Filière Technologies du vivant
Orientation Chimie analytique
Travail de Bachelor
Diplôme 2019
Lopez Alexandro
Analyse des composés phénoliques dans les
vins et dans les extraits de rafle par
HPLC-DAD-MS
Professeur
F r a n k a Ka lm a n
Expert
A g n è s D i e n e s - N a g y
Date de la remise du rapport
Trava il de dip lôme
| é d i t i o n 2 0 1 9 |
Filière Technologie du vivant Domaine d’application Chimie analytique Professeur responsable Dr. Franka Kalman Franka.kalman@hevs.ch Partenaire Agroscope ChanginsObjectif du projet
L’objectif du projet est d’établir une méthode analytique permettant de quantifier par DAD-FLD et potentiellement MS une partie des flavanols et des proanthocyanidines de type B et C présent dans les extraits de rafles et les vins.
Méthodes | Expérie nces | Résultats
Après une identification des molécules d’intérêts dans le vin avec ajout de rafles, une méthode de quantification par HPLC-FLD a été développée pour l’analyse des flavanols et des proantocyanidines. La méthode a permis une bonne séparation des composé suivants : catéchine, épicatéchine, procyanidine B1, B2, B3 et C1. La sensibilité de la méthode permet une injection directe des échantillons. Les LOD obtenus sont : Catéchine = 0,07 mg/L ; Epicatéchine = 0,51 mg7L P.B1 = 0,42 mg/L ; P.B2 = 0,53 mg/L ; P.B3 = 0,91 mg/L ; P.C1 = 0,90 mg/L. La quantification de ces substances a montré une bonne linéarité dans le domaine de concentration observé dans les échantillons. La pré-validation de la méthode donne une précision inférieure à 10% d’erreur ainsi qu’une exactitude inférieure à 15%. Un SOP a été rédigé pour la mise en place d’une analyse de routine des extraits de rafle.
Dans les échantillons de vins, on observe une coélution de P.B3 avec un composé inconnu. Une optimisation de la méthode sera nécessaire pour pouvoir effectuer la quantification par MS.
Proanthocyanidols de type B1. Dimère (épicatéchol-(4β→8) -catéchol)
Proanthocyanidols de type C1. Trimère. Epicatéchol(4β→8) -épicatéchol--(4β→8) -épicatéchol
Analyse des composés phénoliques dans les
vins et dans les extraits de rafle par
HPLC-DAD-MS
4
REMERCIEMENT
Je tiens à remercie le professeur Franka Kalman pour m’avoir mis en contact avec l’Agroscope de
Changins ainsi que ma superviseuse, Mme Agnès Diènes pour, son accueil et son intérêt marqué
concernant l’avancement du projet durant toute la période passée en laboratoire. J’ai énormément
apprécié le thème abordé durant ce travail de Bachelor et pris beaucoup de plaisir à la réalisation de
ce travail.
Je remercie également les collaborateurs en rapport avec le projet pour leur soutien indéfectible et
leur bonne humeur tout au long du projet.
TABLE DES ABRÉVIATIO NS
HPLC : Chromatographie à haute performance (High performance liquid chromatography)
UHPLC : Chromatographie à ultra haute performance (Ultra High performance liquid chromatography) LC : Chromatographie en phase liquide (Liquid chromatography)
MS : Spectrométrie de masse (Mass spectrometrie)
MRM : Surveillance de la réaction principale (Main reaction monitoring)
DMRM : Surveillance dynamique de la réaction principale (Dynamic Main reaction monitoring) P.B1 : Proanthocyanidine B1
P,B2 : Proanthocyanidine B2 P.B3 : Proanthocyanidine B3 P.C1 : Proanthocyanidine C1
DAD : Détecteur à barrette diode (Diode array detector) FLD : Fluorescence détecteur (Fluorescence detector) Rs critique : Résolution critique
tR : temps de rétention
r : coefficient de corrélation
LOQ : limite de quantification (limits of quantification) LOD : Limite de détection (limits of detection)
S/N : rapport signal sur bruit de fond
m/z : rapport masse molaire du composé sur sa charge CE : Energie de collision
Préc. ion : Ion précurseur Prod. ion : Ion produit (fragment)
EIC : Chromatogramme de l’ion extrait (Extracted ion chromatogram) SOP : Mode opératoire (Standard operation procedure)
5
TABLE DES MATIÈRES
Remerciement ... 4
Table des abréviations ... 4
1. Introduction ... 7
1.1 Introduction ... 7
1.2 Phénoloïdes (acide phénols ... 7
1.3 Flavonoïdes (flavanols) ... 8
1.4 Tanins (condensé) ... 8
1.5 Objectif et pré-requis de la méthode ... 9
1.6 Littérature et stratégie ... 9 2. Matériel et méthode... 10 2.1 Equipements : ... 10 2.2 Matériels : ... 10 2.3 Réactifs : ... 11 2.4 Standards ... 11 2.5 Echantillons :... 11 2.6 Méthode : ... 13
2.6.1 Méthode d’essai Agroscope : ... 13
2.6.2 Méthode de quantification développée ... 15
3. Résultats ... 16
3.1 1ère partie : Observation et choix des composés a quantifier ... 16
3.1.1 Echantillon de Vins (T2, M1, M2, M4 et M5) ... 16
3.1.2 Echantillon d’extrait de rafle (Syrah et Merlot) ... 19
3.1.3 Comparaison Standard échantillons vins et extrait de rafle ... 21
3.2 2ème partie : Separation des composés identifiés... 24
3.2.1 Préparation run HPLC ... 24
3.2.2 Colonne PFP ... 25
3.2.3 Colonne C18 ... 27
3.2.4 Résume optimisation séparation ... 29
3.3 3ème partie : Analyse extrait de rafle (Gamaret, Syrah et merlot) et préparation a la quantification 29 3.3.1 Comparaison FLD/ MS entre extrait de rafle et T2 ... 30
3.3.2 Confirmation éligibilité des standards par Scan MS et détection FLD ... 32
3.3.3 Optimisation séparation échantillon témoin T2 ... 33
3.3.4 Préparation quantification par DMRM ... 35
3.4 4ème partie : Quantification et validation de la méthode ... 36
3.4.1 Calibration, LOD et LOQ ... 36
3.4.2 Facteur de réponse ... 38
3.4.3 Quantification Echantillons de vin et extraits de rafle par FLD ... 39
3.4.4 Quantification Echantillons de vin et extraits de rafle par DMRM ... 41
3.4.5 Quantification ajout dosé ... 43
3.4.6 Validation de la méthode ... 44
4. Conclusion et perspectives ... 46
4.1.1 Conclusion ... 46
6
5. Sources... 48
5.1.1 Livre et publication ... 48
5.1.2 Autre ... 48
6. Annexes ... 49
6.1.1 Annexe 1 Facteur de réponse ... 49
6.1.2 Annexe 2 Analyse spectrale extrait de rafle de Gamaret et vin de Gamaret ... 51
6.1.3 Annexe 3 Plan de validation ... 66
7 OH O OH OH O H H3C S CoA O
Figure 2 : Structure de l'acétyle CoA, qui induit la voie acétate
Figure 1 : Structure de l'acide shikimique
OH OH O OH O H O H OH O
Figure 5 : Acide vanillique Figure 4 : Acide salicylique Figure 3 : Acide gallique
O H O CH3 OH O
1. INTRODUCTION
1.1 INTRODUCTION
Les composés phénoliques sont des substances qui jouent un rôle important dans les vins. Ils sont
principalement responsables des différences entre les vins rouges et les vins blancs, notamment la
couleur, ainsi que leurs saveurs [1]. Ces composés proviennent des différentes parties de la grappe du
raisin et sont extraits lors de la vinification. Ces substances ont montré des propriétés comme
bactéricides, antioxydantes et comme un moyen de protection du consommateur par rapport aux
maladies cardio-vasculaires [2]. Les composés phénoliques découlent directement des métabolites
secondaires des végétaux, plus précisément de deux voies biosynthétiques : la voie shikimate et la voie
acétate. Les composés phénoliques sont donc des dérivés non azotés, dont le ou les cycles aromatiques
sont issus du métabolisme de l’acide shikimique et/ou d’un polyacétate (voir figure 1 et 2) [3].
Les composés traités dans ce travail de diplôme appartiennent aux familles issues de la voie
shikimique. Les familles de la voie shikimique sont les suivantes : phénoloïdes, phénylpropanoïdes,
xanthonoïdes, stilbénoïdes, flavonoïdes et les tanins [3]. Les composés traités appartiennent aux
familles suivantes : les phénoloïdes, les flavonoïdes et les tanins.
1.2 PHÉNOLOÏDES (ACIDE PHÉNOLS)
Les phénoloïdes sont issus de la voie shikimate et subissent un clivage enzymatique conduisant aux
acides phénols qui peuvent par la suite subir une décarboxylation menant aux phénols correspondants
[3]. La structure de base des acides phénols est l’acides benzoïques à laquelle est ajoutée un ou
plusieurs groupe hydroxyle. Ces acides phénols sont très communs parmi les végétaux. On les trouve
sous forme libre, sous forme d’ester, ou encore sous forme d’hétérosides [1,3]. Les images suivantes
sont des exemples de structures d’acides phénols courants (figure 3,4 et 5) :
8 C6' C4 O C4 O OH O
Figure 8 : Structure des chalcones
Figure 7 : Structure des dihydroflavanols. La formation des flavanols passe par la perte ou réduction du groupe carbonyle
O OH O H OH OH OH C4 O O H OH OH O H H R1 R2 C8 O O H OH OH O H H R3 R4 C4 C2 O O H OH O H O H OH C7 C8 O OH O O H O H OH
Figure 9 : Structure de flavandiols-3,4 avec la leucopélargonidine
Figure 11 : Procyanidine de type B B1 : R1 = OH; R2 = H; R3 =H; R4 = OH B2 : R1 = OH; R2 = H; R3 = H; R4 =H B3 : R1 = H; R2 = OH; R3 = H; R4 = OH B4 : R1 = H; R2 = OH; R3 = OH; R4 =H
Figure 10 : Procyanidine de type A
1.3 FLAVONOÏDES (FLAVANOLS)
Les flavonoïdes sont tous issus des chalcones qui sont le fruit d’une triple condensation enzymatique
d’acétate (voie mévalonate) sur l’acide cinnamique ou un de ses dérivés (voie shikimate). Les chalcones
sont les précurseurs biosynthétiques de l’ensemble de la famille. En fonction de la présence d’un
groupe hydroxyle en position 6’ et de sa réactivité, la cyclisation des chalcones peut donner lieu à 15
catégories de molécules distinctes. La catégorie traitée dans ce travail est celle des flavanols. La
structure des flavanols-3 est un cycle dihydropyranyle qui a la particularité de posséder un carbone en
position 4 pouvant devenir facilement un carbocation, ce qui permet aux flavanols-3 d’être les
précurseurs des proanthocyanidine ou tanins condensés [1,3]. Les images suivantes montrent les
structures de différentes molécules en rapport avec les flavanols (figure 6, 7 et 8) :
1.4 TANINS (CONDENSÉ)
Les tanins sont des molécules phénoliques hydrosolubles relativement volumineuses. Les masses
moléculaires des tanins sont comprises entre 600 à 3500 Da. Les tanins condensés ou
proanthocyanidine sont des polymères flavaniques constitués d’unité de flavanols-3 ou de
flavandiols-3,4 (Dimère, trimère, oligomère). Dans le cas des dimères, ces derniers sont liés par une ou deux
liaisons flavaniques [3]. L’unité monomère est généralement un flavanol-3 et deux types de liaisons
prédominent. Elles sont notées A et B. La type B se caractérise par une liaison C4-C8 entre les deux
unités composant le tanin et la type A à elle aussi cette liaison, mais dispose aussi d’une liaison éther
entre les carbones C5 et C7. Dans le cas des trimères, les trimères du type C ont les deux liaisons
interflavanes correspondant au type B et les trimères du type D ont une correspondance au type A et
une au type B [1].
C4 O OH O H OH OH OH Figure 6 : Structure de la catéchine qui est un flavanols -39
1.5 OBJECTIF ET PRÉ-REQUIS DE LA MÉTHODE
L’objectif du projet est d’établir une méthode analytique permettant de quantifier par DAD-FLD et
potentiellement MS une partie des flavanols et des proanthocyanidine de type B et C présents dans
les vins et les extraits de rafles. La méthode développée doit être facile à effectuer et ne doit pas
nécessiter une préparation de l’échantillon. La méthode doit également être conforme aux exigences
de méthodes requises par le département qualité des vins. Les exigences sont décrites dans les points
suivants :
• L’exactitude : une exactitude (recouvrement) entre 80 et 120% est exigée
• Précision : une précision (coefficient de variation sur les concentrations) <10% est exigée
• Robustesse : la méthode développée doit permettre une utilisation en routine et la plus simple
possible
1.6 LITTÉRATURE ET STRATÉGIE
Tout d’abord, avant de chercher dans la littérature une quelconque approche, il est nécessaire de se
pencher sur le matériel disponible. En effet, le laboratoire dispose d’un système chromatographique
complet (UHPLC et HPLC, les deux disposant d’un FLD et d’un DAD) et de colonne. Le choix de phase
mobile est un paramètre clé pour l’analyse des composés phénoliques (composé polaire et
hydrosoluble) [4,5]. Le laboratoire dispose de colonne C18, HILIC et PFP (fluorophényle propyle). Si l’on
regarde la littérature, l’analyse des polyphénols se fait aussi bien avec les phases inverses (C18 et plus
rarement avec PFP) que les phases HILIC, la différence se faisant avec le choix de l’éluant.
Le point de départ du travail est une méthode d’essai réalisé par Agroscope [6] qui utilise une C18
comme support chromatographique. Lors de l’avancement du travail, d’autres essais de séparation
seront tentés avec un autre type de colonne.
10
2. MATÉRIEL ET MÉTHODE
2.1 EQUIPEMENTS :
Système chromatographique No. 1 : UHPLC Séries 1290 avec DAD, FLD et MS
• Pompe : G4220A ; No. DEBAA05262 • TCC : G1316C ; No. DEBAC01573 • Sampler : G4426A ; No. DEBAP06279 • Thermostat : G133OB ; No. DEBAK11108 • DAD : G4212A ; No. DEBAF04140 • FLD : G1321B ; No. DEABO02629 MS :
• Agilent Triple Quad LC/MS 6460 ; Model : G6460C ; No. SG15257310 • Source ESI AJS; G1958; No. G60460
Système chromatographique No. 2 : HPLC Série 1200 avec DAD
• Pompe : G1312B ; No.DE63056120 • TCC : G1316A ; No. DEACN16344 • Sampler : G1329A ; No. DE64764668 • Thermostat : G1330B ; No. DEBAK16131 • DAD : G1315C ; No. DE73456961 Colonne :
• Colonne 1 : Zorbax Eclipse Plus C18 ; 1,8µm ; 4.6 mm x 100 mm ; Ref. 959964-902 ; Lot : DO4016
• Colone 2 : Restek Pinnacle DN-PFP ; 1,9µm ; 2.1 mm x 50 mm ; Ref. 15110669T ; Lot : 150910P
Logiciel :
• LC/MSD ChemStation Agilent Technology, V. B04.03-SP2 • QQQ quantitative Analysis Agilent Technology, V.03.1 • Drylab4, Molnare-Institute, V.4.2.1.4
• Optimizer, Agilent Technology, V.2.1
2.2 MATÉRIELS :
• Agitateur planaire IKA KS 3000
• Balance Mettler-Toledo PG2002-S ; 2100 g ± 0.01g • Balance Mettler-Toledo XP204 ; 200 g ± 0.01 mg
• Micropipette Socorex ACURA 835 ; 10-100 µl ; 20- 200 µl; 0,2- 2 ml • Filtre Nylon Whatman 4mm; 45 µm
• Ballon jaugé en verre de 20 ml ± 0,02 ml 10 ml ± 0.04 ml ; 5 ml ± 0.04 ml
• Vials HPLC en verre EcoLine 1,5 ml (Part Number : 11BW-EL ; 1.5 ml Crimp Neck Vial 32 x 11.6 mm clear, wide opening , pk. 100 : Country of Origin. India
11
2.3 RÉACTIFS :
Tableau 1: réactifs utilisés
Nom
Provenance
Fournisseur
Pureté
Lot
CAS
Acétonitrile
USA
J.T.Baker
HPLC
grade
1805491891
75-05-8
Acide
formique
Allemagne
Fluka
>98%
SZBG2510H
64-18-6
Ethanol
Absolue
Allemagne
Merck
HPLC
grade
K49284483 729
1.00983.2511
Eau bi-osmosé
-
Agroscope
-
-
-
Acide
tartrique
Allemagne
Merck
>99,5%
K2364078 657
87-69-4
NaOH 1M
-
Agroscope
-
-
-
Bisulfite de
sodium
Allemagne
Merck
97-100%
K2765843 867
7757-83-7
2.4 STANDARDS
Tableau 2 : Standards utilisés
Nom
Provenance
Fournisseur
Pureté
Lot
CAS
Procyanidine A2
France
Extrasynthese
>99%
04
41743-41-3
Procyanidine A1
France
Extrasynthese
>95%
01
103883-03
Procyanidine B1
France
Extrasynthese
>90%
49
20315-25-7
Procyanidine B2
France
Extrasynthese
>90%51
29106-49-8
Procyanidine B3
France
Extrasynthese
>95%
01
23567-23-9
Procyanidine C1
France
Extrasynthese
>90%
01
37064-30-5
(-) Epicatéchine
Allemagne
Sigma-Aldrich
>90%
BCBH5714V
490-46-0
Acide Gallique
Allemagne
Sigma-Aldrich
97.5-102%
071M0031V
149-91-7
(+) Catéchine
Allemagne
Fluka
>99%
288649/1
7295-85-4
2.5 ECHANTILLONS :
Tableau 3 : Liste des échantillons analysés
Nom
Provenance
Date
Extrait de rafle (Syrah)
Agroscope
22.05.2019
Extrait de rafle (Merlot)
Agroscope
22.05.2019
Extrait de rafle (Gamaret)
Agroscope
27.05.2019
M1 10% rafle ajout
Gland
05.10.2017
M2 20% rafle ajout
Gland
05.10.2017
M4 10% vendange entière
Gland
05.10.2017
M5 20% vendange entière
Gland
05.10.2017
12 Les échantillons de Gamaret ont été obtenus selon le procédé suivant (fig. 12) :
Figure 12 : Schéma de l'obtention des échantillons [7] M1 : Vendange éraflée + 10% en poids de rafle rajouté
M2 : Vendange éraflée + 20% en poids de rafle rajouté
M3 : Vendange éraflée + 30% en poids de rafle rajouté (non disponible) M4 : Vendange éraflée + 10% de vendange entière
M5 : Vendange éraflée + 20% de vendange entière T2 : Témoins (100% vendange éraflée)
Les échantillons d’extrait de rafle ont été obtenus en suivant le procédé suivant [8] : Préparation solution mère
• Remplir un ballon jaugé 1 L d’environ 500 ml d’eau déminéralisée • Ajouter 6 g d’acide tartrique
• Ajouter 40 ml d’une solution d’NaOH 1M • Ajouter 100 mg de sodium métabisulfite • Compléter au trait de jauge avec de l’eau Extraction des rafles
• Prélever environ 7g de rafles et déposez-les dans un erlenmeyer de 125 ml • Ajouter 80 ml de la solution mère et fermer le bouchon
• Mesurer le pH de l’échantillon avant la phase d’ajout d’éthanol
• Déposer l’erlenmeyer dans l’agitateur IKA et rentrer les paramètres suivants :
Temps d’agitation en [h] Vitesse en [rpm] Température en [°C]
288h 10 28
• Abaisser le couvercle de protection et appuyer sur le bouton blanc de l’interface de la machine pour commencer l’agitation
• Ajouter tous les 2 jours 2 ml d’éthanol jusqu’à avoir un volume total d’ajout de 12 ml soit 12 jours d’agitation pour un volume final de 92 ml. Voir tableau N°4
13 Tableau 4 : Concentration final théorique d'éthanol dans l'échantillon
Nombre de jours Ajout éthanol %Ethanol
2 2 2.5 4 4 5 6 6 7.5 8 8 10 10 10 12.5 12 12 15
• Une fois l’ajout terminé, les extrais bruts sont filtrés sur filtre Nylon 45µm.
• L’objectif de cette préparation est de simuler les conditions d’extraction d’un vin lors de l’ajout de rafle.
2.6 MÉTHODE :
Ici sont présentées les deux méthodes principalement utilisées lors de ce travail de Bachelor. Il existe la méthode initiale d’essai d’Agroscope et la méthode finale obtenu à la fin du travail de Bachelor pour la quantification des polyphénols dans les extraits de rafle et dans les vins.
2.6.1 MÉTHODE D’ESSAI AGROSCOPE :
1. Conditions chromatographiques :
Eluant A : H2O / HCOOH 100 :0.1
Eluant B : CH3CN / HCOOH 100 :0.1
Colonne : Eclipse Plus C18 100 x 4.6 mm 1,8 m.
Gradient : Temps [min] A [%] B [%] Débit [ml/min] Pression [bar] 0.0 99 1 1.5 650 10.0 95 5 1.5 650 20.0 85 15 1.5 650 25.0 50 50 1.5 650 26.0 0 100 1.5 650 30.0 0 100 1.5 650
Température : 60°C Stop Time : 30.0 min Post Time : 5.0 min
Volume d’injection : 5.0 l
Détecteur DAD : 280 nm ; 320 nm ; 305 nm ; 360 nm
Détecteur FLD : Excitation 280 nm ; Emission 320 nm
2. Condition MS :
Source (+/-) : Gas Temp. [°C] : 150
Gas Flow [l/min] : 10
Nebulizer [psi] : 25
SheatGas Heater : 400
SheatGas Flow : 12
Capillary [V] : 5000
14 Time segment :
Index Start Time Scan Type Ion Mode Div Valve Delta EMW Store
1 0 Dynamic MRM ESI + Agilent Jet Stream To MS 0 Yes
Time Segment index 1 :
Cpd. Name RT Prec. Ion Prod. Ion Ret. Windows Frag [V] CE [V] Cell Acc [V] Polarity Tyrosol 6.1 137 119 1 135 13 4 - Protocatechuic acide 2.86 153 109 1 125 13 4 - Hydroxytyrosol 3.63 153 123 1 130 13 4 - P-Coumaric acid 11.65 163 119 1 125 13 4 - Gallic acid 1.53 169 125 1 140 13 4 - Vanillic Acid 8.0 167 152 1 110 9 4 - Cafeic acid 7.67 179 135 1 145 13 4 - Ethyl Protocatechuate 19.58 181 108 1 135 13 4 - ferulic acid 15.6 193 134 1 120 13 4 - Ethyl Gallate 13.23 197 124 1 135 13 4 - Cis-Resveratrol 21.2 229 135 1 370 29 4 + Trans-Resveratrol 20.8 229 135 1 370 29 4 + Catechin 6.78 291 139 1 140 13 4 + Epicatechin 12.09 291 139 1 165 13 4 + Cis-coutaric acid 7.31 295 163 1 135 13 4 - Trans-coutaric acid 6.98 295 163 1 135 13 4 - Cis-caftaric acid 4.58 311 179 1 185 9 4 - Trans-caftaric acid 4.44 311 179 1 185 9 4 - fertaric acid 10.37 325 193 1 135 13 4 - Cis-Piceid 14.16 391 229 1 135 13 4 + Trans-Piceid 12.66 391 229 1 135 13 4 + Viniferin Iso 1 12.64 455 361 1 135 13 4 + Viniferin Iso 2 13.07 455 361 1 135 13 4 + Dimer B1 6.68 579 127 1 135 13 4 + Dimer B2 10.86 579 127 1 135 13 4 + Dimer B3 6.68 579 127 1 135 13 4 + Dimer B4 9.08 579 127 1 135 13 4 + Cis-GRP 6.34 616 149 1 135 13 4 - Quercetin 22.9 301 151 1 370 21 4 - Quercetin Glucuronide 18.8 479 303 1 145 13 4 + Kaempferol 23.8 287 153 1 205 30 4 + Kempferol Glucoside 21.3 449 287 1 120 9 4 + Trans-GRP 5.19 616 149 1 135 13 4 - Methylnaringenin 21.38 287 167 1 160 21 4 + Myricetin 20.74 317 151 1 360 25 4 - Pterostilbène 25.99 255 240 1 180 17 4 - E-Viniferin 23.39 455 107 1 170 30 4 + Ampelopsin A 16.63 469 451 1 155 17 4 - E-Vitisin B 24.34 907 559 1 205 21 4 + Hopeaphenol 905 265 1 330 60 4 -
15
2.6.2 MÉTHODE DE QUANTIFICATION DÉVELOPPÉE
1. Conditions chromatographiques :
Eluant A : H2O / HCOOH 100 :0.1
Eluant B : CH3CN / HCOOH 100 :0.1
Colonne : Eclipse Plus C18 100 x 4.6 mm 1,8 m.
Gradient : Temps [min] A [%] B [%] Débit [ml/min] Pression [bar] 0.0 91 9 1.0 600 18.0 83 17 1.0 600 25.0 5 95 1.0 600
Température : 28°C Stop Time : 25.0 min Post Time : 5.0 min
Volume d’injection : 2.0 l
Détecteur DAD : 280 nm
Détecteur FLD : Excitation 280 nm ; Emission 320 nm
16
3. RÉSULTATS
3.1 1
ÈREPARTIE : OBSERVATION ET CHOIX DES COMPOSÉS A QUANTIFIER
Cette partie utilise la méthode d’essai d’Agroscope pour la recherche des composés d’intérêts. Elle est consacrée à la recherche des composés dans les échantillons de vins et les extraits de rafles ainsi que la détermination des longueurs d’ondes de détection les plus sensibles. Cette première partie est effectuée avec le système chromatographique N°1 (voir chapitre 2.1).
3.1.1 ECHANTILLON DE VINS (T2, M1, M2, M4 ET M5)
L’observation des composés par DMRM a montré des problèmes de fenêtres de mesure. Cela est dû au fait que le système ouvre et ferme les intervalles de mesures tous les X mins et lorsqu’un composé arrive entre cet intervalle de fermeture ou d’ouverture, le pic du composé est coupé. Une analyse MRM supplémentaire a été effectuée afin de compléter l’observation de certain pics coupés.
L’observation des composés avec les différentes longueurs d’ondes (DAD : 280 nm, 305 nm, 320 nm et 520 nm ; FLD : Ex 280 nm et Em. 320 nm) a montré que la longueur d’onde à 280 nm n’était pas sélective pour le DAD. En effet, le nombre de pic étant beaucoup trop important, cela rendait difficile l’interprétation MS-DAD (phénomène de coélution et double pic trop important). C’est pourquoi seuls 305, 320 et 520 furent gardés pour le DAD. A noter que 520 nm n’est là que pour renseigner sur la présence d’anthocyane.
Le tableau 5 suivant montre les résultats d’observation de certain composé entre le FLD et DAD : Tableau 5 : Récapitulatif observation DAD et FLD
Observation DMRM /MRM Observation Composé tR [min] FLD DAD
Acide gallique 1.8 - X
Acide Protocatechuique 3.38 - X
Hydroxytyrosol 4.36 X X
Acide trans caftarique 4.32 X X
Acide cis caftarique 4.36 X X
Acide trans coutarique 6.61 - X
Acide cis coutarique 6,97 X X
Catéchine 7.9 / 8.3 X - Dimer1 7.64 X - Dimer2 8,14 X - Dimer3 8,84 X - Dimer 4 10,86 X - Dimer 5 11,64 X - Dimer 6 12,41 X - Acide caféique 9.13 X X Acide fertarique 10.13/10.8 X - Epicatéchine 12.7 / 13.6 X - Acide ferulique 16.7 - X
On constate par cette observation que les proanthocyanidine et les flavanols sont observables en FLD et non en DAD. En ce qui concerne les acides phénols, on remarque que les acides phénols sont tous observables en DAD et seulement une partie en FLD. Les composés affichés dans le tableau 5 sont aussi ceux observé en MS. Suite à une discussion avec le responsable, il a été décidé d’axer la recherche seulement avec le détecteur FLD car il permet l’observation des composés d’intérêt que sont les proanthocyanidine et les flavanols.
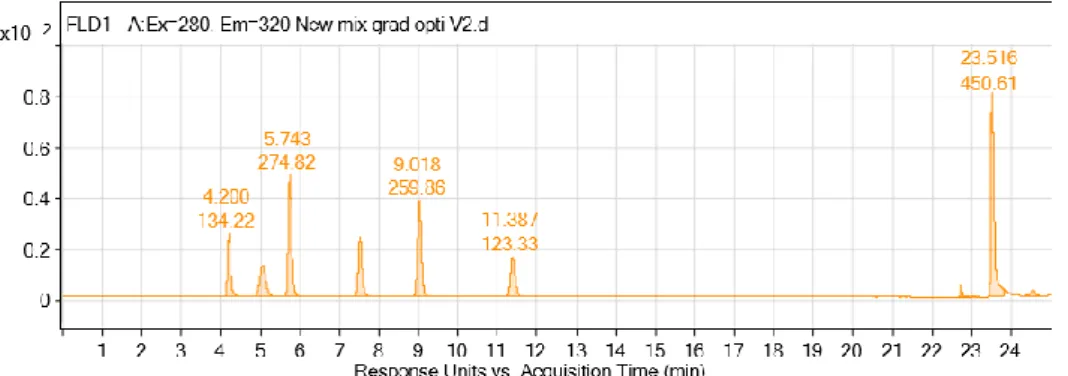
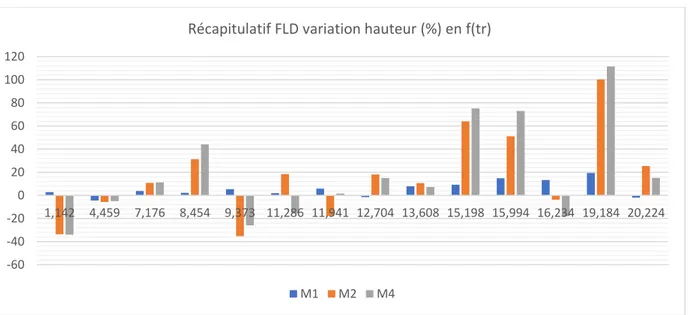
17 La figure 13 suivante montre la variation en hauteur (%) des différents échantillons (M1, M2, M4, M5) par rapport à l’échantillon témoin T2. Il a été décidé de faire la comparaison sur la hauteur car la résolution initiale n’étant pas optimale, des phénomènes de double pic et de coélution apparaissaient, faussant davantage la valeur de l’aire que celle de la hauteur.
Figure 13 : Récapitulatif variation hauteur (%) en f(tr) en FLD
Les tR choisis sont ceux qui présentent une variation significative (>10%). On constate que les tR suivants sont
ceux qui ont la plus forte variation positive : 8.45 min, 11.29 min, 12.7 min, 15.2 min, 16 min et 19.18 min. Ces tR
sont valables pour les échantillons M1, M2 et M4, car le M5 est beaucoup trop concentré. Si l’on enlève M5 du graphique, ont obtient la figure 14 suivante :
Figure 14 : Récapitulatif variation hauteur (%) en f(tr) en FLD
On constate que les tR sont ceux qui ont la plus forte variation positive : 8.45 min, 15.2 min, 16 min et 19.18 min.
Les figures 13 et 14 montrent bien que l’ajout de rafle et/ou de vendange entière influence bel et bien la concentration de certains composés présents dans le vin.
Maintenant que la confirmation que l’ajout de rafle et/ou de vendange a bel et bien un effet sur le vin, une analyse par MS (mode : Product Ion) a été lancée afin d’estimer l’abondance et les fragments des molécules observées en DMRM/ MRM. Les conditions MS pour les paramètres Fragmentar et CE sont les mêmes que celles utilisées dans la méthode d’essai Agroscope.
-100 0 100 200 300 400 500 600 700 800 1,142 4,459 7,176 8,454 9,373 11,286 11,941 12,704 13,608 15,198 15,994 16,234 19,184 20,224 M1 M2 M4 M5 -60 -40 -20 0 20 40 60 80 100 120 1,142 4,459 7,176 8,454 9,373 11,286 11,941 12,704 13,608 15,198 15,994 16,234 19,184 20,224
Récapitulatif FLD variation hauteur (%) en f(tr)
18 L’analyse par Produc Ion a fournis les tableaux de résultats N°6 et 7 suivants :
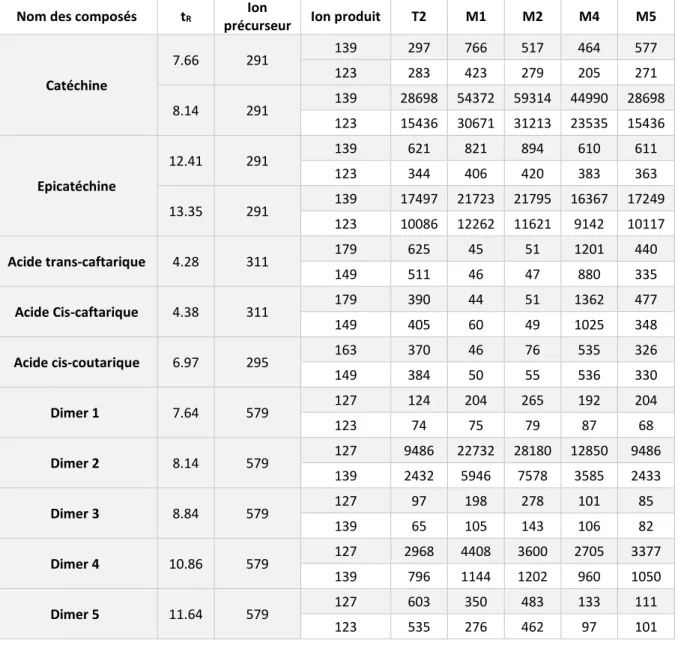
Tableau 6 : Abondance des fragments obtenus à partir des ions précurseurs
Nom des composés tR Ion précurseur Ion produit T2 M1 M2 M4 M5
Acide protocatechuique 3.38 153 109 724 782.8 794.32 745.46 811.5 Acide gallique 1.80 169 125 1148 2740 3447 1494 1419 Hydroxytyrosol 4.33 153 123 810.2 1082 1262 785 739 Acide cafféique 8.86 179 135 8077 9439 9211 7624 7844 Acide férulique 16.51 193 134 189 195 230 153 164 Acide Trans-coutarique 6.61 295 163 1221 86 80 2380 1343 Acide fertarique 9.80 325 193 936 184 240 1109 778 Tableau 7 : Abondance des fragments obtenus à partir des ions précurseurs
Nom des composés tR Ion
précurseur Ion produit T2 M1 M2 M4 M5 Catéchine 7.66 291 139 297 766 517 464 577 123 283 423 279 205 271 8.14 291 139 28698 54372 59314 44990 28698 123 15436 30671 31213 23535 15436 Epicatéchine 12.41 291 139 621 821 894 610 611 123 344 406 420 383 363 13.35 291 139 17497 21723 21795 16367 17249 123 10086 12262 11621 9142 10117 Acide trans-caftarique 4.28 311 179 625 45 51 1201 440 149 511 46 47 880 335 Acide Cis-caftarique 4.38 311 179 390 44 51 1362 477 149 405 60 49 1025 348 Acide cis-coutarique 6.97 295 163 370 46 76 535 326 149 384 50 55 536 330 Dimer 1 7.64 579 127 124 204 265 192 204 123 74 75 79 87 68 Dimer 2 8.14 579 127 9486 22732 28180 12850 9486 139 2432 5946 7578 3585 2433 Dimer 3 8.84 579 127 97 198 278 101 85 139 65 105 143 106 82 Dimer 4 10.86 579 127 2968 4408 3600 2705 3377 139 796 1144 1202 960 1050 Dimer 5 11.64 579 127 603 350 483 133 111 123 535 276 462 97 101
19
Dimer 6 12.41 579
127 6096 7902 7645 4701 6572
139 2251 3217 3079 1691 2353
123 1665 1890 1732 827 1648
L’observation des tableaux 6 et 7 a montré la non-proportionnalité de l’abondance des ions obtenus entre les
échantillons, par exemple entre M1 et M2 (10% et 20% d’ajout de rafle) pour la catéchine aux deux tR, et montre
aussi la faible abondance des ions produits pour l’acide protocatéchuique, l’hydroxytyrosol, l’acide férulique, l’acide fertarique et les acides caftariques.
Pour la majorité des molécules observées, il y avait 1 seul fragment observable avec la fragmentation de la méthode utilisée. Cependant, il y avait 2 à 3 fragments observables pour l’ion précurseur à 579 m/z et ce, à plusieurs temps de rétention différents, ce qui signifie que le ou les dimers sont en quantité. En ce qui concerne les flavanols, l’abondance des ions pour la catéchine et l’épicatéchine est très importante.
Suite à cette observation, il a été décidé de choisir des standards parmi ceux disponible aux laboratoires Agroscope. Les standards suivants ont donc été choisis pour la suite du travail (voir tableau 8) :
Tableau 8 : Tableau récapitulatif des standards utilisés
Standards MW [M+H]+ [M-H]- Procyanidine A2 576.52 577.52 575.52 Procyanidine A1 576.51 577.51 575.51 Procyanidine B1 578.53 579.53 577.53 Procyanidine B2 578.53 579.53 577.53 Procyanidine B3 578.53 579.53 577.53 Procyanidine C1 866.79 867.79 865.79 (-)Epicatéchine 290.271 291.271 289.27 (+)Catéchine 290.271 291.271 289.27 Acide gallique 170.119 169.119 171.119
Parmi les acides phénols analysé, seul l’acide gallique a été retenu, car l’acide gallique disposait d’une abondance supérieure à 1000 pour tous les échantillons testés, contrairement aux autre acides phénols et alcool phénol (hydroxytyrosol).
Le système MS-MS utilisé ne permet pas une identification formelle par la masse des composés observés. En effet, lors de la dernière maintenance du système, la précision de l’appareil était de 0.05 m/z, c’est-à-dire que le système peut seulement confirmer le dixième. Or, pour l’identification des composés étudiés, si l’on prend l’exemple de la catéchine/ épicatéchine, la référence [8] on a trouvé les m/z pratique suivants : 289.0715 ([M-H]
- épicatéchine) et 289.0711 ([M-H]- catéchine). Cependant le système utilisé dans la référence est un Maldi-TOF.
C’est pourquoi dans ce travail, le seul moyen d’identifier formellement les composés est de les comparer avec des standards.
3.1.2 ECHANTILLON D’EXTRAIT DE RAFLE (SYRAH ET MERLOT)
Une fois les standards choisis, il a été décidé de commencer l’observation des composés choisis pour les standards dans les extraits de rafle. Seuls les extraits de rafle de Merlot et de Syrah seront analysés dans cette partie du travail, car l’extrait de rafle de Gamaret n’était pas encore disponible.
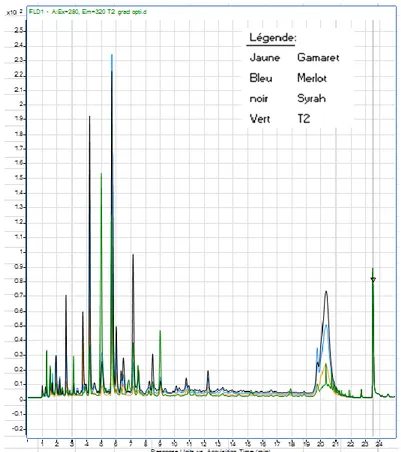
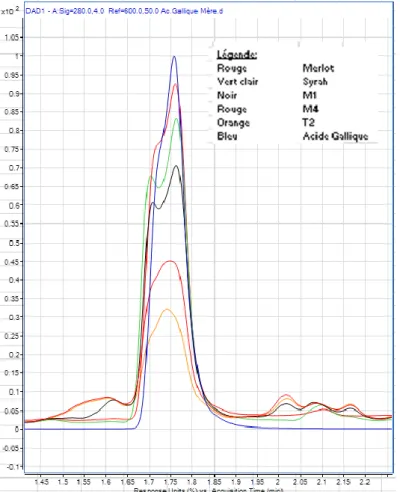
Tout d’abord, la première partie du travail était de déterminer si la méthode d’extraction utilisée (voir chapitre 2.5) était répétable. Les figures 15 et 16 suivantes montrent les overlays des triplicatas effectués sur chaque cépage (Merlot et Syrah) :
20 Figure 15 : Overlay FLD, extrait de rafle de Syrah Figure 16 : Overlay FLD, extrait de rafle de Merlot
La méthode a montré une différence d’intensité relativement importante pour la plupart des pics entre les triplicatas pour les deux échantillons. Cette observation montre que la méthode utilisée pour l’extraction doit encore être optimisée. Cependant, la méthode montre une extraction de composés relativement importante (grand pic large et intense).
La seconde partie du travail était de comparer les standards choisis pour les vins avec les échantillons d’extraits. Une comparaison par spectre MS a été faite. Le tableau 9 suivant illustre cette comparaison :
Tableau 9 : Récapitulatif observation standard-Extrait
Standards MW [M+H]+ [M-H]- Correspondance tR [min] Ionisation Procyanidine A2 576.52 577.52 575.52 8.145/16.202 +/- Procyanidine A1 576.51 577.51 575.51 8.145/16.202 +/- Procyanidine B1 578.526 579.526 577.53 8.145/16.202 +/- Procyanidine B2 578.53 579.53 577.53 8.145/16.202 +/- Procyanidine B3 578.53 579.53 577.53 8.145/16.202 +/- Procyanidine C1 866.79 867.79 865.79 11.08 /11.6 +/- (-) Epicatéchine 290.271 291.271 289.27 13.37 + (+) Catéchine 290.271 291.271 289.27 13.37 + Acide gallique 170.119 169.119 171.119 1.79 +/-
21 Le tableau 9 représente les résultats des overlays des spectre +/- ESI (f(m/z) des standards) avec les chromatogrammes obtenus en FLD. Maintenant que les m/z des standards choisis ont pu être confirmés dans les échantillons d’extraits de rafle, les solutions standards ont été préparées afin de permettre une comparaison FLD et DAD des échantillons de vins et d’extrait de rafles.
3.1.3 COMPARAISON STANDARD ÉCHANTILLONS VINS ET EXTRAIT DE RAFLE
Les solutions mères des standards ont été faites de la manière suivante (voir tableau 10) : Tableau 10 : Récapitulatif pesé et concentration des solutions mères
Concentration Solution Mère Masse [mg] Volume dilution [ml] Conc. Final [g/L] Ac.gallique 10,0 10 1 Catechin 10,1 20 0,505 Epicatechin 10,3 20 0,515 Procyanidin B1 8,4 10 0,84 Procyanidin B2 7,6 10 0,76 Procyanidin B3 7,0 10 0,7 Procyanidin C1 8,0 10 0,8 Les solutions ont ensuite été complétées au trait de jauge avec de l’éthanol.
Les solutions de standard ont donné avec la méthode d’essai Agroscope les tR suivant (voir tableau 11) :
Tableau 11 : Récapitulatif tR des standards
Standard DAD 280 nm [min] FLD [min] Ac.gallique 1,758 - Catéchine 8,154 8,192 Epicatéchine 13,312 13,35 Procyanidine A2 19,557 19,59 Procyanidine A1 16,93 16,96 Procyanidine B1 8,126 8,161 Procyanidine B2 12,385 12,42 Procyanidine B3 8,022 8,059 Procyanidine C1 15,745 15,78
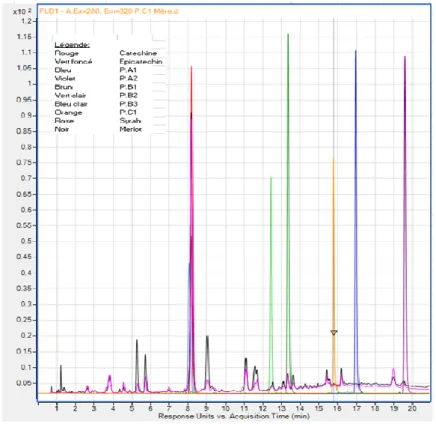
On constate déjà que la méthode utilisée ne permet pas une bonne séparation des standards de catéchine, P.B1 et P.B3. Les comparaisons par overlay qui vont suivre se feront sans M2 et M5 car ces deux échantillons ne sont que M1 et M4 en plus concentré. Il est donc inutile de répéter le travail une seconde fois. Les 3 images suivantes (figures 17 à 20) montrent les overlays des standards avec les échantillons :
22 Figure 17 :Overlay FLD (Ex. 280 nm et Em. 320 nm) Syrah, Merlot et Standard
23 Figure 19 : Overlay DAD 280 nm, échantillons et acide gallique
On constate que pour les extraits de rafle et les vins, les standards P.A1 et P.A2 ne correspondent à aucun pic. Le standard d’acide gallique (figure 19) se superpose aux pics des échantillons, bien qu’un phénomène de coélution soit visible pour les échantillons. Pour les standards suivants : P.B1, P.B2, P.B3 et P.C1, une comparaison unitaire a été effectuée car soit leur séparation était mauvaise dans les échantillons, soit leur concentration était très faible. De cette comparaison, il ressort que le standard P.C1 se superpose aux échantillons, bien que la concentration relativement faible des échantillons ne permette pas une bonne observation. Le standard P.B3 se superpose aux extraits et ne se superpose pas aux vins. Le standards P.B2 se superpose aux échantillons de vins et des extraits, mais la concentration faible des extraits rend l’observation difficile. Le standard de P.B1 se superpose avec tous les échantillons. Quant aux standards de catéchine et épicatéchine, ils se superposent tous les deux avec tous les échantillons. Le tableau N°12 suivant résume la comparaison :
Tableau 12 : Récapitulatif de la comparaison des échantillons avec les standards
Standard Superpose Intensité
Oui Non Haute Faible
Ac.gallique X X
Catéchine X X
Epicatéchine X X
Procyanidine A2 X - -
24
Procyanidine B1 X X
Procyanidine B2 X X
Procyanidine B3 X X X
Procyanidine C1 X X
La comparaison des échantillons avec les standards a permis de montrer que la méthode actuelle ne permet pas une quantification de ces substances. La séparation entre la catéchine, P.B1 et P.B3 doit être améliorée. C’est pourquoi, après discussion avec le responsable de travail, il a été décidé d’effectuer cette optimisation de méthode en utilisant le logiciel Drylab. Une solution de mélange de standards contenant tous les standards à l’exception de P.A1 et P.A2 a donc été faite, et servira de repère lors du suivi d’optimisation.
3.2 2ÈME PARTIE : SEPARATION DES COMPOSÉS IDENTIFIÉS
L’optimisation de méthode par Drylab s’est faite sur le système chromatographique N°2 (voir chapitre 2.1) car le système N°1 était indisponible (analyse prioritaire et maintenance de l’appareillage).
La préparation du mélange mix est la suivante : On prélève 100µl de chaque solution mère (voir tableau 10) à l’exception de l’acide gallique (20µl) que l’on met dans un vial HPLC (soit un volume total de 620µl). On complète ensuite avec 380µl d’eau osmosée pour obtenir 1 ml de solution totale. Le tableau 13 suivant donne les nouvelles concentrations.
Tableau 13 : Récapitulatif concentration solutions mix
Concentration Solution Fille Conc. Final [mg/L] Ac.gallique 20 Catechin 50,5 Epicatechin 51,5 Procyanidin B1 84 Procyanidin B2 76 Procyanidin B3 70 Procyanidin C1 80
3.2.1 PRÉPARATION RUN HPLC
Pour faire fonctionner correctement Drylab, il faut effectuer plusieurs runs sous certaines conditions. Dans notre cas, le but est de trouver les bonnes conditions de séparations en fonction du gradient et de la température, soit un mode de fonctionnement en 2D (2 paramètres qui varie). Le logiciel demande alors 4 run spécifiques afin de pouvoir calculer le modèle 2D. A noter que Drylab ne peut pas simuler des résultats pour d’autres phases stationnaires utilisées que celle utilisée pour les runs. Il en va de même pour l’éluant utilisé.
Après une recherche des bonnes conditions d’élution (diminuer le phénomène de fronting et de tailling) du mélange mix avec les 2 types de colonnes, les conditions initiales d’injection et de débit sont les suivants :
C18 : 2µl injection, 1ml/min PFPP : 1µl injection, 0.5 ml/min
25 Les paramètres des runs sont présentés dans le tableau 14 suivant :
Tableau 14 : Conditions des runs a effectuer
Nombre de run Condition chromatographique Run1 Gradient de 10 min, 1-95% ACN ,30°C
Run2 Gradient de 30 min, 1-95% ACN ,30°C
Run3 Gradient de 10 min, 1-95% ACN ,50°C
Run4 Gradient de 30 min, 1-95% ACN ,50°C Paramètre colonne utilisé :
• PFP : 1,9µm ; 2.1 mm x 50 mm • C18 : 1,8µm ; 4.6 mm x 100 mm
L’objectif de cette optimisation est d’obtenir un Rs critique >1.5, soit une résolution entre les composés les moins résolus, supérieure à 1.5.
3.2.2 COLONNE PFP
Les résultats obtenus après l’intégration des 4 runs et l’identification des pics, ont donné pour la résolution maximum, le tableau N°15 et la figure N°20 suivants :
Tableau 15 : Résultats simulés pour les conditions initiales de la colonne
ID Nom tR[min] Aire Largeur Rs
1 Ac. gallique 0.79 54.21 0.08 28.32 2 Catechine 3.46 35.63 0.10 0.53 3 P.B3 3.52 35.82 0.10 0.66 4 P.B1 3.58 48.89 0.10 4.55 5 P.B2 4.02 37.10 0.09 0.53 6 Epicatechine 4.07 48.70 0.10 3.57 7 P.C1 4.40 44.59 0.09 0.00
Figure 20 : Chromatogramme simulé pour les conditions de colonne initial
On constate que les conditions physiques actuelles de la colonne ne suffisent pas à assurer une résolution minimale de 1.5 entre les substances. Cependant, Drylab est capable de simuler d’autre résultats en fonction des paramètres de la colonne (dimension, taille des particules, porosité). Le tableau 16 suivant résume les résolutions obtenues en fonction des paramètres testés.
26 Tableau 16 : Récapitulatif paramètres testés
Diamètre des particules Diamètre interne Longueur Rs critique ≥1.5 1.9 µm 2.1 mm 50 mm x 2.1 mm 100 mm x 3 µm 2.1 mm 100 mm x 2.1 mm 150 mm x 4.6 mm 50 mm x 4.6 mm 100 mm (1.45) 4.6 mm 150 mm 1.88 5 µm 4.6 mm 150 mm 1.61 4.6 mm 200 mm 1.86 4.6 mm 250 mm 2.06
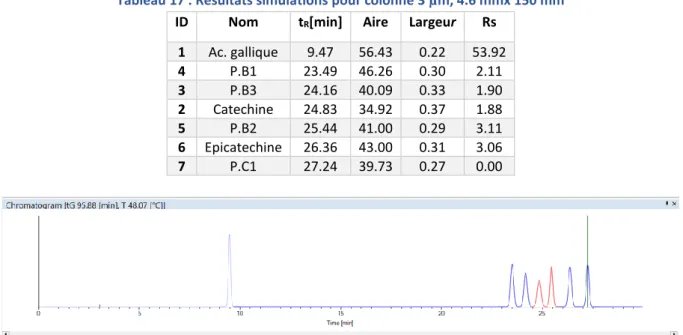
La figure 21 et le tableau 17 suivants illustrent un exemple de simulation obtenue pour la colonne à 3 µm, 4.6 mmx 150 mm pour un Rs critique de 1.88 avec les conditions d’élution suivantes : gradient linéaire ACN de 1-95% en 95.88 min à 48.07 °C. (Dernier pic à 27 min).
Tableau 17 : Résultats simulations pour colonne 3 µm, 4.6 mmx 150 mm
ID Nom tR[min] Aire Largeur Rs
1 Ac. gallique 9.47 56.43 0.22 53.92 4 P.B1 23.49 46.26 0.30 2.11 3 P.B3 24.16 40.09 0.33 1.90 2 Catechine 24.83 34.92 0.37 1.88 5 P.B2 25.44 41.00 0.29 3.11 6 Epicatechine 26.36 43.00 0.31 3.06 7 P.C1 27.24 39.73 0.27 0.00
Figure 21 : Chromatogramme simulé pour colonne PFP 3 µm, 4.6 mmx 150 mm Les 3 autres colonnes suivantes montrent une résolution suffisante :
• 5 µm, 4.6mm x 150 mm ; Rs critique 1.61, gradient linéaire 1-95% ACN en 98.74 min à 48°C (Dernier pic à 29 min).
• 5 µm, 4.6mm x 200 mm ; Rs critique 1.86, gradient linéaire 1-95% ACN en 129 min à 48°C (Dernier pic à 56 min).
• 5 µm, 4.6mm x 250 mm ; Rs critique 2.06, gradient linéaire 1-95% ACN en 179 min à 47.4 °C (Dernier pic à 47 min).
Si un travail de quantification doit être mené avec ce type de colonne, il faudra choisir parmi les quatre colonnes présentées ci-dessus.
27
3.2.3 COLONNE C18
Les résultats obtenus après l’intégration des 4 runs et l’identification des pics, ont donné pour la résolution maximum, le tableau N°18 et la figure N°22 suivants :
Tableau 18 : Résultats simulés pour les conditions initiales de la colonne
ID Nom tR[min] Aire Largeur Rs
1 Acide gallique 4.48 56.97 0.08 53.68 2 P.B1 8.43 48.59 0.07 7.61 3 P.B3 8.96 43.18 0.07 6.05 4 Catechine 9.40 40.94 0.07 12.32 5 P.B2 10.26 48.98 0.07 10.77 6 Epicatechine 10.98 44.30 0.07 9.09 7 P.C1 11.57 49.89 0.06 0.00
Figure 22 : Chromatogramme simulé pour les conditions de colonne initiales
Le Rs critique de 6.05 est obtenue avec les conditions d’élution suivantes : gradient linéaire ACN de 1-95% en 51.2 min à 28.00°C. (Dernier pic à 12 min).
Le test pratique donne les résultats suivants (figure 23 et tableau 19) :
Tableau 19 : Résolution pratique obtenue selon les conditions optimales de Drylab
Composé tR Aire Largeur Rs calculé
Acide gallique 4.25 60.51 0.053 74.8 P.B1 7.89 49.03 0.043 6.8 P.B3 8.35 41.36 0.091 5.9 Catéchine 8.76 37.01 0.046 17.2 P.B2 9.56 46.41 0.046 14.2 Epicatéchine 10.23 42.23 0.046 11.8 P.C1 10.80 45.69 0.051 X
28 La Rs critique est de 5.9 soit une différence de 0.15 (2.5%) par rapport à la simulation Drylab.
La résolution a été calculée avec l’équation 1 suivante :
𝑅𝑠 = 2 𝑥 (𝑡𝑟2 − 𝑡𝑟1)
(𝑤1 + 𝑤2)
La run test a démontré l’efficacité de Drylab. En effet, la simulation est très proche (2.5% de différence) du résultat pratique.
Une fonction de Drylab permet de déterminer le gradient optimal (optimal en fonction du temps de run et de Rs critique). Le logiciel donnait un Rs critique de 10.3 avec les conditions d’élution suivantes : gradient linéaire ACN de 9-17% en 18 min et 17-95 % en 30 min à 28.00°C. (Dernier pic à 11.5 min). Ce test a aussi été effectué, mais en modifiant le gradient : 9-17% en 18 min et 17-95 en 8 min à 28.00°C. Etant donné que le gradient initial est le même, la résolution devrait être inchangée. Le tableau 20 et 21 montrent les nouvelles conditions d’élution ainsi que les résultats pratiques obtenus :
Tableau 20 : Condition d'élution gradient segmenté
Temps [min] % éluant B (ACN) 0 9
18 17
25 95
Tableau 21 : Résolution pratique obtenue selon les conditions optimales de gradient de Drylab
Composé tR Aire Largeur Rs calculé
Acide gallique 1.80 60.49 0.052 50.5 P.B1 4.20 49.08 0.043 12.8 P.B3 5.05 41.27 0.090 10.1 Catéchine 5.74 37.12 0.046 39.0 P.B2 7.52 46.45 0.045 33.7 Epicatéchine 9.02 42.27 0.044 49.4 P.C1 11.39 45.64 0.050 X
Le Rs critique obtenu est supérieur au gradient initial proposé par Drylab. De plus le temps d’analyse est écourté Cet essai de nouveau gradient a été effectué sur le système chromatographique N°1 de nouveau disponible. Le chromatogramme suivant a été obtenu (voir figure 24) :
Figure 24 : FLD (Ex 280 nm et Em 320 nm) Séparation mélange de standard aux conditions optimales La Rs critique est de 10.1 soit une différence de 0.3 (3%) par rapport à la simulation Drylab.
29 Dans le cas où un changement des dimensions de la colonne serait possible, plusieurs simulations ont été effectuées, en changeant les paramètre de la colonne, (dimension, taille des particules). Le tableau N°22 suivant résume les résolutions obtenues en fonction des paramètres testés :
Tableau 22 : Rs critique en fonction des paramètres testés
Diamètre des particules Diamètre interne Longueur Rs critique ≥1.5 1.8 µm
4.6 mm 50 mm 3.73
2.1 mm 50 mm X
2.1 mm 150 mm 3.4
On constate que diminuer la longueur de colonne permet d’obtenir un Rs critique acceptable. Mais changer le diamètre interne tout en conservant la longueur de 50 mm ne permet pas d’obtenir une bonne séparation. Quant à la colonne de 150 mm de long, elle fournit un Rs critique inférieur à la première du tableau N°22.
3.2.4 RÉSUME OPTIMISATION SÉPARATION
Après observation des résultats obtenus pour la colonne PFPP, il a été décidé de ne pas continuer le développement de méthode avec ce type de colonne, car, les quatre colonnes trouvées grâce à la simulation proposent un temps d’analyse trop long. De plus, le maximum trouvé est de 2.06 ce qui est 3 fois moins élevé que celui trouvé avec la colonne C18. Après observation des résultats, la colonne C18 utilisée semble pleinement capable de réaliser cette séparation, car la zone de résolution critique supérieure à 1.5 correspond à environ 2/3 de la surface de la carte de résolution calculé par Drylab. Les deux runs test ont permis de confirmer la fiabilité de ces simulations. En effet la différence du Rs critiques était de 2.5% par rapport au Rs pratique calculé pour le premier gradient et de 3% pour le gradient segmenté. Un essai avec le MeOH comme éluant aurait aussi été une possibilité, cependant une récente publication montre que le MeOH n’est pas l’éluant optimal pour le type de composés recherchés [10].
Le gradient segmenté a été choisi pour commencer la quantification. Ce gradient segmenté a l’avantage de proposer un très bon Rs critique et surtout, il permet un gain de temps important.
3.3 3ÈME PARTIE : ANALYSE EXTRAIT DE RAFLE (GAMARET, SYRAH ET MERLOT) ET
PRÉPARATION A LA QUANTIFICATION
Dans cette partie, l’observation des différences entre les trois extraits obtenus par FLD ainsi que la confirmation de la bonne superposition des standards avec les échantillons a été faite avec la nouvelle méthode d’élution. Une observation supplémentaire des m/z des standards a été effectuée, ainsi qu’une brève analyse MS des pics similaires et différents entre les extraits et l’échantillon témoin de vins (T2).
Comme demandé par le responsable de Bachelor, les analyses de l’extrait de Gamaret ont été réalisées avec la même méthode que celle utilisée avec les autre extraits (méthode d’essai Agroscope, Ch. 2.6.1). Cependant, durant la saisie des données (comparaison entre les extraits), le fait d’avoir entrepris une maintenance de
l’appareillage (système chromatographique N°1) ainsi qu’un nettoyage de la colonne a affecté les tR obtenus pour
le nouvel extrait, ce qui rendait difficile la comparaison. Par souci de bien faire, les 3 extraits ont été analysé avec la méthode nouvellement obtenue. Il s’en est suivi une analyse complète des extraits, similitudes et différences entre les pics en FLD, ainsi qu’une comparaison avec les standards à disposition.
30 Figure 27 : FLD ; 1ère partie chromatogramme ;
méthode du gradient segmenté
Figure 26 : FLD ; 2ème partie chromatogramme ;
méthode du gradient segmenté
3.3.1 COMPARAISON FLD/ MS ENTRE EXTRAIT DE RAFLE ET T2
Les extraits ont montré globalement au niveau des tR des pics le même profil. En effet, il y a de grosses différences
d’intensité entre les extraits, mais pas de différence flagrante quant au tR des pics pour chaque extrait.
Cependant, il y a bien des pics qui sont différents par rapport à T2 et ce, pour tous les cépages testés. Les figures N°25-29 et le tableau N°23 illustrent mes propos :
31 Figure 28 : FLD ; 3ère partie chromatogramme ;
méthode du gradient segmenté
Figure 29 : FLD ; 4ère partie chromatogramme ;
méthode du gradient segmenté
Par l’observation de ces chromatogrammes, on constate qu’il y a bien des différences entre le T2 et les extraits.
Cependant, de la 1ère à la 3ème partie du chromatogramme (figure 26 à 27 ; 0-14 min), la majorité des pics se
superpose bien entre les extraits et le T2. Les m/z ainsi que les tr de ces pics sont présentés dans le tableau N°23 suivant :
Tableau 23 : Présentation des pics d’intérêt (similaires et différents) entre les extraits et T2 ainsi que les m/z majoritaires au tr indiqué
Pic Ionisation Pic Ionisation
No tR [min] -ESI [m/z] +ESI [m/z] No tR [min] -ESI [m/z] +ESI [m/z]
1 1,25 191 215 13 5,10 577 579 149 231 369 118 2 1,50 149 130 14 5,70 295 291 191 203 147 3 1,60 149 132 15 6,00 865 867 321 4 1,69 149 203 16 6,40 865 441 132 118 5 1,92 149 171 17 6,50 576 118 865 118 165 149 6 2,10 164 166 18 6,85 577 169 167 7 2,30 315 166 19 7,20 229 231 149 155 447 118 8 2,60 315 118 20 7,55 577 579 149 334 397 118 9 2,90 149 130 21 8,50 652/229 Syrah 231
32 161 544/652 Syrah 118 10 3,10 111 243 22 9,00 289 291 173 118 463 11 3,80 203 205 23 10,70 865 118 311 493 12 4,21 577 579 24 12,30 577 579 118
3.3.2 CONFIRMATION ÉLIGIBILITÉ DES STANDARDS PAR SCAN MS ET DÉTECTION FLD
Cette partie compare la même table de standards déjà utilisés et présentés dans le chapitre 3.2.1. Le tableau N°24 suivant résume cette comparaison :
Tableau 24 : Correspondance m/z avec les standards utilisés
Correspondance
Standards MW [M+H]+ [M-H]- Extrait T2
Procyanidine B1 578,526 579,526 577,53 multiple multiple
Procyanidine B2 578,53 579,53 577,53 multiple multiple
Procyanidine B3 578,53 579,53 577,53 multiple multiple
Procyanidine C1 866,79 867,79 865,79 multiple multiple
(-) Epicatéchine 290,271 291,271 289,27 multiple multiple
(+) Catéchine 290,271 291,271 289,27 multiple multiple
Acide gallique 170,1195 171,1195 169,1195 1 1
Une correspondance multiple signifie que le chromatogramme EIC en fonction du m/z choisi se superpose à plusieurs pics des échantillons. La figure N °30 suivantes illustre mes propos :
Figure 30 : Overlay chromatogramme +EIC 579 m/z avec les chromatogrammes FLD des extraits de rafles Dans le tableau N°25 est présenté le résumé de la comparaison des overlays des standards avec les échantillons :
33 Tableau 25 : Récapitulatif comparaison tR standards et échantillons
Composé M1 M4 T2 Syrah Merlot Gamaret Ac.gallique X X X X X X Catechine X X X X X X Epicatechine X X X X X X P.B1 X X X X X X P.B2 X X X X X X P.B3 X X X P.C1 X X X X X X
Dans le tableau N°25, M2 et M5 sont absents car ces deux échantillons ne sont que M1 et M4 en plus concentrés. Les proanthocyanidines B2 et C1 ont été observées, cependant, dans les extraits ces concentrations sont très
faibles. La proanthocyanidine B3 a montré un tR différent pour les échantillons de vins.
3.3.3 OPTIMISATION SÉPARATION ÉCHANTILLON TÉMOIN T2
Une observation de la méthode d’élution utilisée a montré que l’acide gallique présentait un léger phénomène de coélution dans les échantillons de vins. Une série de tests de gradient segmenté a été effectuée sur l’échantillon témoin de vin (T2) afin de séparer l’acide gallique. La figure N°31 suivante récapitule les gradients testés :
Test gradient 1 : Test gradient 5 : Test gradient 9 :
T [min] %B T [min] %B T [min] %B
0 10 0 7 0 3
15 17 15 17 20 18
25 95 25 95 40 95
Test gradient 2 : Test gradient 6 :
T [min] %B T [min] %B
0 10 0 7
20 17 20 17
25 95
Test gradient 3 : Test gradient 7 :
T [min] %B T [min] %B
0 5 0 3
15 17 15 18
25 95 35 95
Test gradient 4 : Test gradient 8 :
T [min] %B T [min] %B
0 5 0 3
20 17 20 18
25 95 35 95
Figure 31 : Gradients segmentés testés
Le seul test concluant pour la séparation de l’acide gallique est le test N°3. Cependant, il a été observé par la comparaison de l’évolution de l’élution des pics en fonction des gradients testés que le pic de la
34 proanthocyanidine B3 était impur. En effet, le pic de P.B3 se fondait dans un pic bien plus grand lorsque l’on utilise le gradient segmenté initial. Ce phénomène n’a été observé que dans les échantillons de vin (M1, M4 et T2). Le test N°3 présentait le même problème. Après discussion avec le responsable, il a été décidé de commencer la quantification avec le gradient segmenté initial malgré le phénomène de coélution de l’acide gallique et la séparation de P.B3. La méthode chromatographique utilisée pour la quantification est donc la suivante :
Eluant A : H2O / HCOOH 100 :0.1
Eluant B : CH3CN / HCOOH 100 :0.1
Colonne : Eclipse Plus C18 100 x 4.6 mm 1,8 m.
Gradient : Temps [min] A [%] B [%] Débits [ml/min] Pression [bar] 0.0 91 9 1.0 600 18.0 83 17 1.0 600 25.0 5 95 1.0 600
Température : 28°C Stop Time : 25.0 min Post Time : 5.0 min
Volume d’injection : 2µl solution de calibration/ LOQ/LOD/Ctrl et 2 à 10µl pour les échantillons
Détecteur DAD : 280 nm
Détecteur FLD : Excitation 280 nm ; Emission 320 nm
Les chromatogrammes obtenus par l’utilisation de cette méthode sur l’échantillon témoin T2 et l’extrait de Gamaret sont présentés ci-dessous (figure 32 et 33) :
35 Figure 33 : FLD ; Vin de Gamaret (T2)
3.3.4 PRÉPARATION QUANTIFICATION PAR DMRM
L’objectif secondaire du travail de Bachelor était d’effectuer une quantification par MS (DMRM). Pour ce faire, les standards ont d’abord été analysés via le logiciel Optimizer par injection directe dans le MS. Ce logiciel va permettre de déterminer les conditions de fragmentation optimale (Fragmentor et CE) en fonction de l’ion précurseur choisi et de l’ajout (H+ ; H-).
Le tableau N°26 récapitule les résultats obtenus via le logiciel :
Tableau 26 : Résumé des valeurs obtenus pour les fragments les plus abondants
Composé Précurseur ion Product ion Fragmentar Energie de collision tR [min]
Acide gallique 169 (-H) 125 71 12 1.80 Catéchine 291 (+H) 139 106 12 5.74 Epicatéchine 291 (+H) 139 111 8 9.02 P.B1 577(-H) 289 152 24 4.20 P.B2 577(-H) 289 157 24 7.52 P.B3 577(-H) 289 162 23 5.00 P.C1 864 (-H) 575 230 20 11.38
On conste que pour l’acide gallique, catéchine, épicatéchine et P.C1, l’ionisation positive convient mieux pour l’obtention des fragments. Pour les autres composés, c’est le phénomène inverse qui est observé. Les paramètres de fragmentation ayant été déterminés, la quantification par MS peut donc débuter.
36
3.4 4ÈME PARTIE : QUANTIFICATION ET VALIDATION DE LA MÉTHODE
Dans cette partie, la quantification des échantillons a été effectuée par le biais de courbe de calibration. Le facteur de réponse des différents composés a été lui aussi déterminé ainsi que la LOQ et LOD propre à chaque composé, analysé.
3.4.1 CALIBRATION, LOD ET LOQ
La méthode chromatographique utilisée et la méthode MS sont celles présentées aux chapitre 3.3.3 et 3.3.4 L’estimation de la calibration s’est faite par comparaison des aires entre tous les échantillons, et la solution mix de standard effectuée dans le chapitre 3.2.
Il a été observé que par rapport aux aires des échantillons, le domaine de concentration de chaque composé devait être le suivant (tableau N°27) :
Tableau 27 : Domaine de concentration observé
Composé quantifiés Domaine de concentration Acide gallique 10-150 mg/L Catéchine 10-100 mg/L Epicatéchine 5-80 mg/L P.B1 34-255 mg/L P.B2 7,5-75 mg/L P.B3 15-210 mg/L P.C1 10-40 mg/L
Les courbes de calibrations ont été effectuées avec les solutions mères des standards (tableau 10, Ch. 3.1.3). Les tableau N° 28 montrent les concentration courbes de calibration :
Tableau 28 . Concentration courbes de calibration pour chaque composé
Concentration [mg/L] P.B1 P.B2 P.B3 P.C1 Acide gallique Catéchine Epicatéchine Std1 33,6 7,6 14 9,6 10,2 10,2 5,25 Std2 84 15,2 42 16 20,4 30,6 10,5 Std3 168 45,6 70 24 51 51 21 Std4 252 76 140 32 102 81,6 52,5 Std5 336 152 210 40 153 102 84 Std6 X X 280 80 204 153 126 Les linéarités suivantes ont été obtenus (Tableau N°29) :
Tableau 29 : Linéarité FLD et DMRM des calibrations
Composé Droite r Courbe DMRM r P.B1 1,574866x - 6,23157 0,99968 7,647226 + 1,115207 ln(x) – 0,051412 ln(x)^2 0,99982 P.B3 1,930521x - 5,39297 0,99969 6,742410 + 1,450248 ln(x) – 0,076897 ln(x)^2 0,99995 P.B2 1,978415x - 5,62095 0,99996 6,249588 + 1,296517 ln(x) – 0,065460 ln(x)^2 0,99989 P.C1 1,322110x - 4,15791 0,99883 2,658356 + 1,794237 ln(x) – 0,104253 ln(x)^2 0,99807 Catéchine 5,040223x - 1,015141 0,99991 9,305708 + 1,112708 ln(x) -0,044993 ln(x)^2 0,99994 Epicatéchine 5,098697x - 0,98506 0,99984 10,103530 + 0,925764 ln(x) – 0,024678 ln(x)^2 0,99995 Acide gallique 5,142758x - 6,4922 0,99994 7,987136 + 0,604620 ln(x) -0,007819 ln(x)^2 0,99987
37 Des contrôles ont été ajoutés afin de garantir la bonne linéarité des courbes obtenues. Le tableau N°30 suivant montre les concentrations des échantillons de contrôle :
Tableau 30 : Concentration échantillon de contrôle
Ctrl Concentration [mg/L] P.B1 84 P.B3 70 P.B2 76 P.C1 40 Acide gallique 61,2 Catechine 61,2 Epicatechine 63 Les recouvrements suivants ont été obtenus (tableau N° 31) :
CTRL Moyenne concentration [mg/L] FLD Moyenne Recouvrement FLD Moyenne concentration [mg/L] MS Moyenne Recouvrement MS P.B1 88,0 105 84,0 100 P.B3 69,40 99 73,47 105 P.B2 78,98 104 78,72 104 P.C1 39,38 98 40,81 102 Catechine 78,7 96 88,4 108 Epicatechine 50,2 96 62,2 118 Acide gallique 100,1 98 106,5 104
Les recouvrements sont tous <10% pour les échantillons FLD. Cependant le recouvrement en MS de la catéchine est >10%.
Les LOQ et LOD ont été estimés par l’observation du S/N de la solution de mélange de standard, calculé par le logiciel Agilent. Selon les estimations, les concentrations calculées suivantes ont été obtenues (tableau N°30) :
Tableau 31 : Concentration LOD et LOQ
LOD Concentration [mg/L] LOQ Concentration [mg/L] P.B1 0,42 P.B1 1,68 P.B3 0,91 P.B3 2,80 P.B2 0,53 P.B2 1,52 P.C1 0,90 P.C1 3,20 Catechine 0,15 Catechine 0,24 Epicatechine 0,16 Epicatechine 0,32 Ac.gallique 2,00 Ac.gallique 2,00
Les solutions de LOD et LOQ ont été injectées, 3 fois chacune afin d’obtenir la moyenne des S/N. Les S/N suivants ont été obtenus (tableau N°32) :
38 Tableau 32 : Résumé S/N obtenu pour chaque composé
LOD Moy. S/N] LOQ Moy. S/N] P.B1 3,1 P.B1 11,3 P.B3 4,3 P.B3 11,2 P.B2 4,4 P.B2 10,4 P.C1 3,3 P.C1 10,7 Catechine 6,9 Catechine 15,4 Epicatechine 5,6 Epicatechine 14,8 Ac.gallique 702,3 Ac.gallique 702,3
Les valeurs limites de quantification obtenues sont très proches du S/N désiré (S/N≥10 pour la LOQ et S/N≥3 pour la LOD), à l’exception de la LOQ pour l’acide gallique. En effet, il est nécessaire de faire une nouvelle solution de LOQ et LOD pour ce composé. Le S/N obtenu pour l’acide gallique a montré que l’on pourrait descendre jusqu’à une concentration théorique d’environ 0.03 mg/L pour la LOQ et 0.09mg/L pour la LOD.
3.4.2 FACTEUR DE RÉPONSE
Le facteur de réponse a été déterminé pour chaque composé en divisant l’aire du pic par sa concentration. Un facteur de réponse moyen a été déterminé avec les concentrations des standards des courbes de calibration. Le tableau N°33 suivant résume ces résultats :
Tableau 33 : Résumé facteur de réponses obtenus en fonction des composés analysés
Composé Facteur réponse 1 moyen [mg/L]
Facteur réponse 2 moyen [µM/L]
Rapport 1 Fcat / Fautre composé
Rapport 2 Fcat / Fautre composé
Acide gallique 4,95 0,84 1,03 1,75 Catéchine 5,07 1,47 1,00 1,00 Epicatéchine 5,04 1,46 1,01 1,01 P.B1 1,51 0,87 3,36 1,69 P.B2 1,72 0,99 2,95 1,48 P.B3 1,80 1,04 2,82 1,41 P.C1 1,13 0,98 4,49 1,51
L’obtention du rapport entre le facteur de réponse de la catéchine et le facteur de réponse d’un des composés analysé permettra de seulement utiliser la courbe de calibration de la catéchine ou un étalon interne de catéchine pour pouvoir déterminer la concentration en équivalent catéchine d’un composé analysé. Le calcul se fait de la manière suivante :
•
Conc. eq catéchine =
Aire composé analysé x Rapport des facteur de reponse x Conc.etalon catechineAire etalon catechine
Ce calcul est valable si l’on souhaite déterminer la concentration des substances mentionnées ci-dessus, ainsi que pour un volume d’injection égal au volume d’injection de l’étalon
Une image représentant l’ensemble des évolutions des facteurs de réponses en fonction de la concentration en mg/L et µM/L se trouve à l’annexe 1. Tous les composés analysés y sont illustrés.
![Figure 12 : Schéma de l'obtention des échantillons [7]](https://thumb-eu.123doks.com/thumbv2/123doknet/14334397.498515/12.892.224.715.166.442/figure-schéma-obtention-échantillons.webp)
![Tableau 8 : Tableau récapitulatif des standards utilisés Standards MW [M+H] + [M-H] - Procyanidine A2 576.52 577.52 575.52 Procyanidine A1 576.51 577.51 575.51 Procyanidine B1 578.53 579.53 577.53 Procyanidine B2 578.53 579.53 577.5](https://thumb-eu.123doks.com/thumbv2/123doknet/14334397.498515/19.892.104.788.131.208/récapitulatif-standards-utilisés-standards-procyanidine-procyanidine-procyanidine-procyanidine.webp)
![Tableau 18 : Résultats simulés pour les conditions initiales de la colonne ID Nom t R [min] Aire Largeur Rs](https://thumb-eu.123doks.com/thumbv2/123doknet/14334397.498515/27.892.101.791.251.551/tableau-résultats-simulés-conditions-initiales-colonne-aire-largeur.webp)