tionreliesonaphosphorylation-dependentinteractionwiththeSCF(betaTrCP) ubiquitinligase.MolCellBiol2001;21(6):2192–202.
LiJ,LeeAS.StressinductionofGRP78/BiPanditsroleincancer.CurrMolMed 2006;6(1):45–54.
LiaoPC,TanSK, LieuCH,Jung HK.Involvementof endoplasmicreticulumin paclitaxel-inducedapoptosis.JCellBiochem2008;104(4):1509–23.
LozanoJ, MenendezS,MoralesA,EhleiterD,LiaoWC,Wagman R,etal.Cell autonomousapoptosisdefectsinacidsphingomyelinaseknockoutfibroblasts.J BiolChem2001;276(1):442–8.
LuL,HanAP,ChenJJ.Translationinitiationcontrolbyheme-regulatedeukaryotic initiationfactor2alphakinaseinerythroidcellsundercytoplasmicstresses.Mol CellBiol2001;21(23):7971–80.
MannMJ,HendershotLM.UPRactivationalterschemosensitivityoftumorcells. CancerBiolTher2006;5(7):736–40.
McCulloughKD,MartindaleJL,KlotzLO,AwTY,HolbrookNJ.Gadd153sensitizes cellstoendoplasmicreticulumstressbydown-regulatingBcl2andperturbing thecellularredoxstate.MolCellBiol2001;21(4):1249–59.
MehrpourM,EsclatineA,BeauI,CodognoP.Overviewofmacroautophagyregulation inmammaliancells.CellRes2010;20(7):748–62.
MilaniM,RzymskiT,MellorHR,PikeL,BottiniA,GeneraliD,etal.TheroleofATF4 stabilizationandautophagyinresistanceofbreastcancercellstreatedwith Bortezomib.CancerRes2009;69(10):4415–23.
MorselliE,GalluzziL,KeppO,VicencioJM,CriolloA,MaiuriMC,etal.Anti-and pro-tumorfunctionsofautophagy.BiochimBiophysActa2009;1793(9):1524–32.
NishitohH,MatsuzawaA,TobiumeK,SaegusaK,TakedaK,InoueK,etal.ASK1 isessentialforendoplasmicreticulumstress-inducedneuronalcelldeath trig-geredbyexpandedpolyglutaminerepeats.GenesDev2002;16(11):1345–55.
NotteA,LeclereL,MichielsC.Autophagyasamediatorofchemotherapy-induced celldeathincancer.BiochemPharmacol2011;82(5):427–34.
NotteA,NinaneN,ArnouldT,MichielsC.Hypoxiacounteractstaxol-induced apo-ptosisinMDA-MB-231breastcancercells:roleofautophagyandJNKactivation. CellDeathDis2013;4:e638.
OgataM,HinoS,SaitoA,MorikawaK,KondoS,KanemotoS,etal.Autophagy isactivatedforcellsurvivalafterendoplasmicreticulumstress.MolCellBiol 2006;26(24):9220–31.
OhokaN,YoshiiS,HattoriT,OnozakiK,HayashiH.TRB3,anovelERstress-inducible gene,isinducedviaATF4-CHOPpathwayandisinvolvedincelldeath.EMBOJ 2005;24(6):1243–55.
OyadomariS,MoriM.RolesofCHOP/GADD153inendoplasmicreticulumstress.Cell DeathDiff2004;11(4):381–9.
PanY,XiaoJ,LiangG,WangM,WangD,WangS,etal.Anewcurcuminanalogue exhibitsenhancedantitumoractivityinnasopharyngealcarcinoma.OncolRep 2013;30(1):239–45.
PierreM,DeHertoghB,GaigneauxA,DeMeulderB,BergerF,BarekeE,etal. Meta-analysisofarchivedDNAmicroarraysidentifiesgenesregulatedbyhypoxiaand involvedinametastaticphenotypeincancercells.BMCCancer2010;10:176.
RashevaVI,DomingosPM.Cellularresponsestoendoplasmicreticulumstressand apoptosis.Apoptosis2009;14(8):996–1007.
RohwerN,CramerT.Hypoxia-mediateddrugresistance:novelinsightsonthe functionalinteractionofHIFsandcelldeathpathways.DrugResistUpdat 2011;14(3):191–201.
Romero-RamirezL,CaoH,NelsonD,HammondE,LeeAH,YoshidaH,etal.XBP1is essentialforsurvivalunderhypoxicconditionsandisrequiredfortumorgrowth. CancerRes2004;64(17):5943–7.
RonD,WalterP.Signalintegrationintheendoplasmicreticulumunfoldedprotein response.NatRevMolCellBiol2007;8(7):519–29.
RouschopKM,WoutersBG.Regulationofautophagythroughmultipleindependent hypoxicsignalingpathways.CurrMolMed2009;9(4):417–24.
RouschopKM,vandenBeuckenT,DuboisL,NiessenH,BussinkJ,SavelkoulsK, etal.Theunfoldedproteinresponseprotectshumantumorcellsduringhypoxia throughregulationoftheautophagygenesMAP1LC3BandATG5.JClinInvest 2010;120(1):127–41.
RzymskiT,MilaniM,SingletonDC,HarrisAL.RoleofATF4inregulationofautophagy andresistancetodrugsandhypoxia.CellCycle2009;8(23):3838–47.
RzymskiT, Milani M, Pike L, BuffaF, Mellor HR, Winchester L, et al. Reg-ulation of autophagy by ATF4 in response to severe hypoxia. Oncogene 2010;29(31):4424–35.
SchonthalAH.Pharmacologicaltargetingofendoplasmicreticulumstresssignaling incancer.BiochemPharmacol2013;85(5):653–66.
SemenzaGL.HIF-1:upstreamanddownstreamofcancermetabolism.CurrOpin GenetDev2010;20(1):51–6.
ShajahanAN,RigginsRB,ClarkeR.TheroleofX-boxbindingprotein-1in tumori-genicity.DrugNewsPerspect2009;22(5):241–6.
Shannon AM, Bouchier-Hayes DJ, Condron CM, Toomey D. Tumour hypoxia, chemotherapeuticresistanceandhypoxia-relatedtherapies.CancerTreatRev 2003;29(4):297–307.
ShenJ,ChenX,HendershotL,PrywesR.ERstressregulationofATF6localizationby dissociationofBiP/GRP78bindingandunmaskingofGolgilocalizationsignals. DevCell2002;3(1):99–111.
ShenS, Kepp O, Kroemer G. The end of autophagic cell death? Autophagy 2012;8(1):1–3.
SongJ,QuZ,GuoX,ZhaoQ,ZhaoX,GaoL,etal.Hypoxia-inducedautophagy con-tributestothechemoresistanceofhepatocellularcarcinomacells.Autophagy 2009;5(8):1131–44.
SzegezdiE,LogueSE,GormanAM,SamaliA.Mediatorsofendoplasmicreticulum stress-inducedapoptosis.EMBORep2006;7(9):880–5.
SzegezdiE,MacdonaldDC,NiChonghaileT,GuptaS,SamaliA.Bcl-2familyonguard attheER.AmJPhysiolCellPhysiol2009;296(5):C941–53.
TraversKJ,PatilCK,WodickaL,LockhartDJ,WeissmanJS,WalterP.Functionaland genomicanalysesrevealanessentialcoordinationbetweentheunfoldedprotein responseandER-associateddegradation.Cell2000;101(3):249–58.
UranoF,WangX,BertolottiA,ZhangY,ChungP,HardingHP,etal.Couplingofstress intheERtoactivationofJNKproteinkinasesbytransmembraneproteinkinase IRE1.Science2000;287(5453):664–6.
VannuvelK,RenardP,RaesM,ArnouldT.Functionalandmorphologicalimpactof ERstressonmitochondria.JCellularPhysio2013;228(9):1802–18.
WalterP,RonD.Theunfoldedproteinresponse:fromstresspathwaytohomeostatic regulation.Science2011;334(6059):1081–6.
WangJ,YinY,HuaH,LiM,LuoT,XuL,etal.BlockadeofGRP78sensitizesbreast cancercellstomicrotubules-interferingagentsthatinducetheunfoldedprotein response.JCellMolMed2009;13(9B):3888–97.
WellingtonCL,EllerbyLM,HackamAS,MargolisRL,TrifiroMA,SingarajaR,etal.
Caspasecleavageofgeneproductsassociatedwithtripletexpansiondisorders generatestruncatedfragmentscontainingthepolyglutaminetract.JBiolChem 1998;273(15):9158–67.
WhiteE.Deconvolutingthecontext-dependentroleforautophagyincancer.Nat RevCancer2012;12(6):401–10.
WinterSC,BuffaFM,SilvaP,MillerC,ValentineHR,TurleyH,etal.Relationofa hypoxiametagenederivedfromheadandneckcancertoprognosisofmultiple cancers.CancerRes2007;67(7):3441–9.
YeJ,KoumenisC.ATF4,anERstressandhypoxia-inducibletranscriptionfactor anditspotentialroleinhypoxiatoleranceandtumorigenesis.CurrMolMed 2009;9(4):411–6.
YerlikayaA,KimballSR,StanleyBA.PhosphorylationofeIF2alphainresponseto 26Sproteasomeinhibitionismediatedbythehaem-regulatedinhibitor(HRI) kinase.BiochemJ2008;412(3):579–88.
YorimitsuT,KlionskyDJ.Eatingtheendoplasmicreticulum:qualitycontrolby autophagy.TrendsCellBiol2007;17(6):279–85.
YuS,ZhuK,LaiY,ZhaoZ,FanJ,ImHJ,etal.atf4promotesbeta-cateninexpression andosteoblasticdifferentiationofbonemarrowmesenchymalstemcells.IntJ BiolSci2013;9(3):256–66.
ZengL,LuM,MoriK,LuoS,LeeAS,ZhuY,etal.ATF6modulatesSREBP2-mediated lipogenesis.EMBOJ2004;23(4):950–8.
ZhangH,Bosch-MarceM,ShimodaLA,TanYS,BaekJH,WesleyJB,etal. Mitochon-drialautophagyisanHIF-1-dependentadaptivemetabolicresponsetohypoxia. JBiolChem2008;283(16):10892–903.