ﺎﻨﺘﻤﻠﻋ ﺎﻣ ﻻﺇ ﺎﻨﻟ ﻢﻠﻋ ﻻ ﻚﻧﺎﺤﺒﺳ
ﻢﻴﻜﳊﺍ ﻢﻴﻠﻌﻟﺍ ﺖﻧﺃ ﻚﻧﺇ
:ﺔﻳﻵﺍ :ﺓﺮﻘﺒﻟﺍ ﺓﺭﻮﺳ
31
À Allah
Tout puissant
Qui m’a inspiré
Qui m’a guidé dans le bon chemin
Je vous dois ce que je suis devenu
Louanges et remerciements
Pour votre clémence et miséricorde
À
FEU SA MAJESTE LE ROI
HASSAN II
À
SA MAJESTÉ LE ROI
MOHAMED VI
Chef Suprême et Chef d’Etat-Major Général des Forces Armées
Royales
Roi du MAROC et garant de son intégrité territoriale
À
SON ALTESSE ROYALE
LE PRINCE HÉRITIER
MOULAY EL HASSAN
À
SON ALTESSE ROYALE
LE PRINCE MOULAY RACHID
Que Dieu le protège.
À
A
Monsieur le Médecin Général de Brigade
Mohammed ABBAR
Professeur d’urologie
Inspecteur du Service de Santé des Forces Armées Royales.
En témoignage de notre grand respect,
Et notre profonde considération
A
Monsieur le Médecin colonel major
El Mehdi ZBIR
Professeur en Cardiologie
Directeur de l’HMIMV –Rabat.
En témoignage de notre grand respect
Et notre profonde considération
A
Monsieur le Médecin Colonel Taoufiq AMEZIANE
Professeur de Médecine Interne
Directeur de l’E.R.S.S.M
En témoignage de notre grand respect
Et notre profonde considération.
A
Monsieur le Médecin Général
Abdelhamid HDA
Professeur agrégé de Cardiologie
Ancien Inspecteur du Service de Santé des Forces Armées
Royales
et Directeur de l'E.R.S.S.M
En témoignage de notre profond respect, notre profonde
considération et sincère admiration.
A
Monsieur le Général de Corps d’Armée
Abdelfattah LOUARAK
Inspecteur Général des Forces Armées Royales
En témoignage de notre grand respect
Et notre profonde considération et sincère admiration
A
Monsieur le Médecin générale brigade
Mohammed ABBAR
Inspecteur du Service Santé
En témoignant de notre grand respect
Et notre profonde considération
A
Monsieur le Médecin colonel major
El Mehdi ZBIR
Directeur de l’Hôpital Militaire d’Instructions Mohamed V
- Rabat
En témoignage de notre grand respect
Et notre profonde considération
A
Monsieur le Médecin colonel major
Elbaaj Mohammed
Directeur de l’Hôpital Militaire Moulay Ismail - Meknes
En témoignage de notre grand respect
Et notre profonde considération
A
Monsieur le Médecin général de brigade
BOULAHYA Abdellatif
Directeur de l’Hôpital Militaire Avicenne – Marrakech
En témoignant de notre grand respect et notre profonde
considération
A
Monsieur le Médecin Colonel
Taoufiq AMEZIANE
Directeur de l’Ecole Royale du Service de Santé Militaire
En témoignage de notre grand respect
Et notre profonde considération.
A
Monsieur le Médecin colonel
El Matar Abderrahmane
Commandant le Groupement Formation et Instruction
de l’ERSSM
En témoignage de notre grand respect
Et notre profonde considération
A ceux qui me sont les plus chers
A ceux qui ont toujours cru en moi
A ceux qui m’ont toujours encouragé
ﻲﻣﺃ ﺮﻴﺨﻟﺎﺑ ﻲﺘﻴﻧﺩ ﺕﺀﺎﺿﺃ ﺔﻌﻤﺷ ﺖﻧﺃﻭ ،ﻱﺩﺍﻮﻟﺍ ﻲﻓ ﺖﺘﺒﻧ ﺓﺮﻫﺯ ﺖﻧﺃﻭ ،ﺎﻬﺤﻳﺭﻭ ﺔﻨﺠﻟﺍ ﺮﻴﺒﻋ ﺖﻧﺃ ﻲﻣﺃ ﺔﻛﺮﺒﻟﺍﻭ . ﻡﻷﺍ ﺖﻧﺃ ،ﹰﺎﻧﺎﻨﺣﻭ ﹰﺎﺒﺣ ﻪﻨﻣ ﻱﻮﺗﺭﺃ ﻱﺬﻟﺍ ﻊﺒﻨﻟﺍﻭ ،ﻲﺗﺎﻴﺣ ﺀﻲﻀﻳ ﻱﺬﻟﺍ ﺭﻮﻨﻟﺍ ﺖﻧﺃ ﺔﺒﻴﺒﺤﻟﺍ ﻲﻣﺃ ﺃ ﻚﺑ ﻲﻟ ﹰﺎﺌﻴﻨﻬﻓ ،ﻡﺎﻧﻷﺍ ﻦﻴﺑ ﺎﻬﺑ ﺮﺨﺘﻔﻳﻭ ،ﺎﻬﻣﺍﺪﻗﺃ ﺖﺤﺗ ﺔﻨﺠﻟﺍ ﺖﻌﺿ ﻭ ﻲﺘﻟﺍ ﺔﻤﻴﻈﻌﻟﺍ ﻡﻷﺍ ﺎﻬﺘﻳ . ﺔﻤﻠﻛ ﺖﻧﺃ ﺮﺸﻧ ﹰﺍﺩﺭﻭ ﺎﻳ ،ﺮﺤﺒﻟﺍ ﺓﺆﻟﺆﻟ ﺎﻳ ،ﺀﺎﻤﺴﻟﺍ ﻲﻓ ﻊﻤﻠﺗ ﺔﻤﺠﻧ ﺎﻳ ،ﻲﺑﺭﺩ ﺭﺎﻧﺃ ﹰﺍﺮﻤﻗ ﺎﻳ ،ﻩﺍﻮﺘﺤﻣﻭ ﻩﺎﻨﻌﻣ ﺖﻧﺃ ﺏﺎﺘﻛ ﻲﻓ ﻩﺎﻨﻏﺄﻓ ﻖﻓﻷﺍ ﻲﻓ ﻪﺒﻴﻃ . ﻲﺑﺃ ﻞﺟﺃ ﻦﻣ ﺮﻬﺳﻭ ﺐﻌﺗ ﻦﻣ ﻰﻟﺇ ،ﻢﻠﻌﻟﺍ ﻖﻳﺮﻃ ﺪﻬﻤﻴﻟ ﻲﺑﺭﺩ ﻦﻋ ﻙﺍﻮﺷﻷﺍ ﺪﺼﺣ ﻦﻣ ﻰﻟﺇ ﻲﺟﺮﺨﺗ ﻱﺪﻫﹸﺃ ﻟﺇ ،ﹼﻞﻤﻳ ﻢﻟ ﻭ ﻲﺘﺣﺍﺭ ﺔﻔﺧ ﻭ ﺐﻀﻨﻳ ﻻ ﺐﺣ ﻪﺘﻠﻣﺎﻌﻣ ﻲﻓ ﻦﻣ ﻰﻟﺇ ،ﹼﻞﻜﻳ ﻢﻟ ﻭ ﺲﻴﻔﻨﻟﺍ ﻭ ﻲﻟﺎﻐﻟﺎﺑ ﻰﺤﺿ ﻦﻣ ﻰ ﻰﻠﻋ ﻙﺮﻜﺷﺃ ،ﹰﺎﻤﻠﺣ ﻡﺎﻨﻤﻟﺍ ﻲﻓ ﻥﺎﻛ ﻮﻟ ﻰﺘﺣ ﹰﺎﺣﺮﻓ ﻲﻨﺣﺮﻔﺗ ﻪﺘﻳﺅﺭ ﻦﻣ ﻰﻟﺇ ،ﻲﻀﻘﻨﺗ ﻻﻭ ﻢﺘﺗ ﻻ ﻞﻇ ﻭ ﻚﺘﻴﺑﺮﺗ ﻚﻴﻧﺎﻔﺗ ﻰﻠﻋ . ﻲﺑﺃ ﻲﺗﺪﺟ ﻙﺪﻴﻔﺣ ﺐﻠﻘﺑ ﺖﻟﺯﺎﻣ . ﺪﻌﺑ ﻲﺗﻮﻤﺗ ﻢﻟ . ﻚﺣﻭﺭ ﻰﻠﻋ ﻡﻼﺳ . ﺨﻟﺍ ﺕﺎﻨﺟ ﻰﻟﺇ ﷲﺍ ﻥﺫﺈﺑ ﺪﻠ . ﺔﻤﻳﺮﻛ ﺕﺎﻤﻠﻜﻟﺍ ﻞﻈﺗﻭ ،ﻚﺘﺑﻮﺒﻴﻃ ﻭ ﻚﻧﺎﻨﺣ ﺎﻬﺑ ﻒﺻﻷ ﻑﻭﺮﺤﻟﺍ ﻲﻋﺪﺘﺳﺃ ،ﺔﻴﻧﺎﺜﻟﺍ ﻲﻣﺃ ﻭ ﻯﺮﺒﻜﻟﺍ ﻲﺘﺧﺃ ﺕﺎﻤﻠﻜﻟﺎﻛ ﺖﺴﻴﻟ ﺕﺎﻤﻠﻛ ﺝﺎﺘﺤﻳ ﻚﻔﺻﻭ ﻥﺄﻛ ﻭ ﺓﺰﺟﺎﻋ . ﺎﻧﺃ ﻭ ﻲﻟ ﻪﺘﻣﺪﻗ ﺎﻣ ﻞﻛ ﻰﻠﻋ ﻙﺮﻜﺷﺃ ﻱﺮﻜﻓ ﻭ ﻲﺘﻠﻴﺨﻣ ﺕﹼﺬﻏ ﺔﻴﻟﺎﻳﺮﺳ ﺺﺼﻗﻭ ،ﺐﺣ ﻭ ﻒﻄﻋ ﻦﻣ ﺮﻴﻐﺻ . ﺪﻤﺤﻣ ﺐﻴﺼﻨﻟ ﻚﻨﻣ ﻚﺘﻔﺼﻟ ﻥﺇ ﺮﻴﺒﻜﻟﺍ ﻲﺧﺃ ﺎﻳ … ﻚﻧﺎﻨﺣ ﻲﻓ ﺮﻴﺒﻜﻟﺍ ﺥﻷﺍ ﺖﻧﺄﻓ … ﻲﻓ ﻰﺘﺣ ﺮﻴﺒﻛ ﻚﻟﺎﺻﻭ … ﻙﺮﺒﺻ ﻲﻓ ﺮﻴﺒﻛ … ﺀﻲﺷ ﻞﻛ ﻲﻓ ﺮﻴﺒﻛ .
ﻥﺎﻧﺪﻋ ﻯﺭﺃ ﻦﻛﺃ ﻢﻟ ﺍﺮﻴﻐﺻ ﺖﻨﻛ ﻦﻴﺣ ،ﻲﻟ ﺔﺒﺴﻨﻟﺎﺑ ﻢﻟﺎﻌﻟﺍ ﺖﻧﺃ ،ﺀﺎﻴﺷﻷﺍ ﻢﻈﻋﺃ ﻲﺧﺃ ﺎﻳ ﻚﺑ ﻲﻨﻄﺑﺮﻳ ﻱﺬﻟﺍ ﺑﺃﻭ ﺎﻤﺋﺍﺩ ﻞﻈﻴﺳ ﻱﺬﻟﺍ ﻲﺧﺃ ﺖﻧﺃ ،ﻚﻴﻓ ﻻﺇ ﻢﻟﺎﻌﻟﺍ ﻯﺭﺃ ﺪﻋﺃ ﻢﻟ ﺕﺮﺒﻛ ﻦﻴﺣﻭ ،ﻚﻟﻼﺧ ﻦﻣ ﻻﺇ ﻢﻟﺎﻌﻟﺍ ﺍﺪ ﻡﺍﻭﺪﻟﺍ ﻰﻠﻋ ﻡﺪﻘﺘﻟﺍ ﻮﺤﻧ ﻲﻨﻌﻓﺪﻳﻭ ﺮﻴﺼﻤﻟﺍ ﻲﻨﻛﺭﺎﺸﻳ ﻲﻌﻣ . ﻚﻟ ﺎﻧﺃ ﻭ ﺀﺎﺨﺴﺑ ﻰﻘﻴﺳﻮﻤﻟﺍ ﺐﺣ ﻲﻌﻣ ﺖﻤﺳﺎﻘﺗ ﻦﺘﻤﻣ ﺪﺟ . ﻝﺍﻮﻧ ﻙﺭﺎﺜﻳﺇ ﻭ ﻚﺋﺎﻄﻋ ﻢﺠﺤﺑ ﻚﺒﺣﺃ ،ﻚﺋﺎﻔﺻﻭ ﻚﺋﺎﻘﻧ ﻢﺠﺤﺑ ﻚﺒﺣﺃ ،ﺊﻴﺷ ﻞﻛ ﻲﻓ ﻝﺍﻮﻧ ﺎﻬﻤﺳﺈﻛ . ﺪﻤﺣﺃ ﻭ ﻲﻨﺘﻤﻬﻟﺃ ،ﺓﺎﻴﺤﻠﻟ ﻚﺗﺮﻈﻧ ﻭ ﺶﻴﻌﻟﺍ ﻲﻓ ﻚﺑﻮﻠﺳﺃ ﻲﻨﻤﻬﻠﻳ ،ﺊﻴﺷ ﻞﻛ ﻲﻓ ﻲﺗﻭﺪﻗ ﺖﻧﺃ ﻭ ﺖﻨﻛ ﻭ ﻲﻨﺗﺪﻧﺎﺳ ﺪﺋﺍﺪﺸﻟﺍ ﺖﻗﻭ ﻪﻴﻠﻋ ﻲﺴﻔﻧ ﺪﻨﺳﺃ ﹰﻼﺒﺟ ﺖﻟﺯﺎﻣ . ﺀﺎﺟﺭ ،ﺪﻤﺼﻟﺍ ﺪﺒﻋ ،ﻥﻻﺰﻏ ،ﺔﻨﻴﻣﺃ ،ﻦﻳﺪﻟﺍ ﺭﻮﻧ ﻲﺗﺍﻮﺧﺃ ﻭ ﻲﺗﻮﺧﺈﺑ ﻢﻜﻣﺎﻤﺘﻫﺍ ﻰﻠﻋ ﺍﺮﻜﺷ ﻭ ﻢﻜﺗﺪﻧﺎﺴﻣ ﻭ ﻢﻜﻤﻋﺩ ﻰﻠﻋ ﺍﺮﻜﺷ . ،ﻞﻴﻋﺎﻤﺳﺍ ،ﻦﻴﻣﺃ ،ﺲﻧﻮﻳ ﺀﺎﻳﺮﻛﺯ ، ﻝﻼﻋ ، ﻒﺳﻮﻳ ﺰﻨﻛ ﹰﺎﻤﺋﺍﺩ ﻖﻳﺪﺼﻟﺍ ﻦﻜﻟﻭ ﻖﻳﺪﺻ ﹰﺎﻤﺋﺍﺩ ﺲﻴﻟ ﺰﻨﻜﻟﺍ ﻢﻜﻌﻣ ﻥﻮﻛﺃ ﻱﺪﺣﻭ ﻲﻨﻧﺃ ﻮﻟ ﺎﻤﻛ ﺮﻘﻔﻟﺍﻭ ﻰﻨﻐﻟﺍ ﻲﻓ ،ﻖﻴﹼﻀﻟﺍﻭ ﺔﻌﺴﻟﺍ ﻲﻓ ،ﻥﺰﺤﻟﺍﻭ ﺡﺮﻔﻟﺍ ﻲﻓ ،ﺀﺍﺮﻀﻟﺍﻭ ﺀﺍﺮﺴﻟﺍ ﻲﻓ ﻲﻌﻣ ﻥﻮﻘﺒﺘﺳ ﻭ ﻢﺘﻨﻛ . ﻢﻜﺒﺣﺃ
ﺔﺠﻳﺪﺧ ﻡﻼﺳ ﻰﻠﻋ ﻦﻣ ﻲﻨﺘﻌﻤﺟ ﻢﻬﺑ ﻰﻠﺣﺃ ﺕﺎﻗﻭﺃ ﻲﺗﺎﻴﺣ ﻭ ﺪﻌﺳﺃ ﻲﻣﺎﻳﺃ ... ﻡﻼﺳ . ﺓﻭﺮﻣ ﻮﺻ ﻭ ﻚﺣﻭﺭ ﻝﺎﻤﺟ ﺎﻣﺃ ،ﺩﺭﻭ ﻞﻘﺤﻛ ﺓﺮﻄﻋﻭ ،ﺾﻴﺑﺃ ﻦﻴﻤﺳﺎﻳ ﻦﺼﻐﻛ ﺖﻧﺃ ﺔﻘﻴﻗﺭ ﹰﺎﻔﺻﻭ ﻪﻟ ﺪﺟﺃ ﻼﻓ ﻚﺗ ﹰﺍﺮﻴﺒﻌﺗ ﻻﻭ . ﺮﻴﺒﻋ ﻞﺒﻠﺒﻟﺍ ﺮﻴﻔﺻ ﺕﻮﺻ ﻭ ﻲﻬﺘﻨﺗ ﻻ ﺺﺼﻗ ﻭ ﻝﺍﻮﻃ ﻡﺎﻳﺃ ﻲﺣﻭﺭ ﻭ ﻲﺒﻠﻗ ﺔﻴﻨﻏﺃ ﻑﺮﻌﺗ ،ﻩﻮﺟﻮﻟﺍ ﺞﻬﺑﺃ ﻭ ﺔﺒﺣﻷﺍ ﺏﺮﻗﺃ . ﻒﺻﻮﺗ ﻻﻭ ﺪﻌﺗ ﻻ ﻚﻨﺳﺎﺤﻣ ﻭ ﻑﺮﺗ ﻲﺗﺎﻴﺣ ﻲﻓ ﻙﺩﻮﺟﻭ ﻭ ﻒﻐﺷ ﻚﺘﻘﻓﺭ ﻭ ﻑﺮﺷ ﻚﺘﺒﺤﺻ . ﻡﻼﺣﺃ ﻝﺎﻤﺟ ﺡﻭﺮﻟﺍ ﻭ ﺐﻠﻘﻟﺍ ﻭ ،ﺮﻬﻈﻤﻟﺍ ﻚﺤﺿ ﻰﺘﺣ ﺀﺎﻜﺒﻟﺍ ﻭ ﺕﺎﻗﻭﺃ ﻦﻟ ﺲﻨﹸﺗ ﻑﺎﺼﻧﺇ ﻙﺭﺪﻗﺃ ﻭ ﻚﻣﺮﺘﺣﺃ ﻭ ﻚﺒﺣﺃ ﺭﺪﻘﺑ ﻙﺩﺎﻨﻋ ﻭ ،ﻚﺘﻴﺒﺼﻋ ﺍﺮﻜﺷ ﻰﻠﻋ ﻞﻛ ﻭ ﻱﺃ ﻲﺷ
A mes chers neveux et nièces
Youssef , hamza, ahmed, latifa, yasmine, rime, israe, aymane, maha, lina,
nour et l’adorable petite ghita
A ma grande famille
Aucun langage ne saurait exprimer mon respect et ma considération pour
votre soutien et encouragements. Je vous dédie ce travail en
reconnaissance de l’amour que vous m’offrez quotidiennement et votre
bonté exceptionnelle. Que Dieu le Tout Puissant vous garde et vous
A mes ami(e)s imane, ahlam ,loubna, asmae, hamza merzaq, aïmane,
anas, nidal, ghandi et karima, oualid, zakariae, ismail et sara, nouhaila,
sarra, sara et lahssen, salma el bakkali, meryem, hicham lamaani et lina,
houssame et saloua, zineb et wafae nouioura, oumaima, hamza, insaf,
abderrahim, youssef chadid et ses camions, ghalmane, abdellah, abdou,
othmane, aya, dalila, malakout, suki, rabie, najib hassad
En souvenir des moments merveilleux que nous avons passés et aux liens
solides qui nous unissent.
Un grand merci pour votre soutien, vos encouragements, votre aide.
J’ai trouvé en vous le refuge de mes chagrins et mes secrets.
Avec toute mon affection et estime, je vous souhaite beaucoup de réussite
et de bonheur, autant dans votre vie professionnelle que privée.
Je prie Dieu pour que notre amitié et fraternité soient éternelles…
Les voix du chœur et le chœur philharmonique du Maroc
Je vous remercie d’avoir rendu mes rêves réalité, je vous remercie pour
toute l’énergie positive que vous m’avez donnée.
A nôtre maître et président de thèse
Professeur BOUHOUCHE AHMAD
Professeur de Génétique Humaine
Vous m’avez accordé un grand honneur en acceptant de présider le jury
de notre thèse.
Veuillez, Cher Maître, trouver dans ce modeste travail l’expression de ma
haute considération et mon profond respect pour avoir guidé les premiers
A nôtre maître et rapporteur de thèse
Professeur CHERRAH YAHIA
Professeur de Pharmacologie
Vous m’avez fait un grand honneur en acceptant de me confier ce travail.
Je vous remercie de votre patience, votre disponibilité et de vos précieux
conseils dans la réalisation de cette thèse. Votre compétence, votre
dynamisme et votre rigueur ont suscité une grande admiration et un
profond respect. Vos qualités professionnelles et humaines me serve
d’exemple. Veuillez croire à l’expression de ma profonde reconnaissance et
A nôtre maître et juge de thèse
Professeur ALAOUI KATIM
Professeur de Pharmacologie
Je vous remercie de votre enseignement et je vous suis très reconnaissants
de bien vouloir porter intérêt à ce travail que vous avez aimablement
accepté de juger. Veuillez accepter, cher maître, dans ce travail l’assurance
A nôtre maître et juge de thèse
Professeur ELMOSTAFA EL FAHIME
Professeur de biologie moléculaire
Je vous remercie de l’honneur que vous m’avez fait en acceptant de juger
ce travail et pour votre accueil chaleureux, votre disponibilité, votre aide
et vos remarques instructives.
Veuillez trouver dans ce travail, Cher Maître, l’expression de mon estime
et de ma considération.
Liste des abréviations
Liste des abréviations
5-FU : 5-fluouracile
ABCB : ATP Binding Cassette Subfamily B
ABCC : ATP Binding Cassette Subfamily C
ABCP : ATP Binding Cassette transporter in placenta
ADN : Acide désoxyribonucléique
ADNc : Acide désoxyribonucléique complémentaire
AINS : Anti-inflammatoire non stéroïdien
ApoE : Apolipoprotéine E
ARN : Acide ribonucléique
ARNm : Acide ribonucléique messager
ASC : Aire sous la courbe
ATP : Adénosine triphosphate
AVK : Anti vitamines K
AVP : Acide valproïque
BCRP : Breast Cancer Resistance Protein
BDKRB2 : Bradykinin Receptor B2
CAP : Cholesterol and Pharmacogenomics
CIPC : Clinical Pharmacogenetics Implementation Consortium
COX : Cyclooxygénase
CRP : Crotéine C réactive
CYP : Cytochromes P
dHPLC : Denaturing High Performance Liquid Chromatography
DL : Déséquilibre de liaison
DPD : Dihydropyrimidine déshydrogénase
DRD : Récepteurs dopaminergiques D-like
E2 : Oestradiol
EI : Effet indesirable
ELN : European Leukemia Network
EMA : European Medicine Agency
ERCC : Excision repair cross-complement gène
FDA : Food and drug administration
FISH : Fluorescence in-situ hybridisation
FMO : Monooxygénases à flavines
FRET : Fluorescence Resonance Energy Transfer
GoDARTS : Genetics of Diabetes Audit and Research in Tayside Scotland
GST : Glutathion S-transférases
GWAS : Genome-wide association study
HbA1c : Hémoglobine glyquée A1c
hERG : Human Ether-à-go-go-Related Gene
HMG-CoA : Hydroxy-méthyl-glutamyl-coenzyme A
HMGCR : Hydroxy-méthyl-glutamyl-coenzyme A réductase
IA : Inhibiteur de l’aromatase
ICH : International Conference on Harmonisation of Technical Requirements for
Registration of Pharmaceuticals for Human Use
IEC : Inhibiteurs de l'enzyme de conversion
IFNα : Interféron alpha
IM : Imatinib mésylate
INR : International Normalized Ratio
ISP : Ion Sphere Particule
ITK : Inhibiteurs de tyrosine kinase
LLA : Leucémie lymphoblastique aiguë
LMC : Leucémie myéloïde chronique
ME : Métaboliseurs extensif
MGB : Minor Grove Binder
MI : Métaboliseur intermédiaire
ML : Métaboliseurs lent
MM : MisMatch
MTHFR : Méthylène tétrahydrofolate réductase
MTX : Méthotrexate
MU : Métaboliseurs ultrarapides
MXR : Mitoxantrone Resistance protein
NAD(P)H+ : Nicotinamideadénine-dinucléotide-phosphate
NAT : N-acétyltransférases
NCBI : National Center for Biotechnology Information
NGS : Next-generation sequencing
NHGRI : Institut national de recherche sur le génome humain
OATP : Organic-anion-transporting polypeptides
OCT : Organic cation transporter
P-gp : P-glycoprotéine
PCR : Polymerase Chain Reaction
PGt : Pharmacogenetic
PM : Perfect Match
PMT : Photomultiplicateur
PRINCE : Pravastatin Inflammation/CRP Evaluation
RCyC : Réponse cytogénétique complète
RCyM : Réponse cytogénétique majeure
RFLP : Restriction Fragment Length Polymorphism
RHC : Réponse hématologique complète
RMM : Réponse moléculaire majeure
Rt-PCR : PCR en temps réel
SBS : Sequencing by synthesis
SCA : Syndrome coronarien aigu
SNC : Système nerveux central
SNP : Single-nucleotide polymorphism
SOLiD : Séquence par OligoLigation et Detection - Applied Biosystems
SSCP : Single Strand Conformation Polymorphisms
SSLP : Simple Sequence Length Polymorphism,
SULT : Sulfotransférase
TILLING : Targeting Induced Local Lesions in Genomes
TPMT : Thiopurine S-Méthyltransférase
TS : Thymédylate synthase
UGT : UDP-glucuronosyltransférases
UMAS : Universal Micro-Array System
VCR : Vincristine
Liste des figures
Figure 1: Localisation des variations génétiques affectant la réponse aux médicaments. ... 3 Figure 2: familles pharmacologiques - Les membres d’une famille pharmacologique ont
en commun l’effet principal tandis que les effets secondaires peuvent différer selon les substances ... 12
Figure 3: Profil de digestion de produits de PCR digérés ou non par l’enzyme de
restriction PstI ... 65
Figure 4: Électrophorégramme du séquençage d’un sujet hétérozygote C/A (double pic) et
d’un sujet homozygote C/C (pic unique) pour le polymorphisme étudié ... 66
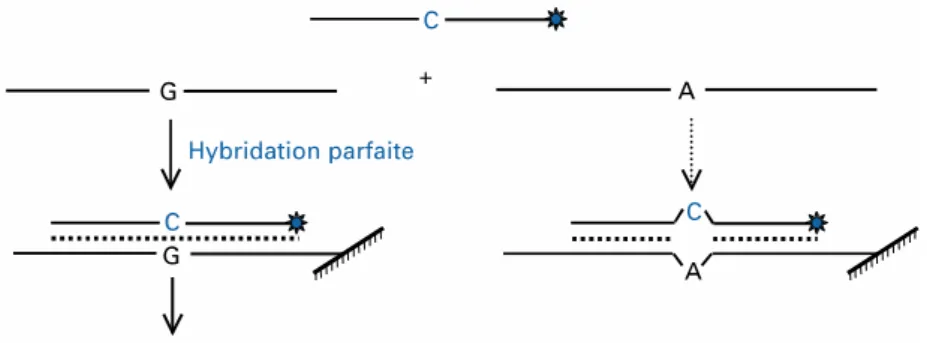
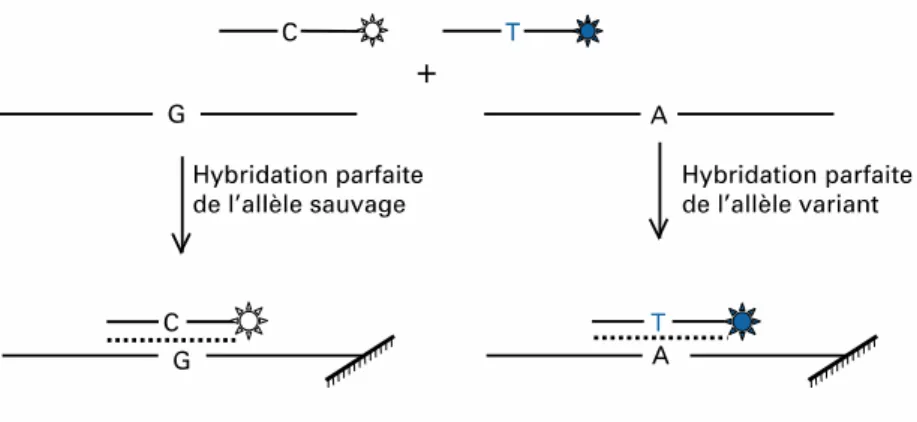
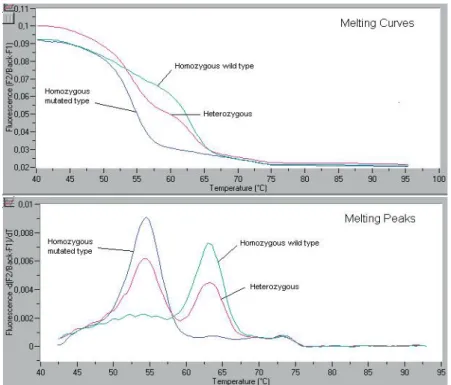
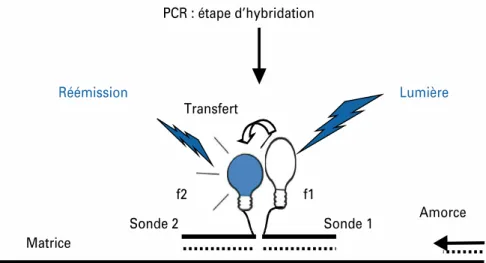
Figure 5: Pincipe du pyroséquençage ... 66 Figure 6: Principe de l’hybridation spécifique d’allèle avec une sonde discriminante ... 67 Figure 7: Principe de l’hybridation spécifique d’allèle avec deux sondes discriminantes ... 68 Figure 8: Principe de la technique d’extension d’amorces ... 68 Figure 9: Résultats de l’étude d’un polymorphisme par mesure du point de fusion par PCR
en temps réel ... 69
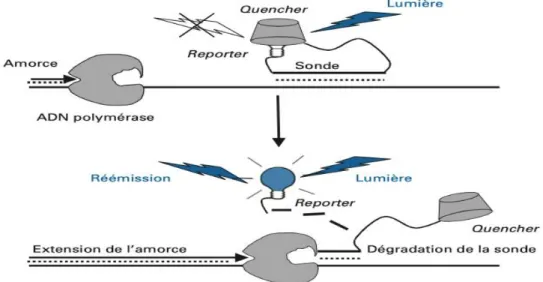
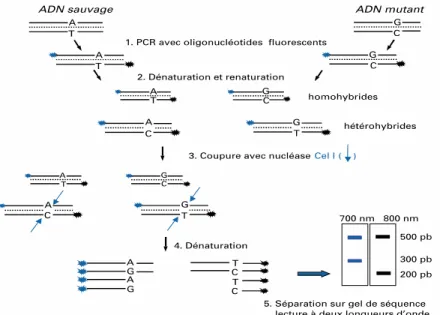
Figure 10: Principe du TaqMan... 70 Figure 11: Principe du FRET ... 72 Figure 12: Principe du fonctionnement des balises moléculaires ... 72 Figure 13: Principe de la technique de TILLING ... 74 Figure 14: Principe de la PCR-SSCP ... 75 Figure 15: Principe de la technique de la dHPLC et résultats d’un chromatogramme ... 76 Figure 16: Principe de la technique de SNP mining ... 77 Figure 17: Résultat d’un génotypage par analyse de STR ... 78 Figure 18: Les différentes étapes du NGS ... 79
Figure 19: Représentation schématique des deux grands principes d’enrichissement de
l’ADN génomique du patient. ... 80
Figure 20: Schéma du principe de séquençage sur une plateforme MiSeq (Illumina). ... 82 Figure 21: Schéma du principe du séquençage sur une plateforme Ion Proton™ (Thermo
Fischer). ... 83
Figure 22: Principe de la technologie des à puces à ADN ... 86 Figure 23: Cinétique et différents niveaux de réponse ou de résistance chez un patient
atteint de LMC PC traitée d’emblée par IM 400 mg/j. ... 99
Figure 24: Proportion de patients en situation de résistance hématologique primaire ou
secondaire à l’IM en fonction des différentes phases de la maladie. ... 100
Figure 25: Proposition d’arbre décisionnel global en 2007 devant une résistance à l’IM... 104 Figure 26: Métabolisme du tamoxifène en endoxifène ... 106
Liste des tableaux
Tableau 1: Définitions de l’échec (*) et des réponses suboptimales () à l’IM chez les
patients porteurs de LMC récemment diagnostiquées et traitées par IM 400 mg/j... 99
Tableau 2: Résumé des résultats des essais de phases I et II avec le nilotinib et le dasatinib
Sommaire
I. Introduction ... 2 II. Rappel pharmacocinétique et pharmacodynamique ... 6
1. Pharmacocinétique ... 6 1.1. Définitions ... 6 1.2. Bases physiologiques des phases pharmacocinétiques ... 6 a) Absorption ... 6 b) Distribution... 8 c) Métabolisme ... 9 C.1) Réactions de phase 1 ... 9 C.2) Réactions de phase 2 ... 10 d) Elimination ... 10 d.1) Elimination rénale ... 11 d.2) Autres voies d’élimination ... 11 2. Pharmacodynamie... 12 2.1. Mécanisme d’action ... 13 a. Action par fixation spécifique ... 13 a.1) Fixation sur une protéine ... 13 a.2) Fixation sur le génome ... 14 a.3) Autres sites de fixation ... 14 b. Action sans fixation dans l'organisme ... 14 c. Action sur des organismes étrangers ... 14 3. Variabilité et conséquences ... 15 3.1. Sources de variabilité de la réponse aux médicaments ... 15 a) Mécanismes des EI et polymorphismes pharmacodynamiques ... 15
b) Mécanismes des EI et polymorphismes pharmacocinétiques ... 17 b.1 Administration et élimination des médicaments ... 17 b.2. Métabolisme de phase I des médicaments ... 17 b.3. Métabolisme de phase II des médicaments ... 18
III Pharmacogénétique et pharmacogénomique ... 21
1. Historique et définition ... 21 1.1. Historique de la pharmacogénétique et de la pharmacogénomique... 21 1.2. Définitions ... 24 2. Les applications cliniques de la pharmacogénétique ... 25 2.1. Amélioration de la sécurité et de l’efficacité des médicaments ... 25 2.2. Codéveloppement de médicaments et de tests de pharmacogénétique ... 26 2.3. Sauvetage de médicaments ... 26 2.4. Surveillance post-commercialisation ... 27 3. La pharmacogénétique au Maroc ... 27
IV. Cibles étudiées en pharmacogénétique ... 31
1. Pharmacogénétique du métabolisme de phase I des médicaments... 31 1.1. Enzymes du cytochrome P450 (CYP)... 31 a) CYP1A2 ... 31 b) CYP2B6 ... 32 c) CYP2C ... 34 d) CYP2D6 ... 39 e) CYP3A ... 42 1.2. Monooxygénases à flavines ... 43 2. Pharmacogénétique des gènes impliqués dans le métabolisme de phase II des médicaments ... 44
2.1. N-acétyltransférases (NATs) ... 45 2.2. UDP-glucuronosyltransférases (UGT) ... 46 2.3. Glutathion S-transférases (GST) ... 48 2.4. Sulfotransférase (SULT) ... 50 2.5. Thiopurine S-Méthyltransférase (TPMT) ... 52 3. Pharmacogénétique des transporteurs ... 53 3.1. Les transporteurs ABC ... 53 a) ABCB1 ... 53 b) ABCG2... 54 3.2. Transporteurs SLC ... 55 a) Polypeptides organiques transporteurs d'anions ... 56 b) Transporteurs de cations organiques... 58 4. Pharmacogénétique des enzymes ... 59 4.1. Vitamine K époxyde réductase VKORC1... 59 4.2. HMGCoA réductase ... 61 4.3. Dihydropyrimidine déshydrogénase ... 62
V. Les techniques d’analyses des polymorphismes génétiques ... 64
1. Techniques d’analyse des polymorphismes de type SNP ... 64 1.1. Techniques d’analyse des SNPs répertoriés ... 64 a) Les polymorphismes de taille de fragments de restriction ou RFLP (Restriction Fragment Length Polymorphism) ... 64 b) PCR et séquençage ... 65 c) Pyroséquençage ... 66 d) PCR et hybridation spécifique d’allèle ... 67 e) Extension d’amorce ... 68
f) Mesure du point de fusion par PCR en temps réel (rt-PCR) ... 69 1.2. Techniques d’analyse des SNPs inconnus... 73 a) Le TILLING ... 73 b) La PCR-SSCP (Single Strand Conformation Polymorphisms) ... 74 c) La chromatographie liquide à haute pression en condition dénaturante (dHPLC) .. 75 d) Le SNP mining ... 76 e) Le séquençage ... 77 2. Les polymorphismes de type microsatellite (Simple Sequence Length Polymorphism, SSLP) ... 77 3. Séquençage à haut débit ... 78 3.1. Construction des librairies ... 80 3.2. Séquençage NGS proprement dit ... 81 4. Puce à ADN ... 84
VI. Une approche par étapes pour la mise en œuvre des tests pharmacogénétiques dans le cadre des soins primaires... 88
1. Étape 1 : Identification du patient ... 90 2. Étape 2 : Commande de tests pharmacogénétiques ... 91 3. Étape 3: Application des résultats des tests pharmacogénétiques ... 92 4. Étape 4 : Information du patient ... 95
VII. Pharmacogénétique de quelques médicaments approuvés par la FDA à test obligatoire ... 98
1. L’imatinib mésylate ... 98 1.1. Fréquence et définitions de la résistance à l’imatinib mésylate ... 98 a) Définition... 98 b) Fréquence ...100 1.2. Mécanismes de résistance à l’imatinib mésylate ...101
a) Modification de la biodisponibilité intracellulaire de l’IM ...101 b) Amplification de BCR-ABL...101 c) Mutations du domaine kinase d’ABL ...101 d) Évolution clonale ...102 e) Cellules souches quiescentes ...102 1.3. Gestion de la résistance à l’imatinib mésylate ...102 2. Tamoxifène...105 2.1. Polymorphismes du CYP2D6 ...105 2.2. Génotypage du CYP2D6 et tamoxifène en pratique clinique ...107 3. Acide valproïque...107 3.1. Les variantes de l'UGT ...107 3.2. Les variantes du cytochrome P450 ...108 4. La vincristine ...109 5. L'anastrozole ...110 5.1. Cytochrome P450 et polymorphismes de nucléotides uniques ...110 5.2. Impact des polymorphismes du CYP19A1 sur l'efficacité de l'activité de l'anastrozole et de l'aromatase ...111 5.3. Influence des SNP sur les effets secondaires associés à l'anastrozole ...112
VIII. Conclusion ...115 Résumés ...117 Bibliographie ...121
1
2
I. Introduction
La réponse aux médicaments varie considérablement d'un patient à l'autre, ce qui est largement attribué aux différences innées entre les individus dans leur capacité à métaboliser et à réagir génotype d'une personne dans les décisions de traitement médicamenteux, dans le but de fournir la thérapie la plus efficace et la plus sûre pour ce patient. Au cours de la dernière décennie, des progrès significatifs ont été réalisés dans notre compréhension de la contribution des différences génétiques en pharmacocinétique et en pharmacodynamie dans la variabilité interindividuelle de la réponse aux médicaments. La pharmacogénomique peut conduire non seulement à une meilleure utilisation des thérapies existantes, mais aussi à la mise au point de nouveaux médicaments basés sur une meilleure compréhension du contrôle génétique des fonctions cellulaires.
Le génome humain est composé d'environ 20 000 gènes codant pour des protéines. Le type de variation le plus courant est, de loin, le polymorphisme d'un seul nucléotide (SNP), qui se définit comme une différence d'une seule base qui existe entre les individus. Plus de 22 millions de SNP ont été signalés dans le génome humain (1). Les SNP qui entraînent une substitution d'acide aminé sont dits non synonymes. Les SNP non synonymes présents dans les régions codantes du gène (par exemple, les exons) peuvent affecter l'activité de la protéine et avoir des conséquences importantes sur les réponses aux médicaments dont le métabolisme, le transport ou les effets cellulaires sont dépendants. Les polymorphismes synonymes n'entraînent pas de substitution d'acides aminés ; cependant, ceux qui se produisent dans une région régulatrice du gène (par exemple, une région du promoteur, un intron) peuvent modifier l'expression du gène et la quantité de protéine produite. Deux SNP ou plus sont fréquemment hérités ensemble, en se basant sur le hasard. Cet effet est appelé déséquilibre de liaison (DL). Un haplotype désigne un ensemble de SNP qui se trouvent en DL. Les autres types de variations comprennent les polymorphismes d'insertion-délétion, les courtes répétitions en tandem et les variations nucléotidiques à bases multiples.
3
Des polymorphismes se produisent couramment pour les gènes codant pour les protéines du métabolisme des médicaments, les transporteurs et les protéines cibles (figure 1). Le métabolisme des médicaments et les génotypes des transporteurs peuvent affecter la biodisponibilité des médicaments sur le site cible ; les génotypes des cibles des médicaments peuvent affecter la sensibilité d'un patient à un médicament. Dans de nombreux cas, les gènes des protéines impliquées dans la disponibilité du médicament ainsi que les gènes des protéines du site cible du médicament influencent conjointement la réponse au médicament.
Les termes "pharmacogénétique" et "pharmacogénomique" sont souvent utilisés de manière interchangeable. Comme les réponses aux médicaments sont généralement déterminées par des protéines multiples plutôt que par des protéines uniques, les tendances récentes des recherches sur les déterminants de la réponse aux médicaments sont passées de la pharmacogénétique à la pharmacogénomique. Toutefois, par simplicité, la pharmacogénétique et la pharmacogénomique seront considérées comme synonymes dans cette thèse.
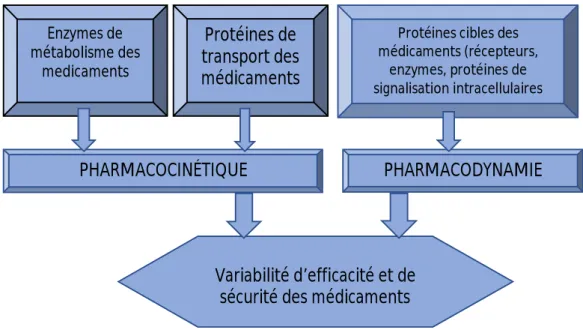
Figure 1: Localisation des variations génétiques affectant la réponse aux médicaments. Celles qui se produisent dans les gènes de métabolisme ou de transport des médicaments peuvent affecter la pharmacocinétique des médicaments, tandis que les SNP dans les gènes codant pour les protéines cibles des médicaments peuvent affecter la pharmacodynamique des médicaments.
Enzymes de métabolisme des medicaments Protéines de transport des médicaments
Protéines cibles des médicaments (récepteurs,
enzymes, protéines de signalisation intracellulaires
PHARMACOCINÉTIQUE PHARMACODYNAMIE
Variabilité d’efficacité et de sécurité des médicaments
4
Malgré les progrès scientifiques réalisés, la médecine personnalisée envisagée il y a de nombreuses années n'est pas encore devenue une pratique courante. Des exceptions à ce retard existent surtout en oncologie et plus récemment en cardiologie, où le génotypage pour déterminer l'efficacité du clopidogrel commence à devenir une routine dans certains centres médicaux universitaires (2)(3). Des exemples de thérapies guidées par le génotype commencent à émerger dans d'autres domaines thérapeutiques. Cependant, des défis importants se posent encore dans les aspects éthiques, socio-économiques, réglementaires, législatifs et de développement de médicaments qui doivent être abordés et résolus avant que la médecine personnalisée puisse être mise en œuvre de manière pratique et satisfaisante dans la pratique clinique à plus grande échelle.
Cette thèse va aborder plusieurs points, d’abord un rappel de la pharmacocinétique et de la pharmacodynamie et les conséquences de leurs variabilités sur la survenue d’effet indésirables, puis défini la pharmacogénétique, précise ses applications cliniques et donne une idée sur la situation actuelle de la pharmacogénétique au Maroc. Elle cite les différentes cibles et biomarqueurs et leurs variants impliqués dans la variabilité de réponse aux médicaments et les techniques d’analyse permettant la détection des polymorphismes génétiques de ces cibles. Ensuit on va détailler les quatres étapes principales et un certain nombre de considérations cliniques qui se posent systématiquement avec l'adoption de tests pharmacogénétiques dans le cadre des soins primaires et qui comprennent l'identification du patient, la commande de tests, l'application des résultats des tests et l'information du patient.
Et finalement et à travers des exemples de médicament dont le test génétique est obligatoire selon la FDA on va illustrer le rôle de pharmacogénétique dans l’identification des médicaments qui seront métabolisés trop rapidement ou trop lentement et ceux qui ont moins de risques de ne pas agir.
5
Rappel pharmacocinétique
et pharmacodynamique
6
II. Rappel pharmacocinétique et pharmacodynamique
1. Pharmacocinétique
1.1. Définition
La pharmacocinétique est traditionnellement définie comme l’étude du sort des médicaments dans l’organisme, depuis leur absorption jusqu’à leur élimination. Cette discipline permet ainsi de relier les doses de médicaments administrées aux concentrations sanguines observées, et de décrire leur évolution en fonction du temps.
1.2. Bases physiologiques des phases pharmacocinétiques
a) Absorption
Dans la majorité des situations rencontrées en clinique, le site d’administration du médicament est distinct de son site d’action : administration par voie orale d’un médicament dont le site d’action pourra être le cœur, les vaisseaux, les reins par exemple. Le site de l’administration, le tube digestif dans la situation présentée ici, n’est qu’un lieu de transit, le médicament étant ensuite véhiculé dans l’organisme par la circulation sanguine. L’absorption correspond donc au passage du médicament de son site d’administration à la circulation sanguine générale, au travers de membranes biologiques.
Avant toute absorption, il est souvent nécessaire que le principe actif soit libéré de la forme galénique : dissolution du comprimé, ou de la paroi de la gélule. Plusieurs phénomènes interviennent ensuite dans l’absorption des médicaments : d’une manière schématique, on peut distinguer des phénomènes d’absorption actifs et des phénomènes d’absorption passifs.
Le transport passif, ou simple diffusion, représente le passage du médicament du milieu le plus concentré au milieu le moins concentré. Cette diffusion suit la première loi de Fick (le flux de diffusion est proportionnel au gradient de concentration), dont la formulation usuelle en pharmacocinétique est la suivante :
k × S × Δc D= L
Avec : D : vitesse de diffusion, k : constante, S : surface d’échange, Δc : différence de concentration entre les deux milieux, L : épaisseur de la membrane.
7
La valeur de la constante k dépend de la taille de la molécule (plus le poids moléculaire augmente, plus la valeur de k diminue), et de sa lipophilie. La lipophilie peut être mesurée en calculant le coefficient de partage huile/eau : ce coefficient représente le rapport des solubilités dans un solvant organique et dans un solvant aqueux. La liposolubilité d’une molécule étant variable selon son état d’ionisation, le pH du milieu et le pKa de la molécule seront aussi des facteurs pouvant influencer la diffusion. En effet, une molécule ionisée est davantage soluble dans l’eau que la même molécule sous sa forme non ionisée, et sera moins à même de passer les barrières biologiques lipidiques. Cette ionisation est sous la dépendance du pH du milieu et de la constante de dissociation entre la forme ionisée et la forme non ionisée. Pour une molécule acide qui se dissout dans l’eau, cette constante de dissociation Ka est rattachée à l’équilibre suivant entre la forme non ionisée AH et la forme ionisée A− :
AH + H 2 O ⇔ A− + H 3 0+ Avec Ka =[ A-][ H 3 0+ ] /[AH ]
En fonction du pH du milieu, et donc de la concentration en [H3O+], l’équilibre sera déplacé dans un sens ou dans l’autre. Ainsi, un médicament acide faible sera d’autant plus sous sa forme non ionisée que le pH sera faible, et donc davantage absorbé. Ce phénomène explique les différences d’absorption entre l’estomac (milieu acide) et l’intestin grêle (milieu basique) selon les médicaments.
Ce mécanisme de transport passif n’est pas saturable. En considérant que la diffusion ne se fait que dans un sens, et en dérivant et reformulant la relation provenant de la loi de Fick, nous obtenons la relation usuelle du phénomène d’absorption :
dQ0/dt = −K × Q0
Avec
dQ0/dt : quantité de médicament diffusant depuis le compartiment extérieur
(compartiment d’absorption) vers le compartiment intérieur (compartiment central), et Q0 : quantité de médicament dans le compartiment extérieur.
8
Le transport actif, relativement rare, consiste en l’utilisation de pompes permettant le passage du médicament au travers d’une membrane, y compris lorsque le gradient de concentration n’est pas favorable, et nécessite un apport énergétique. Ce mécanisme est saturable.
Il existe également d’autres mécanismes d’absorption d’importance moindre, comme le transport facilité ou la pinocytose (4).
Bien entendu, toute perturbation du fonctionnement normal du tube digestif pourra avoir un impact sur l’absorption du médicament : diarrhée ou constipation pourront réduire ou prolonger le temps de présence du médicament dans le tube digestif et donc la fraction de médicament absorbée, une achlorhydrie ou un traitement par inhibiteur de la pompe à protons pourront réduire ou augmenter l’ionisation d’un médicament et son absorption au niveau de l’estomac, etc.
b) Distribution
La distribution, qui correspond à la diffusion du médicament dans les différents tissus de l’organisme, est également influencée par le caractère plus ou moins lipophile du principe actif, ainsi que par son affinité pour différents tissus : os, tissu adipeux, …
Le médicament peut se fixer de manière réversible aux protéines plasmatiques : cette fixation se fait principalement sur l’albumine et l’alpha 1 glycoprotéine acide, l’albumine fixant préférentiellement les médicaments acides, alors que l’alpha 1 glycoprotéine acide fixe davantage les médicaments basiques. On distingue alors la fraction de médicament liée aux protéines et la fraction libre. Le plus souvent, seule la fraction libre peut agir sur son site d’action, diffuser dans les tissus, ou subir les processus de métabolisme ou d’élimination, si bien que l’on peut considérer que la fraction du médicament qui est liée aux protéines plasmatiques constitue une forme de stockage du médicament.
La fixation étant un processus réversible obéissant à la loi d’action de masse, nous pouvons considérer ce phénomène comme un équilibre entre une fraction libre et une fraction liée :
9 Avec :
[protéine] : concentration molaire en protéines non occupées par le médicament, [médicament] : concentration molaire en médicament non lié aux protéines,
[complexe protéine] : concentration molaire en complexe médicament-protéine,
K1 et K2 : constantes de vitesse des réactions.
On peut également définir KA = K1/K2 comme étant la constante d’association, et KD =1/KA. comme étant la constante de dissociation.
Cette fixation aux protéines peut être pratiquement nulle pour certains médicaments (isoniazide par exemple), ou au contraire extrêmement importante pour d’autres principes actifs (liaison à 97% pour la warfarine par exemple).
c) Métabolisme
Le métabolisme correspond à la transformation du médicament par l’organisme. Cette transformation est essentiellement réalisée par des réactions impliquant des enzymes. Pour les médicaments, il est usuel de séparer en deux groupes les réactions impliquées dans le métabolisme : les réactions de phase 1 et les réactions de phase 2. Les réactions de phases 1 précèdent habituellement celles de phase 2 dans la chronologie du métabolisme d’un médicament.
C.1) Réactions de phase 1
Les réactions de phase 1 sont principalement : - des réactions d’oxydation (perte d’électron), - des réactions de réduction (gain d’électron)
- des réactions d’hydrolyse (décomposition du principe actif par l’eau), de création ou rupture de cycles.
D’une manière générale, ces réactions conduisent à la formation de métabolites polaires, plus hydrophiles que la molécule mère. On parle parfois de réactions de « fonctionnalisation » car elles correspondent à l’ajout d’une fonction chimique à la molécule mère : fonction acide, alcool, amine…
10
Certaines enzymes impliquées dans ces réactions de phase 1 appartiennent à la superfamille du cytochrome P450. Cette superfamille comporte trois familles chez l’homme : CYP1, CYP2, CYP3. Ces enzymes sont caractérisées par :
- l’existence de polymorphismes génétiques pour certaines isoformes, 2D6 et 2C19 principalement,
- la possibilité d’avoir leur action stimulée ou inhibée par certains xénobiotiques. A titre d’illustration, citons l’induction du CYP 2C19 et 3A4 par la rifampicine, pouvant fortement réduire l’efficacité des médicaments qui lui sont associés, en augmentant leur métabolisme par ces enzymes (cas de certains médicaments de type antivitamine K)(5). Une situation particulière peut également être décrite : un médicament peut induire son propre métabolisme, conduisant ainsi au fur et à mesure des administrations à une augmentation de sa dégradation.
C.2) Réactions de phase 2
Les réactions de phase 2 sont principalement des réactions de conjugaison, qui correspondent à des réactions, souvent réversibles, entre la molécule mère et une biomolécule de l’organisme de nature variable : acide glucuronique, glutathion, urée… Ces réactions peuvent se produire au niveau des fonctions créées lors de réactions de phase 1. Les métabolites formés sont le plus souvent davantage solubles dans l’eau que la molécule mère, ce qui facilite leur élimination dans les urines.
d) Elimination
L’élimination d’un médicament de l’organisme peut se faire par différentes voies : urinaire, biliaire, respiratoire (dans l’air expiré), cutanée (par la sueur), salivaire, ou dans le lait maternel. Les voies principales sont l’élimination par les reins dans les urines, et l’élimination par le foie dans la bile. Pour un certain nombre de voie d’élimination, un phénomène de réabsorption du médicament peut être rencontré, qui contribue à diminuer ou ralentir l’élimination du médicament. Les mécanismes décrits pour l’absorption sont également retrouvés pour l’élimination (diffusion passive, transport actif, pinocytose…).
11
d.1) Elimination rénale
Les reins sont les principaux organes d’élimination de l’organisme. Ils sont formés d’un ensemble d’unités fonctionnelles appelées néphrons (environ un million de néphrons par rein), comportant deux parties : le glomérule responsable de la filtration du sang, et le tubule dans lequel se produisent des phénomènes d’absorption et d’excrétion passifs et actifs. Les tubules des néphrons rejoignent le canal collecteur, qui se prolonge par l’uretère et la vessie. Seules les molécules de taille inférieure à environ 60 000 Dalton, non liées aux protéines plasmatiques, peuvent filtrer à travers le glomérule. Il est possible de calculer la quantité de médicament filtrée par unité de temps (F) au niveau des glomérules en appliquant la formule :
F = [(1 − p) × C ] × DFG Avec :
p : fraction de médicament liée aux protéines plasmatiques C : concentration sanguine en médicament
DFG : Débit de Filtration Glomérulaire
d.2) Autres voies d’élimination
Le foie, responsable de la plus grande part des réactions de métabolisme décrites précédemment, est également impliqué dans l’élimination de certains médicaments. En effet, après administration par voie orale, les médicaments absorbés par le tube digestif circuleront par la veine porte en direction du foie, où ils pourront être métabolisés (effet de premier passage hépatique) puis rejoindre la circulation générale par la veine cave inférieure, où bien être excrétés dans la bile, au niveau intestinal, pour être éliminés dans les fèces. Cette excrétion biliaire peut être plus ou moins fortement compensée par une réabsorption du médicament dans l’intestin, formant un cycle entre l’intestin, la veine porte, le foie et la bile, appelé « cycle entéro-hépatique » (6).
12
2. Pharmacodynamie
Elle est l’étude de la relation concentration-effet. Elle reflète l’action du médicament sur l’organisme. Celle-ci est intimement liée aux caractéristiques pharmacocinétiques de la molécule d’intérêt. Elle est aussi dépendante de l’environnement, du pH ou de la température par exemple, lors de l’interaction de la molécule active avec sa cible, le plus souvent un récepteur.
On appelle effet pharmacodynamique une modification mesurable et reproductible, fonctionnelle ou organique, provoquée par un médicament dans un système biologique appelé « effecteur ».
Un médicament provoque un ou plusieurs effets pharmacodynamiques, pour des doses qui peuvent être différentes.
Un médicament possède :
- un effet principal, utilisé en thérapeutique
- des effets secondaires (latéraux), qui sont utiles ou indifférents ou gênants ou nuisibles.
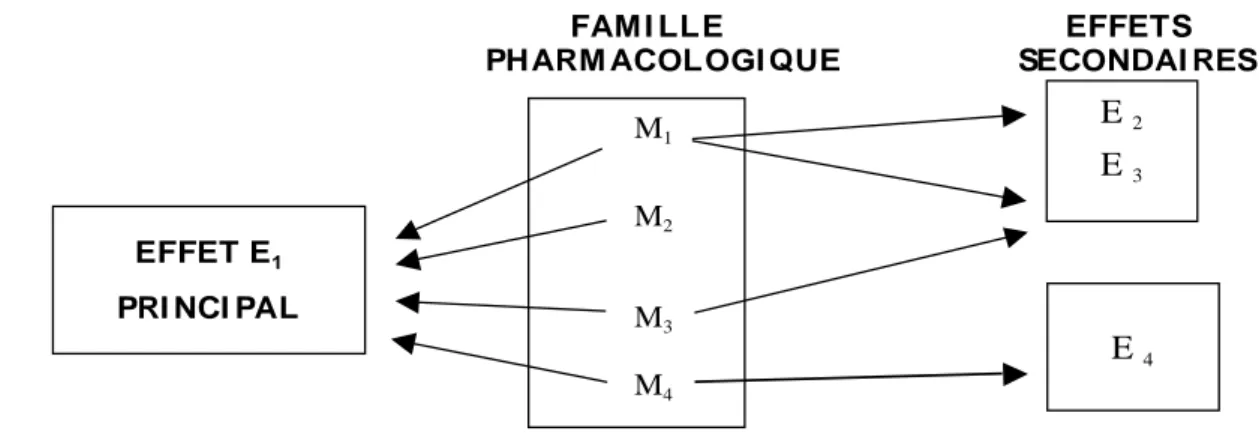
Un même effet pharmacodynamique peut être provoqué par plusieurs médicaments ; l'ensemble de ces médicaments constitue une famille pharmacologique (Figure 2).
FAM I LLE EFFETS
PHARM ACOLOGI QUE SECONDAI RES
M1 M2 M3 M4 E 2 E 3 E 4 EFFET E1
PRI NCI PAL
Figure 2: familles pharmacologiques - Les membres d’une famille pharmacologique ont en commun l’effet principal tandis que les effets secondaires peuvent différer selon les substances.
13
2.1. Mécanisme d’action
Les structures sur lesquelles les médicaments agissent sont appelées « cibles ».
a. Action par fixation spécifique
Les médicaments agissent en général par fixation dans l'organisme : corpora non agunt nisi fixata (EHRLICH). Cette fixation est spécifique du médicament et de son effet. Elle dépend étroitement de sa structure et de ses propriétés chimiques.
La structure moléculaire sur laquelle se fixe le médicament est appelée « récepteur ». Remarque : le terme de récepteur est ambigu. Il est pris ici dans une acception large, celle d’une molécule qui fixe un médicament, quelle qu’elle soit chimiquement ou fonctionnellement.
a.1) Fixation sur une protéine
Dans la plupart des cas, la fixation s’effectue sur une protéine. Il peut s’agir de :
- récepteurs : les récepteurs sont des protéines particulières qui font partie des systèmes physiologiques de communication intercellulaire (transmission de l’information).
Remarque : le terme récepteur est pris ici dans son sens restrictif - enzymes : les médicaments peuvent :
• activer ou inhiber le fonctionnement de l’enzyme (activateurs enzymatiques et inhibiteurs enzymatiques). Leur action peut être réversible ou irréversible
• détourner l’activité enzymatique. C’est le cas des anti-métabolites, faux substrats qui ressemblent au substrat physiologique et qui prennent sa place, mais les produits de la réaction sont inactifs
- transporteurs : les transporteurs sont des protéines qui font passer les ions et les petites molécules physiologiques à travers les membranes cellulaires. On distingue : • des transports passifs (transporteurs, pour un ion ou une molécule dans un sens ; symporteurs, pour plusieurs ions ou molécules ; antiporteurs, pour des échanges d’ions ou de molécules)
14
• des transports actifs, avec dépense d’énergie (pompes). C’est la cible de médicaments qui activent ou inhibent leur fonctionnement - canaux ioniques : les canaux sont des protéines transmembranaires permettant le passage sélectif de certains ions (Na+ , K+ , Ca++ , Cl- ) suivant le gradient électrochimique. Ils peuvent être ouverts ou fermés. Leur ouverture peut être provoquée par un ligand (excitation) ou par un potentiel d’action. Les effets peuvent être la naissance d’un potentiel d’action, une contraction, une sécrétion ou inversement une inexcitabilité cellulaire
- protéines de la structure cellulaire : comme la tubuline, rarement.
a.2) Fixation sur le génome
Des médicaments peuvent se fixer sur le génome (ADN, ARN, protéines associées). Ils peuvent moduler l’expression génétique. Certains peuvent empêcher la prolifération cellulaire. Cette fixation peut être aussi responsable de l’effet mutagène ou cancérigène de certains d'entre eux.
a.3) Autres sites de fixation
Certains rares médicaments se fixeraient ailleurs que sur des protéines ou des nucléotides, par exemple sur les lipides membranaires ou les sels de calcium de la trame osseuse.
b. Action sans fixation dans l'organisme
Ces médicaments agissent grâce à leurs propriétés physiques (volume, pouvoir couvrant, etc.) ou en modifiant celles du milieu extra cellulaire (pouvoir osmotique, équilibre acido-basique, équilibre électrolytique, etc.). Les structures chimiques peuvent être très différentes pour un même effet.
c. Action sur des organismes étrangers
Certains médicaments agissent sur des organismes pathogènes (bactéries, virus, parasites, champignons). Les mécanismes d’action sont semblables à ceux énumérés ci-dessus.(7)
15
3. Variabilité́ et conséquences
3.1. Sources de variabilité de la réponse aux médicaments
La réponse aux médicaments est souvent variable d’un individu à l’autre, tant sur le plan pharmacologique (efficacité) que sur le plan toxicologique (EI), ce qui rend parfois leur maniement délicat surtout quand l’index thérapeutique est faible. Les inefficacités thérapeutiques, plus difficiles à appréhender, posent un problème du même ordre (exemple : un malade sur deux ne tire aucun profit de son traitement antidépresseur) (8). Donc, les variations interindividuelles de réponse aux médicaments représentent un problème médical et de santé publique important. Les médicaments, même prescrits aux doses recommandées, peuvent être une cause importante de morbidité et de mortalité, ou à l’inverse, d’inefficacité chez certains patients. En dehors de problème d’observance, une telle variabilité dépend de facteurs environnementaux (alimentation, interactions médicamenteuses, tabagisme, consommation d’alcool), de l’état du malade (âge, sexe, grossesse, sévérité́ de la maladie, pathologies associées), et enfin de déterminants génétiques (variations génétiques du métabolisme, du transport des médicaments et des cibles pharmacologiques) (9).
a) Mécanismes des EI et polymorphismes pharmacodynamiques
Les substances thérapeutiques exercent leur action par le biais d’interactions spécifiques avec des structures cibles comme des récepteurs ou des canaux ioniques. Ces protéines présentent également le plus souvent des différences d’origine génétique au niveau de leur structure et de leur régulation, avec des répercussions sur l’efficacité d’un traitement (10).
Récepteurs
On connaît un nombre croissant de polymorphismes affectant l’étape pharmacodynamique de l’action du médicament, les exemples les plus classiques sont :
– le polymorphisme codant pour le gène VKROC1 (mutation dans le gène VKROC1 : la sous-unité du complexe de la vitamine K époxyde réductase) responsable de la résistance aux antivitamines K (AVK), pouvant exposer au risque thrombotique et nécessitant des posologies plus élevées (8)(11)(10);
16
– le polymorphisme du gène codant pour SLCO1B1 (“solute organic anion-transporting polypeptide” responsable de l’absorption des statines dans le foie) induisant une myopathie après la prise de simvastatine(10);
– le polymorphisme du gène codant pour BDKRB2 (récepteur beta2 bradykinine) responsable de la toux induit par les inhibiteurs de l’enzyme de conversion (IEC) ;
– le polymorphisme du gène codant pour les récepteurs ostrogéniques responsables d’une déminéralisation sous substitution post ménopausique ainsi que d’une thrombose veineuse après la prise de contraceptifs oraux (12) ;
– le polymorphisme du gène codant pour ERCC1 et pour ERCC2 (“excision repair cross-complement gene” responsable de la réparation croisée complétant le système de réparation par excision de nucléotides de l’ADN endommagé) responsable d’une neurotoxicité après prise de l’oxaliplatine;
– le polymorphisme du gène codant pour MTHFR (méthylène tétrahydrofolate réductase) induisant une toxicité neurologique ou hématologique lors de la prise du méthotrexate (MTX)(13);
– le polymorphisme du gène codant pour les DRD 2, 3 et 4 (récepteurs dopaminergiques D-like) responsables d’une agranulocytose lors de la prise des antipsychotiques, clozapine et halopéridol (11)(14)(15);
– le polymorphisme du gène codant pour les récepteurs sérotoninergiques pouvant induire une prise de poids sous neuroleptiques ;
– le polymorphisme du gène codant pour la thymédylate synthase (TS) induisant une toxicité hématologique lors de la prise de 5-fluouracile (11)(13);
– le polymorphisme du gène codant pour RYR1 (gène codant pour un récepteur ryanodine trouvé dans le muscle squelettique régulant le canal de libération du calcium) induisant une hyperthermie maligne lors de la prise des anesthésiques halogénés (halothane) et du succinylcholine (14)(16).
17
b) Mécanismes des EI et polymorphismes pharmacocinétiques
b.1 Administration et élimination des médicaments
Les molécules vont subir un transport transmembranaire. C’est la phase d’expulsion du produit ou de métabolites conjugués faisant intervenir généralement des transporteurs de la famille des protéines ABC. Des “transporteurs lents” consécutifs à un polymorphisme ont été identifiés pour la P-gp et divers analogues, affectant la pharmacocinétique de nouveaux substrats de ces transports (digoxine, ciclosporine, antirétroviraux, anticancéreux), de même que leur pénétration dans le système nerveux central (tricycliques, antiépileptiques) ou dans les cellules saines ou tumorales (anticancéreux). C’est le cas du polymorphisme de la P-gp responsable de la résistance aux antiépileptiques, aux anticonvulsivants, au tacrolimus et au ciclosporine (12)(14)(15)(17).
b.2. Métabolisme de phase I des médicaments
Il a pour but de créer ou de libérer des fonctions polaires, afin d’augmenter l’hydrophilie de la substance d’origine, c’est-à-dire augmenter sa solubilité en milieu aqueux afin de faciliter son élimination. Les réactions de la phase I sont catalysées par des oxygénases, des réductases, des estérases et des peptidases.
Réactions d’oxydation CYP 450 dépendants
Le système d’oxydation à CYP 450 est un système mono-oxygénase, multienzymatique capable de catalyser diverses biotransformations de médicaments nécessitant une molécule d’oxygène et de NAD(P)H+ (nicotinamide adénine dinucléotide phosphate).
Dans cette phase I CYP 450 dépendants, on trouve les polymorphismes suivants : – le polymorphisme du gène codant pour CYP2C9 induisant un surdosage lors de la
prise des AVK (les sujets homozygotes mutés nécessitent une diminution de posologies plus grande que les sujets hétérozygotes, jusqu’à 50 % de la dose utilisée chez les homozygotes sauvages) (8)(12)(14)(18);
– le polymorphisme du gène codant pour CYP2D6 responsable d’une toxicité chez les métaboliseurs lents (ML) de type cardiotoxicité ou effets atropiniques lors de la prise des antidépresseurs tricycliques (11)(12)(14);
18
– le polymorphisme du gène codant pour CYP2C19 et du gène codant pour CYP2D6 induisant une efficacité réduite chez les métaboliseurs ultra-rapides (MU) lors de la prise du tamoxifène (8)(10);
– le polymorphisme du gène codant pour CYP3A5 responsable de l’augmentation de la concentration en tacrolimus donnant une neurotoxicité (8) ;
– le polymorphisme du gène codant pour CYP2C9 responsable de la toxicité lors de la prise des sulfamides(18).
Réactions d’oxydation CYP 450 indépendants
Ces réactions impliquent d’autres enzymes n’appartenant pas au système CYP450 et qui sont capables d’oxyder des médicaments, on distingue :
Dans cette étape de réactions d’oxydation à CYP 450 indépendants, de réactions de réduction et de réactions d’hydrolyse on peut citer les polymorphismes suivants:
– le polymorphisme du gène codant pour la COX-2 (cyclooxygénase) responsable de la résistance à l’aspirine (19) ;
– le polymorphisme du gène codant pour HMG-CoA (hydroxy-méthyl-glutamyl-coenzyme A) induisant une diminution du LDL-cholestérol lors de la prise de la simvastatine (12);
– le polymorphisme du gène codant pour la DPD (dihydropyrimidine déshydrogénase) induisant une neurotoxicité lors de la prise de 5-FU (13).
b.3. Métabolisme de phase II des médicaments
Les réactions chimiques, catalysées par les enzymes de phase I, produisent des métabolites avec des groupements fonctionnels suivants : -OH, -NH2 , -SH, -COOH préparant
ainsi les composées à la phase II.
Au cours de cette phase II, les médicaments subissent une conjugaison avec d’autres molécules hydrosolubles afin de faciliter leur élimination faisant intervenir des transférases, des acétylases ou encore des glucuronidases. Il existe différents types de conjugaison, les plus répandues chez l’homme sont des réactions de glucuronoconjugaison, de sulfoconjugaison, d’acétylation, de méthylation, de glycine conjugaison et de glutathion conjugaison (8).
19
Étant donné que les enzymes du métabolisme de la phase II sont génétiquement déterminées, l’existence d’une anomalie enzymatique au cours de cette phase peut se traduire par une perturbation des réactions métaboliques avec des conséquences cliniques parfois néfastes ; c’est le cas des polymorphismes suivants:
– le polymorphisme du gène codant pour la NAT*2 (N-acétyl-transférase) responsable d’une hépatotoxicité et d’une neurotoxicité (neuropathies périphériques) lors de la prise de l’INH ;
– le polymorphisme du gène codant pour l’UGT1A1*28 responsable d’une neutropénie, d’une myélosuppression et d’une diarrhée lors de la prise de l’irinotécan (8)(11)(12)(13);
– le polymorphisme du gène codant pour la GST (glutathion-S-transférase) induisant une neurotoxicité lors de la prise de l’oxaliplatine (13);
– le polymorphisme du gène codant pour l’UGT2B7 et du gène codant pour l’UGT1A responsable de l’action antalgique du métabolite de morphine plus renforcée par rapport à celle de la morphine (16).
Il convient de noter que les “biomarqueurs” proposés par la pharmacogénétique sont prédictifs de la réponse aux médicaments et donc utilisables avant l’instauration du traitement (8).
20
Pharmacogénétique
et pharmacogénomique
21
III Pharmacogénétique et pharmacogénomique
1. Historique et définition
1.1.
Historique
de
la
pharmacogénétique
et
de
la
pharmacogénomique
La pharmacogénétique est apparue en tant que science expérimentale dans les années 1950, lorsque des chercheurs, utilisant de nouveaux outils d’analyse des différences interindividuelles en terme de réponse aux médicaments, ont décrit pour la première fois des polymorphismes génétiques liés à la réponse à la succinylcholine ou au métabolisme de l’isoniazide et de médicaments antipaludéens comme la primaquine(20). Friedrich Vogel a été le premier, en 1959, à utiliser le terme de pharmacogénétique pour décrire le concept de la variabilité des réponses basée sur des différences génétiques (21).
Avant cette date, la pharmacogénétique avait ses racines dans trois domaines de recherche : le métabolisme des médicaments, la génétique Mendélienne, et les récepteurs chimiques (22). On peut même remonter jusqu’en 510 avant J.C., où Pythagore découvrait que chez certains individus, la consommation de fèves provoquait l’apparition d’une anémie hémolytique.
Puis, en 1914, Archibald Garrod développe cette observation pour affirmer que les enzymes détoxifient les xénobiotiques afin qu’ils puissent être excrétés sans danger. Toutefois, il remarque que certaines personnes ne possèdent pas ces enzymes et sont, alors, susceptibles de présenter des effets indésirables. Plus tard, l’anémie hémolytique due à la consommation de fèves est identifiée chez les individus présentant un déficit en Glucose-6- phosphate déshydrogénase (G6PD).
Au début du XXème siècle la naissance de la pharmacogénétique continue avec la combinaison de la génétique mendélienne et l’observation des phénotypes. Lucien Cuenot en France, Archibald Garrod et Williams Bateson en Angleterre, suggèrent que le matériel génétique joue un rôle essentiel au niveau des transformations chimiques dans les organismes, et qu’il existe un lien entre le matériel génétique et l’activité des enzymes (20). Garrod propose le concept d’individualité chimique, suite à l’observation de certains cas de porphyrie associée à l’absorption d’hypnotique. Il suggère que les enzymes sont impliquées dans la détoxification des substances chimiques exogènes.