Development of Polymeric
Nanoparticle Vaccines for
mARCHNs
Im
m
unostim
ulation
MAASSxdHUSETTS INSTITUTEby
Pamela A. Basto
0
B.S. Biomedical Engineering
BR
IESThe University of Texas at Austin, 2006
-SUBMITTED TO THE DIVISION OF HEALTH SCIENCES & TECHNOLOGY IN
PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF
DOCTORATE IN PHILOSOPHY IN MEDICAL ENGINEERING MEDICAL PHYSICS
AT THE
MASSACHUSETTS INSTITUTE OF TECHNOLOGY AUGUST 2013
02013 Pamela A. Basto All rights reserved.
The author hereby grants to MIT permission to reproduce and to distribute publicly paper and electronic copies of this thesis document in whole or in part
in any medium now known or hereafter created.
Signature of Author:
4 ealth Science & Technology Institute of Medical Engineering & Science
August 28, 2013
Certified by:
-Rot6t Langer, ScD David H. Koch Institute Professor Thesis Supervisor
Accepted by: - / r /
Enviry Brown, MD, PhD
& /ector, Harvard-MIT Program in Health Sciences & Technology Professor of Computational Neuroscience & Health Sciences & Technology
Table of Contents
List of Figures...5
List of Tables ... 10
D edication ... 11
Acknowledgem ents ... 12
Abbreviations, Symbols & Definitions...14
A bstract...15
Chapter 1: Background... 17
1.1 Introduction... 17
1.1.1 Historical context of vaccines, burden of illness, and potential market...17
1.1.2 Definition of Vaccine... 18
1.1.3 Current Approach for Vaccine Delivery & Limitations: ... 19
1.2 Fundamental Principals of the Immune Response ... 20
1.2.1 Innate Immune Response ... 21
1.2.2 Adaptive Immune Response...22
1.2.3 Cytokines are crucial mediators of immune response ... 24
1.2.4 Immunological differences between mice and humans ... 25
1.3 Toll-like receptor complexes, natural ligands, and synthetic adjuvants ... .... 27
1.3.1 Definition of Toll-like receptors ... .... ... 27
1.3.2 Necessity for Adjuvants in Vaccines: ... . ... 27
1.3.3 Synergy of TLR for vaccine development... ... 29
1.4 Polymeric Nanoparticles for Synthetic Vaccines ... 29
1.4.1 Potential Advantages of Polymeric Nanoparticles as a Vaccine Carrier:...29
Chapter 2: Development of a control releasing R848 nanoparticle vaccine platform ... 32 2.1 Introduction... ... ... ... ... ... ... 32 2.2 Rationale...34 2.3 Methods ... .... 36 2.3.1 Materials: ... 36 2.3.2 Instrumentation:... ... ... 36
2.3.4 Synthesis of PLGA-AF647 and PLGA-AF488 Block Copolymer ... 38
2.3.5 Nanoparticle vaccine formation via double emulsion ... 38
2.3.6 Nanoparticle characterization... 39
2.3.7 Histology...40
2.3.8 In Vitro DC stimulation and Co-culture Assays ... 40
2.3.9 C14 Nanoparticle Biodistribution ... 41
2.3.10 In vivo Nanoparticle Cellular Uptake & Distribution...42
2.3.11 Mouse Fluorescence Imaging ... 42
2.3.12 In vivo antigen-specific T cell proliferation assay ... 42
2.3.13 Serum analysis of free R848 ... 42
2.3.14 ELISAs ... 43
2.3.15 Statistical Analysis...43
2.4 Polymer Synthesis & Characterization ... 43
2.5 Vaccine Nanoparticle Synthesis & Characterization ... 45
2.6 Nanoparticles for Dendritic Cell Activation In Vitro ... 48
2.7 Nanoparticle vaccines stimulate DCs to activate CD8+ cells in vitro...50
2.8 Nanoparticle Vaccine Bio-distribution In Vivo... 51
2.9 Nanoparticle Vaccine Induces Antigen-Specific Cell Proliferation In Vivo ... 52
2.10 R848-Nanoparticles Induce Humoral Response ... 56
2.11 Summary ... 57
Chapter 3. Development of a epitope specific vaccine platform applied for PCSK9 serum level reduction...59
3.1 Introduction...59
3.1.1. Background & Significance of Cardiovascular Disease in the US...59
3.1.2. Current clinical treatments for hypercholesterolemia ... 60
3.1.3. PCKS9 Regulates the LDL Receptor Recycling Pathway ... 61
3.1.3. Current Therapeutics to Inhibit PCSK9... 63
3.2 Rationale ... 66
3.3 Methods ... 67
2.3.1 Materials: ... 67
2.3.2 Biotin-Streptavidin Nanoparticle Platform Formulation & Characterization:
... 68
2.3.3 Assay for determination of available active binding sites: ... 69
2.3.3 PCSK9 Nanoparticle Vaccination: ... 69
2.3.4 WT-PCSK9 & GOF-PCSK9 LDL-R Binding Assay: ... 70
2.3.5: Assays for mPCSK9, LDL-c/HDL-c, and anti-hPCSK9 antibodies:...70
3.3 PCSK9 Epitope Selection ... 70
3.4 Nanoparticle Synthesis & Characterization... 72
3.5 In Vitro LDL-R knockdown with Sera from Immunized Mice ... 78
3.6 In Vivo mPCSK9 Reduction and ot-hPCSK9 IgG Increase ... 80
3.7 Nanoparticle Vaccines have potential to Reduce LDL- Cholesterol In Vivo ... 82
3.5 Summary & Future Directions...85
Chapter 4. Future Directions ... 87
4.1 Sum m ary ... 87
4.2 Next generation vaccines against infectious disease ... 88
4.2 Non-traditional epitopes for cancer vaccine development ... 89
Bibliography ... 93
Appendix... 97
Al. PCSK9 Insert Sequences ... 97
A1. WT Sequence ... 97
A1.2 PCSK9 Mutated sequence ... 98
A2. HER-2-Targeted Nanoparticle-Affibody Bioconjugates for Cancer Therapy* ... 99
A2. Single-Step Assembly of Homogenous Lipid-Polymeric and Lipid-Quantum Dot Nanoparticles Enabled by Microfluidic Rapid Mixing**...107
List of Figures
Figure 1.1 Incentives for Vaccine Research: Strategic Plan 2010-2020. World Health Organization. Taken from 2002 ... 18
Figure 2.1 Selected TLR 7 agonists in order of immunostimulatory potency...33 Figure 2.1 Schematic of nanoparticle immune system activation. ... 36
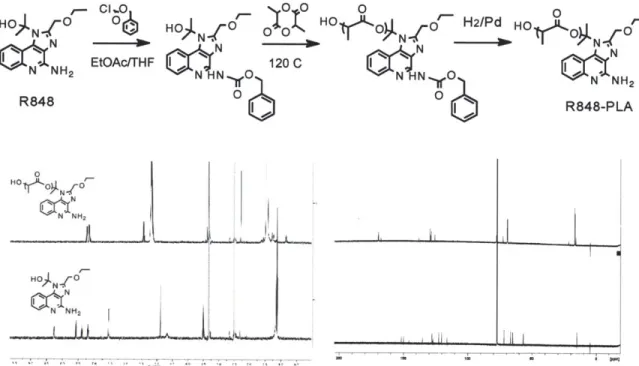
Figure 2.3 (Top) Schematic of protection of R848 followed by lactide ring opening polymerization (Bottom) H' and C" NMR of polymer vs. free R848... 44 Figure 2.3 2D NMR Analysis of R848-PLA conjugate (Top) COSY (Bottom left) HMBC (Bottom right) HSQC... 45
... 46
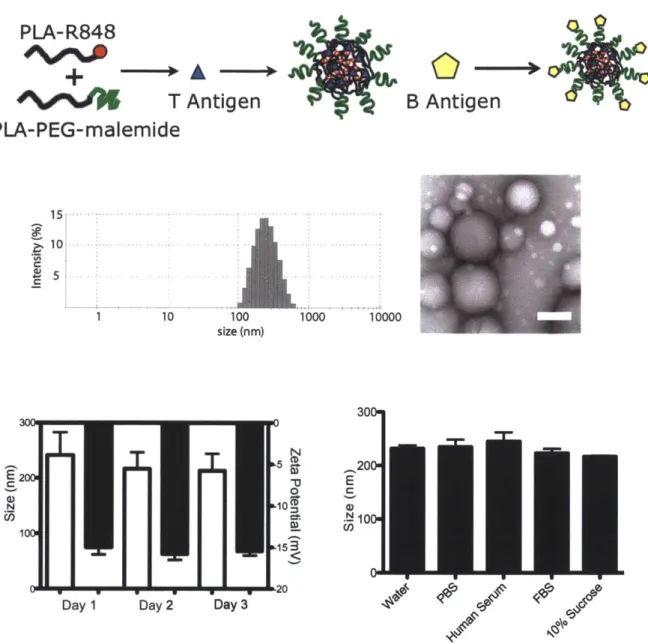
Figure 2.4 Nanoparticle schematic demonstrates the integration of polymer conjugates. Representative nanoparticle size distribution measured by dynamic light scattering (n = 15 separate batches). Transmission electron microscopy of
nanoparticles, scale bar is 200 nm. Nanoparticle stability demonstrated by size and charge in 7.4 pH H20 measured by DLS. (n=5). Nanoparticle size stability after 3
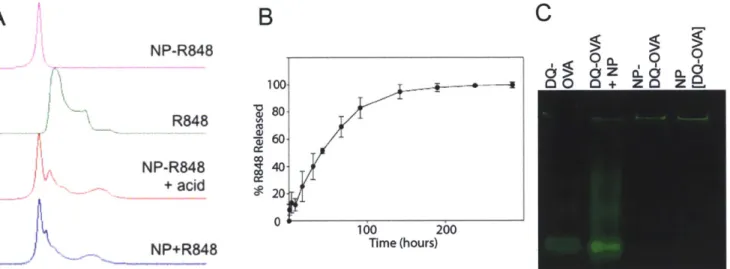
hours in various physiological conditions (n=5)... 46 Figure 2.5 A) R848 encapsulation within nanoparticle vaccines as measured by HPLC. B) Drug release of R848 from nanoparticles formed by w/o/w as measured
by HPLC (n=3). C) Protein gel of nanoparticles and 10 ug of DQ-OVA
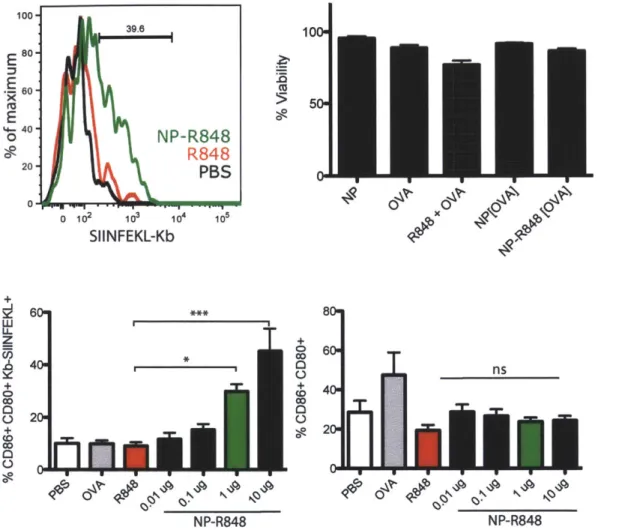
demonstrating encapsulation or surface conjugation of protein... 47 Figure 2.6. A) Histogram of CD80+CD86+ and MHCI-SIINFEKL of 1 ug of R848 conjugated nanoparticles vs. 0.6 ng free R848 and 15 ng of OVA vs. PBS. B) % BMDC viability assessed by 7AAD at 24 hours after 2 hour incubation with nanoparticles or soluble factors. Viability is normalized against BMDCs without treatment. n=6. C) Percentage of CD86+ CD80+ Kb-SIINFEKL+ BMDCs and
CD86+CD80+ BMDCs on day 3 with 2 hour incubation of nanoparticles, 0.6 ng
R848, and 15 ng OVA. OVA and R848+OVA variables matched the equivalent of 1 ug of NP-R848[OVA] as highlighted by the green bar. n=9,10 of three experiments *
p<0.05, *** p<0.001 (one way ANOVA with Bonferroni's post hoc test)... 49
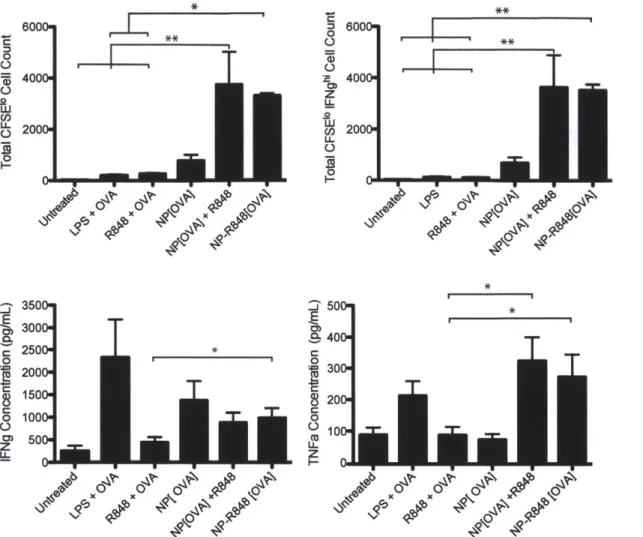
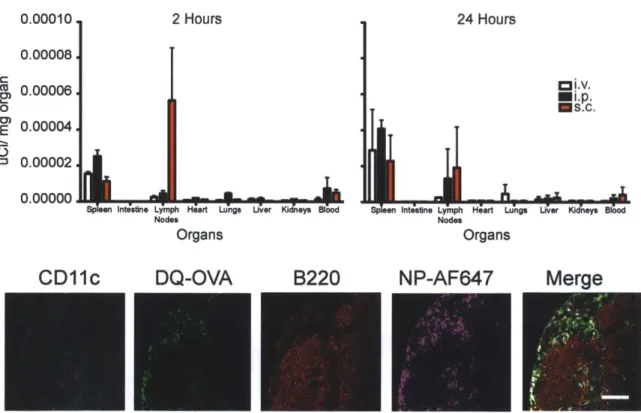
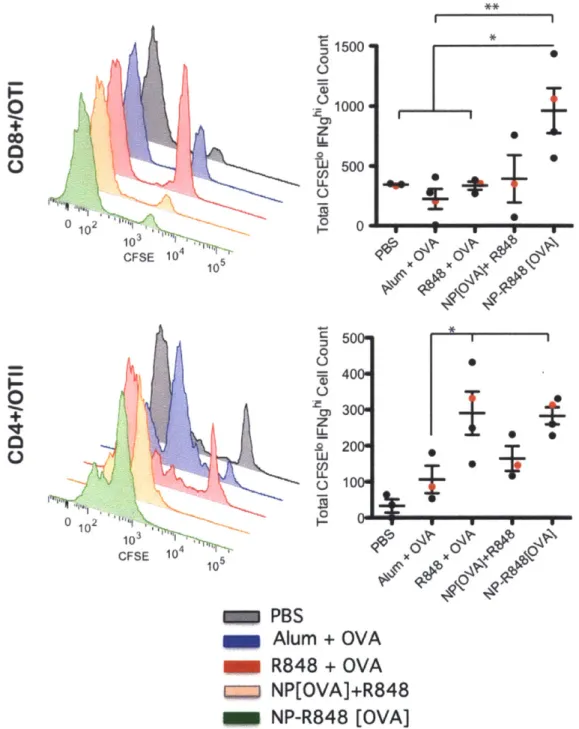
Figure 2.7. Graphs represent mean total cell count +/- s.e.m. of quadruple culture of pooled lymph node cells from one out of 3 independent experiments. Supernatants were collected from each well of co-culture at day 3 and tested by ELISA for IFNg and TNFa concentrations. *p<0.05 by ANOVA with Tukey's post hoc test. Data shown is n=8 of combined three independent experiments... 51 Figure 2.8 (Top) Injection of 0.06 uCi of nanoparticles through different routes of administration: intravenous, intraperitoneal, and subcutaneous. (Bottom) Popliteal lymph node at 1 hour after footpad injection with 100 ug of R848-NPs...52 Figure 2.10 Functional proliferation of adoptively transferred CFSE-labelled Va2+
ANOVA with Tukey's post hoc test. Data representative of 3 (OTI) and 2 (OTII)
experim ents... 54
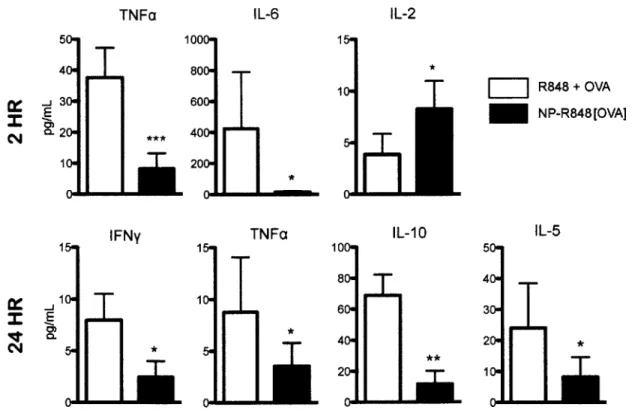
Figure 2.11 Luminex panel on 1:1 dilution of sera taken from mice injected intravenously with 300 ug of nanoparticles or 10 ng of R848 + OVA in soluble form at 2 and 24 hours *p<0.05 ** p<0.01 *** p<0.001 by ANOVA with Tukey's post hoc
test. Data shown is pooled data of n=8 of three experiments... 55
Figure 2.13 Anti-OVA IgG concentration at day 21, day 35, and day 42 after 100 ug day 0 subcutaneous vaccination. **p<0.05 by ANOVA with Tukey's post hoc test. Data shown is n=6,7 of two independent experiments... 56
Figure 2.14 Anti-nicotine antibody concentrations of C57BL/6 mice injected with vaccine formulations at week 0, 2, 4, 8. n=4,5 of 1 experiment. Data by Matteo lannacone, Frank Alexis, and Elena Tonti [34] . ... 57
Figure 3.1 Schematic of PCSK9/LDL-R synthesis and degradation pathway ... 63
Figure 3.2 Three-dimensional ribbon diagrams illustrate the location of the predicted epitopes of all four candidates as recommended by Professor Timothy Springer. ... 72
Figure 3.3 A) Schematic of biotin-streptavidin-biotin-epitope nanoparticle formulation. B) Nanoparticle stability of OVA323.33-biotin- NPs in physiological
conditions. n=3 C) Size and charge of nanoparticle formulation using streptavidin and candidate epitopes in water, n=12 D) TEM of nanoparticle formulation of OVA323.33-biotin- NPs, bar is 100 nm ... 74
Figure 3.4 Assessment of available biotin binding sites on PLGA-PEG-biotin-streptavidin nanoparticles with fluorescein-biotin A) Free biotin was incubated with
100 ug of nanoparticles prior to fluorescein-biotin to find the lowest concentration
for available binding site saturation, n=4 B) Amount of fluorescein-biotin binding after incubation with associated peptide candidates n=6 ... ... 77
Figure 3.5 Vaccination timeline of CFSE labeled OTII cells after adoptive transfer on Day -1 and immunization with nanoparticles on Day 0; followed by histograms of
CFSE labeled OTII cells on Day 3 with varying vaccination protocols...78
Figure 3.6 A) Histograms depicting LDL-R levels on the surface of non-transfected, WT-PCSK9, and GOF-PCSK9 HepG2 cell lines B) Day 38 sera on transfected HepG2 cell lines n=5, 2 experimental repeats. Data by Junghwan Sung ... 80
Figure 3.7 ELISA results for mouse PCSK9 levels and anti-hPCSK9 IgG levels in the serum at day 10 and day 38 after immunization on day 0 and day 28. A) Levels of mouse PCKS9 assessed by ELISA in 1:200 serum dilution at day 10 and day 38.
** p<0.01 by ANOVA with Tukey post test. B) Anti-human PCSK9 IgG levels
demonstrate mice immunized with nanoparticle vaccines with candidate epitopes have increased levels of human PCSK9 specific antibodies. Data representative of two experim ental cohorts ... 82
Figure 3.8 Serum LDL/VLDL and HDL levels in vaccinated mice at day 10, 38, and 212. Day 10 and day 38 are representative of two cohorts, day 212 represents one cohort. n=4,5 ... ... ... 84 Figure 4.1 A) Schematic of nanoparticle vaccine for Chlamydia trachomatis B) Chlamydia loads via qPCR of the uterus on day 6 after challenge after transcervical immunization 4 weeks prior. Data by Georg Stary and Aleksandar Radovic-M oreno. Reproduced from [58,59]... 89
Figure A1.1. Schematic diagram of the formation of drug encapsulated PLA-PEG-Mal nanoparticle-Affibody bioconjugates. Nanoparticle's size diameter (< 100 nm) and distribution was visualized by electron microscopy. The hydrophilic polyethyleneglycol (PEG) chains on the surface reduce the protein absorption on the hydrophobic polymeric surface to form "stealth" nanoparticles. Direct visualization of Affibody conjugation on the surface of the nanoparticle was carried out using fluorescent image of fluorescent Affibody (Alexa Fluor 532; red) conjugated to nanoparticles. After washing the nanoparticle-Affibody bioconjugates, the fluorescent signal increases with an increased amount of fluorescent Affibody (0-20
% Affibody/polymer molar ratio) on the nanoparticle surface confirming the
chemical conjugation efficiency. B) 1H-NMR (proton nuclear magnetic resonance) spectrum represents the PLA-PEG-Affibody bioconjugates. The LH-NMR spectrum shows the protons assigned to the polymer (b = 1-6 ppm) and the presence of
Affibody polypeptide (h = 7-8 ppm) confirming the chemical conjugation of the
Affibody on the polymeric nanoparticles... .... 102
Figure A1.2 Fluorescent microscopy of nanoparticle-Affibody bioconjugates incubated with HER-2 positive cell lines. Capan-1 cells, SK-BR-3 cells and SK-OV-3 cells were grown on chamber slides and incubated in OptiMEM medium supplemented with 5 jpg of NBD fluorescent dye encapsulated into nanoparticles shown in green with (upper panel) or targeted nanoparticle-Affibody bioconjugates (lower panel) for 2 hours prior imaging using fluorescent microscopy at 60X magnification. The cell nuclei and the actin cytoskeleton are stained with blue (4',6-diamidino-2-phenylindole) and red (Alexa-Flour Phalloidin-488), respectively. The deconvoluted fluorescent images represent the mid-cross section of the cells after washing (3 times), permeabilizing and staining steps...103 Figure A1.3 Combined fluorescent images (60X magnification) of a single SK-BR-3 cell to reconstruct a three-dimensional image of the cell. A1-A4 (upper panel) images represent the mid-cross section images of the same SK-BR-3 cell being rotated at 30-degree intervals along the y- axis. A4 represents an image of SK-BR-3 rotated to 90-degree along the y-axis demonstrating particles shown in green (NBD fluorescent dye encapsulated into the nanoparticles) internalized inside the cell. The
cell nuclei and the actin cytoskeleton are stained with blue (4',6-diamidino-2-phenylindole) and red (Alexa-Flour Phalloidin-488), respectively. B1-B4 (lower panel) represents fluorescent images of the same SK-BR-3 (shown in the upper panel) cell without the actin cytoskeleton staining confirming the internalization of the nanoparticle-Affibody bioconjugates inside the cell... 103
Figure A1.4. Cell viability assay (MTS assay) to evaluate the differential toxicity of targeted (Np-Affb) and untargeted nanoparticles (Np) with and without encapsulated paclitaxel (Ptxl). In this assay, the nanoparticle formulations were incubated for 2 hours, cells were subsequently washed and incubated in cell growth media to allow the effect of the drug on the cell cycles before quantifying the nanoparticle formulations toxicities against two cancer cell lines expressing HER-2
(SK-BR-3 and SK-OV-3).ANOVA test "*" p<0.01; "**" p<0.05...105
Figure A2.1 Nanoprecipitation of lipid-polymeric NPs. (a) A representative schematic of input and output streams within hybrid lipid-polymeric nanoparticle formation in microchannels with Tesla structures (b) Illustrative figure of microfluidic synthesized NP component layers (c) TEM image of uranyl acetate stained hybrid NPs after synthesis which highlights differences in density of the core versus near the surface of the NP potentially illustrating the lipid-PEG layer. Bar is labeled at 100 nm (d) Reproducible average size distribution of hybrid NPs generated through microfluidics. Average size is 40 nm. (e) Solvent mixing in the Tesla micromixing structures using fluorescent dye and water at 5 pL/min and 50 jL/min, respectively, shows complete mixing at the fourth turn in the channel (scale
bar: 100pm )...111
Figure A2.2. Characterization of Lipid-PLGA structure (a) Comparison of average
NP size from the product stream with aqueous : organic flow ratios of 10:1 and 5:1
respectively where the input organic stream is either PLGA, PLGA and lipid, or lipid alone. (b) Determination of lipid coverage of polymeric NPs. Zeta potential and size of NPs as the ratio of lipid to PLGA (w/w) is decreased. (c) Size distributions in water and PBS of NPs as the ratio of lipid to PLGA is changed. Complete lipid coverage of polymeric cores is observed at a ratio of lipid to PLGA ratio of 1:10. Above this ratio, the remaining lipid forms other nanostructures such as liposomes and below this ratio, NPs are not stable in PBS due to inadequate lipid coverage. 112 Figure A2.3 Control of NP's physicochemical properties. (a) Control of surface charge and lipid coverage of the hybrid NPs is elucidated by changes in zeta potential of the NPs in PBS using DSPE-PEG with modified functional groups of carboxyl, methyl, and amine (b) Control of NP size by varying PLGA viscosity and concentration in the organic stream. Flow ratios of aqueous to organic streams and rate were kept constant at 10 : 1 at a total flow rate of 55 pL/min. ... 113 Figure A2.4. Slow versus rapid mixing. Comparison of NP size distribution in water and PBS for rapid and slow mixing of lipid and PLGA solutions with lipid : PLGA ratios of 1:1 and 1:10. Under slow mixing conditions without the input of any form of energy, aggregation upon addition of PBS indicates the presence of heterogeneous NPs (i.e. polymeric, lipid, and lipid-polymeric). Under rapid mixing conditions, absence of aggregation upon addition of PBS indicates that only homogenous hybrid lipid-polymeric NPs are formed, except for the 1:1 ratio that results in homogeneous hybrid NPs and liposom es...114 Figure A2.5 NP formation to elucidate stepwise formation of hybrid lipid-polymer NPs within microchannel (a) NP size distribution in water and PBS of particles formed in two-stage manner. PLGA NPs were prepared in the microfluidic mixer,
then washed and placed as an input along with lipid aqueous stream resulting in the generation of hybrid lipid-PLGA NPs. (b) NP size distribution in water and PBS formed through the current one-step microfluidic method...116 Figure A2.6. Preparation of hybrid lipid-QD NPs. (a) Schematic of liposome formation in the Tesla mixer with quantum dots encapsulated within the core (b)
NP distribution of quantum dot encapsulated liposomes formed through the Tesla
mixer (c) TEM image of hybrid lipid-QD NPs stained with 1% phosphotungstic acid aqueous solution showing monodisperse particles with a Z-average size of 60nm. Bar is labeled at 100 nm ... 118
List of Tables
Table 1. Cytokines and their function... 24 Table 2. Comparison of Mouse and Human Immunology. Adapted from [8]... 25
Table 3. Characterization of Nanoparticle Properties ... 48 Table 4. Stages of Development of PCKS9 Therapeutics (Adapted from Do et al.
[52])...65
Table 5. PCSK9 epitope candidates for nanoparticle B antigen ... 71
Table 6. Comparison of available Thermo Scientific Biotin-Binding Proteins...75 Table 7. Clinically available or ongoing clinical trials of cancer vaccine candidates9O
Dedication
Acknowledgements
Completion of my thesis has been a key accomplishment in my life and I am in deep gratitude of several:
Thesis Committee Members: I thank my committee to provide mentorship and for inspiring me to work harder in order to help people in need throughout the rest of my career. My advisor, Professor Langer, whose wisdom, perseverance, support, and outlook on life, I will always carry with me. Bob, thank you for always managing to say the right thing at the right time. Professor Farokhzad has provided years of support and the generation of a supportive learning environment, I feel privileged to have been able to watch you successfully maneuver the clinical translation of your work. I am in gratitude for Professor von Andrian to open his lab in order to gain perspective of a tiny fraction of the immensely large field of immunology. Professor Bhatia has always supported me through HST and serves as a role model on multiple levels. I have had the unique experience of knowing most of my committee's lab members on a personal level and I am fortunate to have had them influence my personal and professional life in addition to science.
In addition to my committee mentors, I wish to specifically note remarkable educators throughout my lifetime that I am honoured with the opportunity to have met: Professor Wolfgang Frey, Professor Rebecca Richards-Kortum, Dr. Michele Follen, Dr. Valerie Pronio-Stelluto, Professor Jeff Drazen, Professor Judah Folkman, Professor Norman Letvin, David Journeay, Ed Davis, Dr. Joel Morrisett, Professor Al Grodzinsky, Professor Rohit Karnik, and Anjali Sastry.
My friends in lab: the nicest window (Ana Jaklenec, Chris Alabi, Abigail
Lytton-Jean, Arturo Vegas, Leon Bellan, Janet Zoldan). And notably the women behind the man: Connie Beal, Ilda Thompson, Bethany Day, Tiffany Greeves, and Tuli Saha. Dr. Frank Alexis and Dr. Aleks Radovic-Moreno have provided their support, expertise, and friendship through the years, I have been fortunate to collaborate with friends in the lab. I am indebted to collaborators who I have been able to work with: Dr. Matteo lannacone, Dr. Ashley Moseman, and Dr. Elena Tonti for the R848-PLA work, Dr. Junghwan Sung, who was my mentor on the PCSK9 project and Dr. Georg Stary, who headed the chlamydia vaccine project. Notably, Rohit Karnik, Archana Swami and Armon Sharei, whom I've been fortunate to work with on other projects outside of this thesis work. All of the members in von Andrian lab have, in some way or another, provided advice and have been a great resource, notably Scott Loughhead, David Alvarez, Michael Flynn, and Carolina Perdomo. MTL, Koch Core, Glenn Paradis, Stuart Levine, Eliza Vasile, Scott Malstrom, all members of these MIT facilities have provided remarkable support and expertise.
This work could not have been completed without a cohort of talented undergraduates that I've had the fortune of mentoring through my time here: Zoe Moyer, Jennifer Chu, Claudia Tenen, Nebiyat Tsegaye, Emma Broderick, and Camille Sullivan, all who I look forward to watching their careers unfold.
HST gave me the invitation to an amazing education and the opportunity to be
immersed in the academic/medical epicenter of Boston and has directed my commitment to returning this education, knowledge, and responsibility in hopes of benefitting the
health of others. My friends here at MIT and back home have served as a priceless support system through the inevitable highs and lows of the PhD process. Your friendships have deeply enriched this chapter of my life. I cannot possibly list all the friendships that I have gained through my time here, but you all know who you are. My friends from Texas, thank you for helping me remember my beginnings and that the things I do, those invisible to the CV, matters. Officer Sean Collier, I, and the MIT community, owe sincere gratitude to your service and efforts for your years here at MIT.
I would also like to thank friendship and remember Ms. Salee Aparece. Thank you x
10A6.
Lastly, but most importantly, my family has been my greatest support throughout my life and I wouldn't have been able to be here without them. Both of my parents, coming from humble beginnings, opened many doors without being traditionally demanding and impressively without knowing how. My sister has always been a motivation to be a role model and I am thrilled that our careers have surprisingly aligned. Lastly, Nathan Reticker-Flynn, has been the continuous source of scientific & emotional support, endless bouts of laughter, and has served as a role model citizen and scientist.
Financial support for this work came from the National Science Foundation Graduate Fellowship, Center for Cancer Nanotechnology Excellence, the Koch Institute, the National Institute of Health, the Prostate Cancer Foundation Grant, and the Harvard-MIT Division of Health Science & Technology. Special thank you to David Koch, who gave our lab resources and a world-renowned facility.
Abbreviations, Symbols & Definitions
APCs- Antigen Presenting Cells
BSA- Bovine serum albumin CTL- Cytotoxic T lymphocyte
DCs- Dendritic Cells
DLS- Dynamic light scattering FACS- Flow cytometry
HIV- human immunodeficiency virus
IFNg- Interferron gamma
LDL-c- low density lipoprotein cholesterol LDL-R -low density lipoprotein receptor
LPS- Lipopolysaccharide
MHC I/II- Major histocompatibility complex
NP- nanoparticle
o/w- oil-in-water OVA- ovalbumin
PCSK9- Proprotein convertase subtilisin/kexin type 9 PEG- poly(ethylene glycol)
PLA- poly(lactic acid)
PLGA- poly(lactic co-glycolic acid)
ssRNA- single stranded RNA
TEM- transmission electron microscopy Th- T helper
TLR -Toll Like Receptor
Abstract
Vaccines have revolutionized medicine by increasing the life expectancy of children and substantially decreasing the morbidity of multiple infectious diseases worldwide. Over several decades, we have acquired significant gains in the understanding of the underlying mechanisms involved in developing protective immunity, yet vaccine development has progressed comparatively slowly. This thesis serves to explore two polymeric nanoparticle platforms to demonstrate the therapeutic potential of synthetic nanocarriers as vaccines with the aim of 1) providing greater spatiotemporal release of
small molecule adjuvant to secondary lymphoid sites and 2) providing a tunable surface for loading B cell antigen epitopes in a specific conformation to drive epitope-specific antibody response.
In recent decades, TLR mechanisms have been elucidated and novel agonists have been developed, yet our generation still has not seen paramount progress in the clinical translation of these agonists due to risks of systemic toxicity and off target effects. In the first section, we synthesized 223±18 nm poly(lactic-co-glycolic acid)- poly(ethylene glycol)/ poly(lactic acid)-R848 (PLGA-PEG/PLA-R848) nanoparticle vaccine that is designed to deliver a combination of antigen and control release of a small molecule adjuvant R848 (tl/2= 42 hours) to drive a potent antigen-specific immune response. Using
ovalbumin as a model protein, this vaccine is able to enhance antigen presentation and co-stimulatory molecules on dendritic cells and subsequently enhanced proliferation of antigen-specific naive CD8+ cells in vitro. Upon vaccination, our delivery system is able to increase cell-mediated and humoral response in comparison to its soluble form, thereby illustrating the potential to bring novel small molecule adjuvants to the clinics.
In the second section, we developed a nanoparticle vaccine platform that allows selective orientation of peptide epitopes to enhance B cell response in an application that has therapeutic potential for treatment for cardiovascular disease (CVD). Utilizing epitopes discovered through in silico modeling for human PCSK9, a plasma protein that plays an important role in LDL cholesterol (LDL-c) levels in the blood, our nanoparticle allows selective orientation through biotin-streptavidin conjugation. Upon vaccination with CPG, selected synthetic epitopes conjugated to polymeric nanoparticles trended to reduce serum LDL-c and serum PCSK9 in murine models. Additionally, antibodies in the serum showed promise to increase LDL-receptor levels in HepG2 cells transfected in with WT-hPCSK9 and GOF-hPCSK9 separately suggesting that this vaccine has the potential to reduce risks of CVD.
These studies demonstrate that designing polymeric nanoparticles for applications to stimulate the immune system can help define new, cost-effective treatment options in applications for prophylaxis against infectious diseases that are unresponsive to traditional routes of vaccination or for immunotherapy against cardiovascular disease and cancer.
Thesis Supervisor:
Robert Langer, Sc.D.
Title: David H. Koch Institute Professor, Massachusetts Institute of Technology
Thesis Readers:
Omid Farokhzad, M.D.
Title: Associate Professor of Anesthesia, Brigham & Women's Hospital Ulrich von Andrian, M.D., Ph.D.
Title: Mallinckrodt Professor of Immunopathology, Harvard Medical School
Thesis Committee Chair:
Sangeeta Bhatia, M.D., Ph.D.
Title: John J. and Dorothy Wilson Professor of Health Sciences & Technology and
Chapter 1: Background
1.1 Introduction
1.1.1 Historical context of vaccines, burden of illness, and potential market
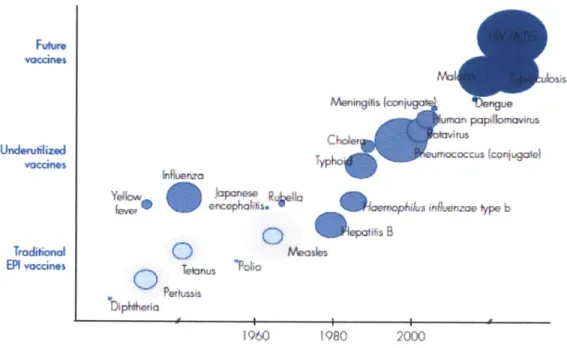
Vaccines have revolutionized the field of medicine and significantly decreased worldwide childhood mortaility rate from their first discovery with inoculation of cowpox to small pox in 1770 to Louis Pasteur's rabies vaccine, Jonas Salk's polio vaccine in 1950s, and Maurice Hilleman's influenza vaccines. The World Health Organization estimates that vaccination, both active and passive, assists in the prevention of three million deaths in children annually and protects another quarter-million from permanent disability [1]. Even with the significant impact that vaccines have had on human health, vaccine development has been slow to there are several infectious diseases that have been proven elusive to protection by vaccines, such as malaria and HIV, as well as chronic diseases with an underlying immunological basis, i.e. cancer and autoimmune disease. Advancements in the understanding of novel biological mechanisms and development of targeted nanoparticle delivery carriers capitalizing on these findings can help introduce more efficient vaccines for prophylaxis and treatment [2].
According to the CDC, malaria had caused 216 million episodes and 655,000 deaths and direct cost of 12 billion. Approximately one third of the world's population is infected with tuberculosis. In 2011, nearly 9 million people reported infected with tuberculosis. Even though society has curbed the overall morbidity and eradicated several infectious diseases, there are still voids in available prophylaxis and compliance for patients in many developing countries, which can be addressed with utilizing relatively recently discovered immunological mechanisms to advance vaccine development.
Fuhtr.
Vaccines
Meningitis coniuga U
1--n papilnomavirus C ha
UndemuizJ . Woccus (conitgale)
Yelkw Po*w Rte t
a encephoi- mps hinfvenzoe type b
oqePa
ii BTraditional
QMeasle
EPI vaccines
o
enu *~O~U Polio1960 1980 2000
Figure 1.1 Incentives for Vaccine Research: Strategic Plan 2010-2020. World Health Organization.
1.1.2 Definition of Vaccine
A vaccine is defined as a biological preparation that can improve immunity to a
particular disease. They can be prophylactic, to curb the risk of future infection by a pathogen of interest, or therapeutic, such as in the case of cancer vaccines. The term vaccine stems from the usage of cow pox in 1796 by Edward Jenner to inoculate patients in order to prevent smallpox infection, Latin vaccina adapted from Latin term vaccin-us, from vacca cow. With the exception of water sanitation, the utilization of vaccines in medicine has been heralded as the key turning point in medicine and has reduced mortality and increased population growth.
Vaccines typically are composed of the antigen of interest combined with an immunologic adjuvant to stimulate and increase the immune responses to the vaccine
without having a specific antigenic effect. The addition of adjuvant allows a reduction of the amount of antigen needed for memory and allows for the reduction in repeat vaccinations. The word adjuvant derives from the Latin word adjuvare, meaning to help or aid. An immunologic adjuvant is defined as any substance that acts to accelerate, prolong, or enhance an antigen-specific immune response when utilized with specific targeted antigens.
1.1.3 Current Approach for Vaccine Delivery & Limitations:
Some commercially available vaccines utilize purified components of pathogen lysates, such as surface carbohydrates or recombinant pathogen-derived proteins that are sometimes fused to other molecules, particularly proteins that can confer adjuvant activity. Experimental vaccines undergoing clinical trials and development include: dendritic cell (DC) vaccines, recombinant vectors, DNA vaccines, T-cell receptor peptides, and synthetic vaccines.
Another method of vaccination, largely at the clinical trial stage, is the injection of antigen-carrying cells, such as DCs or other leukocytes or modified patient-derived tumor cells [3]. Numerous clinical trials using such approaches have been published or are currently ongoing, particularly for immunotherapy against a variety of cancers due to the genetic and phenotypic heterogeneity [4]. To date, most clinical results with DC-based vaccines have shown only marginal therapeutic benefits. Moreover, DC-based vaccines are typically aimed at eliciting cell-based (cytotoxic) immunity and may be less suitable for the induction of high titers of neutralizing antibodies [5].
Vaccine approaches currently used in the clinics are separated into these types: attenuated, killed, toxoid, subunit, conjugate. Vaccines that utilize live attenuated or
inactivated pathogens typically yield a vigorous immune response, but their use has limitations. For example, live vaccine strains can sometimes cause infectious pathologies, especially when administered to immune-compromised recipients. Moreover, many pathogens, particularly viruses, undergo continuous rapid mutations in their genome, which allow them to escape immune responses to anti-genically distinct vaccine strains
[6]. However, most or all pathogens are thought to possess certain antigenic determinants
that are not easily mutated because they are associated with essential functions. Antibodies directed against these conserved epitopes, rather than more variable, non-essential epitopes can protect against highly mutable viruses, such as HIV-1 [7]. Killed vaccines utilize micro-organisms that have been inactivated via chemicals, heat, or radiation. Vaccines based on live or killed intact pathogens do not necessarily promote the recognition of these critical epitopes, but may essentially "distract" the immune system to focus its assault on highly variable determinants. In theory, a synthetically engineered vaccine that mimics the highly immunogenic particulate nature of viral particles, but presents selectively essential, immutable epitopes could yield more potent neutralizing antibody and effector T responses than intact micro-organisms.
There are numerous factors that may affect vaccine efficacy between patients such as:
1) disease, 2) strain, 3) vaccination schedule, 4) individual non responder, 5) ethnicity/ age/ genetics of the patient.
1.2 Fundamental Principals of the Immune Response
To understand the basics of the cells mentioned in this thesis, a brief overview of basic immunological principals serves as a foundation. Higher order organisms possess
multiple layers of defense against foreign pathogen invasion including surface barriers (physical, chemical, and biological), the innate and adaptive immune systems.
1.2.1 Innate Immune Response
Innate response is considered as the first order of defense from pathogens and is the
dominant system of host defense for most organisms. This response characterized as non-specific and does not render long-lasting immunity against a particular pathogen. The response is usually triggered when microbes are recognized by pattern recognition receptors and commonly involves inflammation for leukocyte recruitment as well as the complement system activation. In regards to cellular barriers of the innate immune response, innate leukocytes include phagocytes (macrophages, neutrophils, dendritic cells), mast cells, eosinophils, basophils, and natural killer cells. Phagocytes engulf pathogens to clear invading pathogens and activate the adaptive immune response. Neutrophils are the most abundant type and migrate via chemotaxis to the site of infection. Macrophages act as scavengers for worn cells and antigen presenting cells. Dendritic cells (DC) are notably in tissues that remain in contact with the external envoironment such as the skin, nose, lungs, stomach, and intestines and are rightfully termed as "professional antigen-presenting" cells. Mast cells, located in connective tissues and mucous membranes, assists in regulating the inflammatory response and is commonly associated with allergy and anaphylaxis. Natural killer (NK) cells destroy host cells that have been compromised such as tumor cells and virus infecting cells by identification of cells with low levels of cell-surface marker MHCI, which can be a result of viral infections. Normal host cells typically have enough MHCI to be recognized by killer cell immunoglobulin receptors (KIR) that turns off the response of NK cells.
1.2.2 Adaptive Immune Response
The adaptive immune response evolved in higher order vertabrates to allow for a
stronger immunoprotective response and immunological memory against a specific antigen in a process involving recognition of specific non-self antigens called antigen presentation. The cells of the adaptive immune system of special types of leukocytes, named lymphoctyes. The major group of lymphocytes are B cells and T cells, derived from hematopoietic stem cells in the bone marrow. B cells are involved in the humoral immune response and the generation of antibodies. T cells are associated with the cell-mediated response. Both B and T cells carry receptor molecules that can recognize specific targets, T cells recognizing a "non-self" target.
B cells and T cells are the major types of lymphocytes and can recognize specific targets. T cells recognize pathogens after small fragments have been processed and presented on MHC molecules located on antigen presenting cells, the cells that process the antigen.
There are two major subtypes, CD8+ killer T cells, and CD4+ helper T cells. CD8+ cells recognized antigen peptides, typically 8 amino acids long, coupled to MHCI and CD4+ cells recognize those that are coupled to MHCII, typically peptide chains ranging from 10-12. A third subtype are y8 T cells which can recognize intact antigens that are not bound on MHC receptors. CD8+ killer T cells are a subtype of T cells that targets cells infected with viruses for elimination. These cells are activated by recognition of a MHC:antigen complex combined with the co-receptor CD8. Activated CD8+ cells release cytotoxins such as perforin and granulysin both leading towards apoptosis. Helper T cells indirectly control the innate and adaptive immune response by directing other cells to kill or clear pathogens. CD4+ cells express T cell receptors that recognize antigen
bound to MHCII along with the CD4+ coreceptor. Although CD8+ cells can be activated
by one MHC-peptide complex, CD4+ T cell activation is weaker and needs 200-300
receptors bound by MHC antigen. Cytokine signals from CD4+ T cell activation can enhance microbicidal function of macrophages, killing function of CD8+ cells, and upregulation of CD40L (CD154) which provides the extra-stimulatory signals required to activate antibody-producing B cells.
B cells are the cell types involved with humoral protection. They have antibodies on the surface that bind to a specific foreign antigen. This antigen/antibody complex is taken up by the B cell and processed and subsequently presented on MHCII molecules for assistance in activation by a matching helper T cell which releases lymphokines (IL-2,
IL-3, IL-4, IL-5, IL-6, GMCSF, and IFNg) that activates the B cell to divide into plasma
cells which secretes several antigen-specific antibodies. The antibodies circulate in the blood/ lymph to bind specifically to pathogens expressing the antigen and mark them for destruction by complement activation. Antibodies can neutralize these challenges through binding to bacterial toxins and/or interfere with the pathogen surface molecule used to help infect cells. Typically antibodies interact with only a small region of the antigen, for a peptide typically 5-12 amino acids.
It is known that B and T cells are initially localized in distinct anatomic regions, the superficially located B follicles and the surrounding paracortex and deep cortex. Upon challenge, antigen-specific B cells in follicles as well as CD4 T cells in the T cell area become activated and then migrate toward the border zone between the two compartments. B cells that have phagocytosed lymph-borne antigens process the acquired material and begin to present antigenic peptides in MHC class-II surface molecules that
are then recognized by the activated CD4+ T cells (the TFH cells). Antigen-recognition allows the TFH cells to provide help to B cells, which constitutes a potent survival signal and triggers the formation of germinal centers (GCs) within B follicles. The GC reaction promotes class-switch recombination, affinity maturation of antigen-specific antibodies and the formation of memory B cells and long-lived plasma cells that can produce large amounts of high-affinity antibodies for extended periods of time. Therefore, an ideal vaccine must have several key components that allow antigenic material to be efficiently recognized by both B and T cells and induce vigorous GC reactions.
1.2.3 Cytokines are crucial mediators of immune response
Cytokines are small signaling molecules released by cells and are typically immunomodulatory in nature. The table below summarizes molecules of interest used in our studies.
Table 1. Cytokines and their function
Cytokines Function Producing Cell Recipient Cell Clinical Significance IL-2 growth, proliferation, and differentiation activated T cells CD8+, CD4+, combinatorial therapy of cells towards effector T cells (Treg) for cancers and as
adjuvants, HIV IL-4 ThO-> Th2, B cell and T cell unknown (perhaps T cell, B cell wound repair/ fibrosis
proliferation, differentiation of B cells basophil) in M2 macrophages into plasma cells; decreases the
production of Thi cells, Mp, Ifng and DC IL-12
IL-5 activates eosinophils, stimulates B cell Th2 and mast cells B cell, allergies, allergic
growth and Ig secretion eosinophils rhinitis, asthma
IL-6 production of neutrophils, mediators of T cells, macrophages, cells with iL-6Ra depression, diabetes, the acute phase response (temperature) muscle, osteoblasts, and gp130, B atherosclerosis,
inhibitory effects on TNFa, IL-1, IL- SMCs, adipocytes cells Alzheimer's,
Ira, IL-10 rheumatoid arthritis,
SLE, metastatic cancer
IL-12 Thi differentionation, stimulates Ifg and dendritic cell, Mp NK cells, T cells Autoimmune disease TNFa from T and NK cells, block
formation of new blood vessels
IL-17a production of many cytokines, chemokines, and prostaglandins
IFNg
TNFa
activator of Mp
inflammation & acute phas reaction
Th cells (induced by IL-23) NK and NKT cells, Thl, and activated CD8 cells Mp, CD4+, NK cells, neurons, endothelial cells, myocytes, adipocytes, fibroblasts fibroblasts, endothelial cells, epithelial cells, keratinocytes, \ MP neutrophils, endothelial cells macrophages -phagocytosis epitelial cells,
IL-15 Suppresion may
improve celiac disease allergies and autoimmune disease (rhematoid arthritis, asthma, lupus, graft rejection, psoriasis) autoimmune disease
IBD, Alzheimer's, depression.
proliferation, differentiation, antiproliferative for epithelial cells
cancer cells, Mp cancer, heart disease, marfan's, loeys-dietz
1.2.4 Immunological differences between mice and humans
Although murine models have pervasive use to study immune reactions, there are some noted differences between mouse and human that play a role in clinical translation of immunotherapies. Human blood have a neutrophil:lymphocyte composition of 50-70% neutrophils: 30-50% lymphocytes versus mice blood composition of 10-25% neutrophils to 75-90% lymphocytes [8].
Table 2. Comparison of Mouse and Human Immunology. Adapted from [9]
Mouse Human
Neutroyhts in blood 10-25% 50-70%
Lymphocytes in blood 75-90% 30-50%
TLR2 expression on PBL TLR3
TLR9
TLR1O
Sialic acid Neu5GC expression
CD4 on macrophages
Predominant T cells in skin and mucosa
y/8 T cells respond to phospho-antigens CD1 genes FcaRI FcyRUA, C Serum IgA Ig classes Ig CDR-H3 region
IFN-a promotes Thi
differentiation
Th expression of IL-10
1,4 and IFN-y expression by cultured Th CD28 expression on T cells B7-H3 effects on T cells P-selectin promoter MHC HI expression on T cells MUC1 on T cells Granulysin CXCR1
IL-8, NAP4, ITAC, MCP4,
HCC-1, HCC-2 MPIF-HCC-1, PARC, eotaxin-2/3
DTH lesions
Constitutive MHC I on EC
EC present Ag to CD4+ T
CD40 on EC
Low (induced on many cells including T cells)
Expressed on DC, Mac. Induced by LPS Expressed on all myeloid cells, plasmacytoid DC and B cells
Pseudogene Widespread Absent
y/8 TCR (dendritic epidermal T cells-DETC) No CDld Absent Absent Mostly polymeric
IgA, IgD, IgE, IgGI, IgG2a*, IgG2b, IgG3, IgM * absent in C57BL/6, /10, SJL and NOD mice, which have IgG2c Shorter, less diverse
No Th2 Either/or On 100% of CD4+ and CD8+ Inhibits activation Activated by TNF and LPS Absent Absent Absent Absent Absent Neutrophil-rich Absent No Absent
Constitutive (but not on T cells)
Expressed by DC. No LPS induction
Expressed only on B cells, plasmacytoid DC and N Widely expressed Absent Present c/f TCR Yes CDIabc,d Present Present Mostly monomeric IgAl, IgA2, IgD, IgE, IgGI, IgG2, IgG3, IgG4, IgM
Longer, more diverse Yes ThI and Th2 Sometimes both On 80% of CD4+, 50% of CD8+ Promotes activation Unresponsive to inflammation Present Present Present Present Present Lymphocyte-rich Present Yes Present
1.3 Toll-like receptor complexes, natural ligands, and synthetic
adjuvants
1.3.1 Definition of Toll-like receptors
Toll like receptors (TLRs) are a class of proteins that play an important role in the innate immune and digestive system. TLRs are defined as a single, membrane-spaning, non-catalytic pattern recognition receptor, usually expressed in sentinel cells such as macrophages and dendritic cells, that recognize TLR have been the important link between the innate and adaptive immune responses via dendritic cells. TLR 3 and 4, present on the surface of monocyte derived dendritic cells, utilize the MyD88 -dependent pathway to produce IL-12, IL-18 for the maturation into type 1 helper T cells and the TRIF pathway to simultaneously up-regulate co-stimulatory molecules for T cell differentiation[ 10].
The activation of specific pattern recognition receptors, Toll-like receptors (TLR), recognize conserved structures with very diverse pathogens such as dsRNA (TLR3),
lipopolysachharide of bacterial cell walls (TLR4), and flagella (TLR5). TLR 7-9
comprises a closely related genetic sub-family whose expression is species dependent, cell type specific, is functionally compartmentalized to the endosome. TLR9 recognizes
CpG in unmethylated bacterial or viral DNA and synthetic CpG oligonucleotides [11].
Recent studies utilizing Toll-like receptor ligands have shown that antigens associated with these ligands can produce exceptionally high antibody titers and rapid immune
responses [12-14].
1.3.2 Necessity for Adjuvants in Vaccines:
Resting dendritic cells typically reside in many different tissue types, including lymph nodes in an immature tolerogenic state. These immature DCs present intermediate to
high-levels of peptide-MHC complexes, but without cytokines or costimulatory molecules that help differentiate T cells into effectors. T cells presented a specific antigen
by immature DCs will begin to proliferate but then die by apoptosis or become
unresponsive to additional activation. Comparatively when DCs acquire antigens when exposed to maturation stimuli, these cells can up-regulate MHC and costimulatory molecules and secrete cytokines. This maturation signal can be triggered via adjuvants. Vaccines used for intramuscular injections are typically administered with an adjuvant carrier, most frequently alum (aluminium potassium sulphate), that is thought to establish a depot for prolonged release of antigenic material, but also exerts immunomodulatory activities, such as skewing toward Th2 responses by mechanisms that are incompletely understood since immunization activates a complex cascade of responses [15, 16]. Recent efforts are focused on utilizing DNA and subunit/ conjugate vaccines where a weak antigen is linked to a stronger immunogens [17, 18]. Most cases, the antigen itself is only very weakly immunogenic, therefore an adjuvant is needed to create a more intense immune response [17]. Adjuvants are typically added to vaccine formulations to enhance the host memory response against a particular antigen [18]. Specific adjuvants can cater the type of immune response generated and therefore one can potentially design the immune response due to selecting the specific combination of immunomodulating materials to be delivered.
There are several companies that have partnered with large pharmaceuticals to advance their discovery in small molecule agonists against toll like receptors such as Idera with Merck utilizing TLR agonists targeted to TLR7, 8, 9. Approvals have been made in Europ for MF59, an adjuvant for flue vaccine in elderly (Fluad, Novartis), and
ASO4, a combination of alum and MPL (GSK) as an adjuvant for viral vaccines such as hepatitis B and HPV.
Adjuvants that are currently in development and use are: Mineral salts - e.g., alumuninium hydroxide ("alum"), aluminium phosphate, calcium phosphate; Oil emulsions - MF59, a detergent-stabilized oil-in-water emulsion; Particulate adjuvants
-virosomes, ISCOMS (structured complex of saponins and lipids); Microbial derivatives -MPLTM) (monophosphoryl lipid A), CpG motifs, modified toxins; Plant derivatives
-saponins (QS-21); Endogenous immunostimulatory adjuvants -cytokines.
1.3.3 Synergy of TLR for vaccine development
As foreign pathogens, ie bacteria and viruses, have various components that would activate TLRs simultaneously, there have been many studies on optimal TLR agonist combinations in order to drive synergistic cell mediated and humoral protection. The synergistic stimulation of TLR 2/6 with TLR 9 enabled for improved protection against influenza in mice [19]. Investigations on TLR7 (R837) combined with TLR 4 (MPL) within one PLGA particle and co-injected with PLGA encapsulated HA protein demonstrated synergistic responses in H5N1 influenza and showed significance when used synergistically compared to individual TLR.
1.4 Polymeric Nanoparticles for Synthetic Vaccines
1.4.1 Potential Advantages of Polymeric Nanoparticles as a Vaccine Carrier:
Polymeric nanoparticles have been widely explored and translated for a variety of medical applications for protein and small molecule delivery including vaccine development [5]. A synthetic nanocarrier-based vaccine delivery system provides a
as a means for tolerization [20, 21]. In design considerations, polymeric nanoparticles have the advantage of being able to directly control the physicochemical properties: size, shape, surface charge, hydrophobicity, and release properties. The drug delivery carrier can accommodate more than one protein simultaneously, i.e. a mixture of purified antigenic proteins from an infectious pathogen or a heterogeneous tumor antigens lysate purified from a patient's malignancy [22] where the precise sequence and composition of these proteins is not known. Inclusion of agents that induce DC activation, such as cytokines or ligands for CD40 or TLRs serves as a means to administer potent adjuvants
[23].
The use of polymeric nanoparticles for vaccine delivery has been investigated due to the same properties discovered in their design for drug delivery: biodegradability, higher surface area for adsorption, controlled and enhanced immunogenicity due to size [13, 21, 24-26]. Tetnus toxin and CpG (adjuvant for TLR 9) were co-encapsulated in PLGA nanoparticles (300 nm) which resulted in the induction of an enhanced antigen-specific T-cell proliferative response and very strong serum IgG in comparison with soluable antigen with CpG [27-30]. These studies also support the use of polymeric nanoparticles as adjuvants themselves: in vitro release studies showed that antigen release from antigen-adsorbed nanoparticles arrived at the lymph node site with more rapid kinetics than antigen-encapsulated nanoparticles. Additionally, higher antibody response was observed with antigen-encapsulated than free antigen and adsorbed antigen- nanoparticles [21]. Size is an important factor affecting the immunogenicity of nanoparticles, as discovered particles below 100 nm and greater than lum were less efficient than nanoparticles within that range [25, 26]. Modification of the surface properties of these
particles also indicated preferential targeting of immune cells as positively charged poly-L-lysine particles showed higher phagocytosis in DCs as compared with negatively charged particles [30].
The concept of using polymeric controlled-release technology for vaccine improvement started in 1979 with the encapsulated with bovine serum albumin in ethylene vinyl acetate. Gupta et al. indicated that a controlled released microparticle platform for tetanus toxoid indicated the beginning of controlled release of antigen via polymeric particle systems for vaccine development thereby offering the combination of spatial and temporal control of these systems [31-33]. The advantages of a particulate system is that one can directly package and direct antigens to APCs, co-encapsulation of multiple antigenic epitopes, and packaging both antigen and adjuvant into one carrier [34].
Chapter 2: Development of a control
releasing R848 nanoparticle vaccine
platform
2.1 Introduction
Development of novel effective targeted vaccines enable localization to crucial regions of the immune system and provide a prolonged boost has the potential to elicit rapid and effective protection, increase patient compliance, and reduce costs for viral and parasitic infections, such as hepatitis, HIV, malaria, cancer, etc [20]. A new method for selective and versatile targeting of antigenic material to antigen-presenting cells (APCs) in lymphoid tissues is desired to increase efficacy of the vaccine. The approach involves a targeting moiety conjugated to a carrier, such as nanoparticles, that will be loaded with one or more molecules that are recognized as antigens by B cells or T cells, or both. These adjuvants boost immune responses by activating APCs to enhance their immunostimulatory capacity, by amplifying lymphocyte responses to specific antigens and by inducing the local release of mediators, such as cytokines from a variety of cell types.
We hypothesize that engineering vaccines to emulate viruses and specifically target important immune cells in combination will provide enhanced protection against viral infection. Development of novel vaccines requires amplification of antigen specific signal through various branches of the immune system. Effective adaptive immune response requires recruitment of T cell response by activating APCs and successfully
priming B cells. In designing this system, polymeric nanoparticles are attractive vehicles for vaccine development due to key characteristics of size, controlled release and stability. Prior literature has shown that cells have a maximal nanoparticle uptake, therefore packaging an antigen and adjuvant within a single particle mimicking a viral particle will induce an immune response [35]. We have engineered and synthesized a nanoparticle delivery system that can efficiently prime B cells and produce potent T cell response through a single packaged dose with immunomodulatory components within and on the surface of the particle. We have shown that these next generation vaccines are able to induce a functional cell mediated response in vitro and in vivo, which can be utilized to drive an immune response against any antigen of interest.
0 N N
Loxordbine CL264 Imiquimod Gardiquimod CL097 0 -10 ug/mL
300 ug/mL 50ng-10 ug/mL 1-5 ug/mL 1-10 ug/mL 0.5-5 ug/mL
Figure 2.1 Selected TLR 7 agonists in order of immunostimulatory potency
TLR7 is predominantly expressed in lung, placenta, and spleen, while TLR8 is expressed in lung and peripheral blood leukocytes. TLR7 and TLR8 agonists have different select target cells and subsequent cytokine profiles. TLR7 agonists activate plasmacytoid DCs (pDCs) and B cells to induce IFN regulated cytokines. Contrastingly, TLR8 agonists activate myeloid DCs, monocytes, and monocyte derived DCs leading towards pro-inflammatory cytokines and chemokines: TNF-a, IL-12, MIP-la [36]. While
mouse TLR8 is non responsive to agonists R848 and R837 (imiquimod), human TLR8 is recognizes only R848 but not R837. Literature has shown that guanosine analogs activate through TLR7 and not TLR8. Natural ligands of TLR7 and 8 have been shown to be single stranded RNA (ssRNA) particularly ssRNAs with poly(U) or GU-rich sequences. Yet, certain siRNA motifs can stimulate with absence of GU content, which suggests existence of other specific sequences [37]. Microbial RNA has low nucleoside modification and has been shown to activate DCs through TLR 3, 7, 8 in comparison to mammalian RNA, which have a higher amount of modified nucleosides [38]. Due to its high potency and short half-life in circulation, R848 has not been actively clinically translated [39-41].
2.2
Rationale
In recent years, TLR mechanisms have been elucidated and novel agonists have been developed, yet our generation still has not seen paramount progress in the clinical translation of these agonists due to risks of systemic toxicity and off target effects when injected in soluble form. We hypothesize that engineering polymeric vaccines to emulate a virus particle with a payload of controlled releasing adjuvant in combination with spatially arranged surface antigen will provide enhanced protection against viral infection and has the potential to serve as an advancement in VLP design. Borrowing concepts from targeted nanoparticles for chemotherapeutic delivery [42], we have engineered nanoparticles to deliver a payload of potent R848 adjuvant in combination with antigen to secondary lymphoid organs of interest to increase immunogenicity of the antigen, increase the therapeutic window of R848, and bypass toxic systemic effects. In designing this system, polymeric PLA-PEG nanoparticles are attractive vehicles for vaccine
development due to key characteristics of size, biodegradability, biocompatibility, ability of controlled release, and stability. Conjugation of R848 to the polymer allows for high reproducibility, controlled and local release into the endosome where TLR7/8 receptors are located, as well as the potential to load higher levels of adjuvant than when attempted to encapsulate in soluble form. Additionally, combination delivery of antigen and adjuvant provides the potential of enhanced APC response[43]. As B cells are better activated via polyvalent antigens presented via a fixed surface, as in a virus-like particle, over soluble form. Thereby vaccine carriers that are able to mimic viral particles by presenting polyvalent conformationally intact antigens on their surface are predicted to stimulate a similarly strong B cell response. The T cell antigen can be derived from the same pathogen against which vaccination is intended. Additionally, the antigen may be taken from an unrelated source, such as an infectious agent to which wide-spread immunity already exists (e.g. tetanus toxoid or a common component of influenza virus, such as hemagglutinin). This method utilizes the presence of memory T cells that have arisen in response to prior infections or vaccinations. These memory cells in general react more rapidly and vigorously to antigen re-challenge and therefore may provide a source of help to B cells.
B cell Antigen Antigen Presenting CD4+
Figure 2.1 Schematic of nanoparticle immune system activation.
2.3 Methods
2.3.1 Materials:
Poly(ethylene glycol)-malemide (Mal-PEG-NH2-3500) was purchased from JenKem
(A5006), poly(DL-lactide-co-glycolide) was purchased from Boehringer Ingelheim (i.v.
0.45), D-L lactide (Purasorb DL) purchased from Purac, C'4 labeled PLGA was custom
made from Moravek Biochemicals. R848, (Invivogen). Imject Alum (77161), N-hydroxysuccinimide (24500), 1 -Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (77149) was purchased from Thermo Scientific/ Pierce. Benzyl chloroformate, Polyvinol Alcohol, and all organic solvents (Sigma Aldrich). DQ-Ovalbumin, Cell Trace CFSE Cell Proliferation Kit, AlexaFluor647 (Life Technologies
D12053, C34554, A30679). MACs kits for mouse CD1 lc+, CD8a+ T cell isolation kit II,
CD4+ T cell isolation kit II (Miltenyi 130-052-001, 130-095-236, 130-095-248).
2.3.2 Instrumentation:
HPLC experiment was conducted using reverse-phase C18 column (Supelco) with the mobile phase velocity of lmL/min with the detection wavelength at 320 nm. The mobile phase consists of 0.1% TFA/water and 0.1 %TFA/ACN/water (60:40). Injection volume is
100uL. 1H & "C NMR was performed on a Bruker Avance-600 spectrometer. TEM was
performed using JEOL JEM200CX operated at a voltage of 200 kV. For sample prep, a droplet at 1 mg/mL was placed onto a carbon-copper TEM grid combined with 3% uranyl acetate for negative staining. Particle size (diameter, nm) and surface charge (zeta potential) measurements were made using a Malvern Nano ZS with ZetaSizer Software