HAL Id: hal-01627471
https://hal.archives-ouvertes.fr/hal-01627471
Submitted on 1 Nov 2017
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
mRNA untranslated regions
Abdel Aouacheria, Vincent Navratil, Ricardo López-Pérez, Norma Gutiérrez,
Alexander Churkin, Danny Barash, Dominique Mouchiroud, Christian Gautier
To cite this version:
Abdel Aouacheria, Vincent Navratil, Ricardo López-Pérez, Norma Gutiérrez, Alexander Churkin, et
al.. In silico whole-genome screening for cancer-related single-nucleotide polymorphisms located in
human mRNA untranslated regions. BMC Genomics, BioMed Central, 2007, 8 (1),
�10.1186/1471-2164-8-2�. �hal-01627471�
Open Access
Research article
In silico whole-genome screening for cancer-related
single-nucleotide polymorphisms located in human mRNA
untranslated regions
Abdel Aouacheria*
†1,4, Vincent Navratil
†1, Ricardo López-Pérez
2,
Norma C Gutiérrez
2, Alexander Churkin
3, Danny Barash
3,
Dominique Mouchiroud
1and Christian Gautier
1Address: 1Laboratory of Biometry and Evolutionary Biology, CNRS UMR 5558, Claude Bernard University Lyon 1, 69622 Villeurbanne, France, 2Servicio de Hematología, Hospital Universitario de Salamanca, Centro de Investigación del Cáncer, Universidad de Salamanca-CISC, Spain, 3Department of Computer Science, Ben-Gurion University, 84105 Beer Sheva, Israel and 4Apoptosis and Oncogenesis Laboratory, Institute of
Biology and Chemistry of Proteins, IBCP UMR 5086 CNRS-UCBL, IFR 128 Biosciences Lyon-Gerland; 7 passage du vercors, 69367 Lyon Cedex 07, France
Email: Abdel Aouacheria* - a.aouacheria@ibcp.fr; Vincent Navratil - navratil@biomserv.univ-lyon1.fr; Ricardo López-Pérez - riclopez@cbm.uam.es; Norma C Gutiérrez - normagu@usal.es; Alexander Churkin - chukin@cs.bgu.ac.il;
Danny Barash - dbarash@cs.bgu.ac.il; Dominique Mouchiroud - mouchi@biomserv.univ-lyon1.fr; Christian Gautier - cgautier@biomserv.univ-lyon1.fr
* Corresponding author †Equal contributors
Abstract
Background: A promising application of the huge amounts of genetic data currently available lies in developing
a better understanding of complex diseases, such as cancer. Analysis of publicly available databases can help identify potential candidates for genes or mutations specifically related to the cancer phenotype. In spite of their huge potential to affect gene function, no systematic attention has been paid so far to the changes that occur in untranslated regions of mRNA.
Results: In this study, we used Expressed Sequence Tag (EST) databases as a source for cancer-related sequence
polymorphism discovery at the whole-genome level. Using a novel computational procedure, we focused on the identification of untranslated region (UTR)-localized non-coding Single Nucleotide Polymorphisms (UTR-SNPs) significantly associated with the tumoral state. To explore possible relationships between genetic mutation and phenotypic variation, bioinformatic tools were used to predict the potential impact of cancer-associated UTR-SNPs on mRNA secondary structure and UTR regulatory elements. We provide a comprehensive and unbiased description of cancer-associated UTR-SNPs that may be useful to define genotypic markers or to propose polymorphisms that can act to alter gene expression levels. Our results suggest that a fraction of cancer-associated UTR-SNPs may have functional consequences on mRNA stability and/or expression.
Conclusion: We have undertaken a comprehensive effort to identify cancer-associated polymorphisms in
untranslated regions of mRNA and to characterize putative functional UTR-SNPs. Alteration of translational control can change the expression of genes in tumor cells, causing an increase or decrease in the concentration of specific proteins. Through the description of testable candidates and the experimental validation of a number of UTR-SNPs discovered on the secreted protein acidic and rich in cysteine (SPARC) gene, this report illustrates the utility of a cross-talk between in silico transcriptomics and cancer genetics.
Published: 03 January 2007
BMC Genomics 2007, 8:2 doi:10.1186/1471-2164-8-2
Received: 14 July 2006 Accepted: 03 January 2007 This article is available from: http://www.biomedcentral.com/1471-2164/8/2
© 2007 Aouacheria et al; licensee BioMed Central Ltd.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Background
Genetic variations contribute to the development and maintenance of complex disorders, such as cancer, through alterations in the structure and/or abundance of individual mRNA molecules. The human transcriptome could therefore be considered as a priority target in the fight against cancer. Transcript sequences represent a key source for the search of aberrantly expressed genes and for the identification of genes whose products are deregulated in malignant cells. Among these transcript sequences, Expressed Sequence Tags (ESTs) are partial single-pass sequences of cDNAs made of mRNA from a particular organ, tissue or cell line. Since cDNA libraries are gener-ated from a wide range of cancerous and normal tissues, ESTs can be used both for measuring relative levels of gene expression [1-6], and for detecting single nucleotide dif-ferences among sequences derived from a same gene [7,8]. It is now widely assumed that human genomic DNA con-tains some level of polymorphism, with single nucleotide polymorphisms (SNPs) being the most common form. Owing to large-scale discovery, SNPs constitute an emerg-ing resource for the study of genetically complex disorders such as cancer [9,10]. SNPs localized within the coding regions of genes could modify the amino acid sequence of the encoded products through non-synonymous substitu-tions that, in turn, may impact protein structure and func-tion [7,11-13]. SNPs present in the untranslated regions of genes (UTR-SNPs) may rather have effects on gene expression by affecting regulatory elements or mRNA sta-bility [14-19]. Yet, biochemical evidences as to how UTR-SNPs located in untranslated portions of mRNAs affect gene function are still scarce. Possible mechanisms for 5'-UTR include mRNA splicing interference, regulation of transcription (e.g., through methylation), translation (e.g., through internal ribosomal entry fragments), or mRNA stability [20,21]. The role of the 3'-UTR of mRNA is seen to be as important as that of the 5'-UTR in regulat-ing gene expression. Indeed, in addition to the well-estab-lished role of the poly-(A) tail, which confers protection to the RNA molecule from degradation by exonucleases, resulting in enhancement of translation, there are a number of motif sequences within the 3'-UTR that regu-late mRNA stability and translational efficiency, including the recently identified microRNA-binding sites [22,23]. In this study, we attempted to use a computational proce-dure to identify novel cancer markers, or polymorphisms that could influence gene expression levels in cancer cells. We decided to focus on UTR-located non-coding poly-morphisms because (i) 5'- and 3'-UTR sequences are known to influence cellular steady-state levels of mRNA; (ii) polymorphisms in these sequences are accessible using EST data; (iii) potential association between UTR-SNPs and cancer phenotype is readily assessable using
library features. We first detected human genetic variants located in UTR regions and associated with cancer, i.e., UTR-SNPs that are statistically over-represented in ESTs derived from cancerous libraries. We then used predictive methods to test the potential effects of the detected poly-morphisms on mRNA folding and putative UTR func-tional elements. This report is a first attempt to use human EST databases as a source for the discovery of cancer-asso-ciated untranslated region polymorphisms at the whole-genome level. Our digital approach was combined to standard laboratory genotyping experiments to propose a set of validated variants in the secreted protein acidic and rich in cysteine (SPARC) gene, a key factor in cell-matrix interactions and possibly tumour aggressiveness [24-27].
Results
We developed an EST-based pipeline to detect cancer-associated UTR-SNPs. Details about the data mining pro-cedures are presented in Table 1.
Pre-selection of candidates for cancer association studies
We first identified genetic variants present in untranslated regions (UTR-SNPs) of human genes using EST sequences from different libraries. Among those, we detected genetic variants associated with cancer (i.e., those that are statisti-cally over-abundant in EST libraries derived from cancer-ous cells). Our predictions relied on the digital count of ESTs rather than libraries because of the frequent lack of precision concerning the origin of the source tissue(s) (both normal and tumoral) and for statistical analysis. Despite several other limitations inherent to the EST methodology (e.g. biased or limited sampling, see discus-sion), this whole-genome scanning strategy had the advantage of being a completely hypothesis-free approach that allowed the ab initio detection of cancer-associated UTR-SNPs present on EST sequences. EST-searches led to the identification of a total of 358 UTR-SNPs (on 269 transcripts) that were present at significantly (p < 0.01) higher allele frequencies in tumour compared to normal tissues, out of which 47 were in 5'-UTR and 311 in 3'-UTR. Some aspects, if not all, of this discrepancy could be explained by the fact that sequencing protocols generate more ESTs matching with the 3' end of genes. Our list of UTR-SNPs that potentially contribute to the cancer
phe-Table 1: Overview of the EST-based data mining strategy.
Total SNPs (after algorithm filtering) ~51,760 Total UTR-SNPs 20,304 UTR-SNPs with p < 0.05 1354 UTR-SNPs with p < 0.01 358 Total SNPs in 5'-UTR 47 Total SNPs in 3'-UTR 311 Identification of tumor-associated polymorphisms located in human mRNA untranslated regions. UTR-located SNPs are referred to as UTR-SNPs. SNP counts in each analytical step.
notype is summarized in Table 2 (see Additional file 1 for the complete set). With respect to the delineation of UTR-SNPs from EST data, we estimated how large the fraction of bona fide SNPs was expected to be after filtering using sets of verified SNPs from dbSNPs. We found that a per-centage of 37.7 % (135/358) of the cancer-associated UTR-SNPs contained in our dataset corresponds to vali-dated UTR-SNPs (see column 6 of Additional file 1). Next, three approaches were used for controlling the false dis-covery rate: Bonferroni and Benjamini & Hochberg multi-ple testing corrections, and a resampling procedure. In practical, these statistical tests provided three different magnitudes of false positive estimation that are useful indicators prior to further analysis; the Bonferroni adjust-ment being more conservative than the Benjamini & Hochberg method and the resampling procedure. The candidate SNPs positive after these stringent multiple test-ing corrections (22/358 after Bonferroni and 104/358 after Benjamini & Hochberg, n = 10,514) are highlighted in Additional file 1. By the resampling procedure, we found that 92 observed p-values fell below the fifth per-centile of the empirical p-value distribution (p < 5.54 10-4).
Association with tumour development
Our list of cancer-associated variants contains a number of genes possibly involved in the cellular capabilities that might be acquired by cancer cells [28], e.g., translationally controlled tumour protein TCTP, IL-4-R, HLA class II
anti-gens, TIMP-3, CD147, CD44, and the jun-B, c-fos, AF4, Ki-Ras and RAF proto-oncogenes. Also included in our list
are 38 novel sequences, i.e., entries for which no annota-tion was available at the time of the study (these tran-scripts are referred to as 'NULL' in Additional file 1). In particular, we identified a ~800 bp- long nucleotide sequence located in the 5'-UTR of ENST00000285718, which contained as many as ten cancer-associated UTR-SNPs. The corresponding gene (encoding a putative pro-line-rich protein) has been mapped to 2q13, a region defined as a tumor amplicon [29]. Furthermore, out of the 269 RNAs with UTR-SNPs, the screen returned 22 hits pre-viously identified as bearing cancer-associated non-syn-onymous coding SNPs (nsSNPs) on the basis of a similar computer-based screen [7]. Among these transcripts exhibiting both associated nsSNPs and cancer-associated UTR-SNPs (highlighted in light grey in Addi-tional file 1) are those encoding Heat shock cognate 71 kDa protein, polyadenylate binding protein (PABP)-3, translationally controlled tumour protein (TCTP), immu-noglobulin gamma FcRIIIA, and dynein light chain 1 (DNCL1, see Table 2).
"Hot spots" for base substitutions were found for some transcripts, either as consecutive SNPs (e.g., 1286 c→a and 1287 t→c for ENST00000234617) or as 'nests' of
SNPs (e.g., 991 g→t, 999t→c and 1005 c→t for ENST00000285718). However, most transcripts (~75%) displayed a unique cancer-associated SNP. We found a variant causing a g→c change at nucleotide 175 in the 5'-UTR of RhoH, a gene prone to aberrant hypermutation activity in lymphomas [30]. Interestingly, determination of the origin of the EST libraries revealed that this UTR-SNP was specific to lymphoid tissues. In addition to the previously reported 4 c→a and 956 t→c alterations in the 5'- and 3'-UTRs of Kruppel-like factor 6 (KLF6), an impor-tant DNA-binding transcriptional regulator [31], our anal-ysis also revealed a 1206 c→t polymorphism in the 5'-UTR of this gene. Owing to the high mutation frequency of KLF6 in a number of pituitary tumors [32], knowledge of these KLF6 polymorphisms may be important for pros-tate cancer diagnosis.
Last, we found among the hits a series of UTR-SNPs con-cerning the SPARC gene, which encodes a multifunctional glycoprotein playing roles in tissue development, remod-elling and fibrosis [24-27]. As a regulator of cell-extracel-lular matrix (ECM) interactions, SPARC is thought to represent a major factor in the ECM remodelling occur-ring duoccur-ring tumour invasion. Our in silico analysis revealed 4 UTR-SNPs located in the 3'-UTR of the SPARC gene, corresponding to 1474 g→a, 1551 g→c, 1922 t→g and 2072 c→t changes, which were significantly associ-ated with the tumoral state. Noteworthy, of all the 'digital' hits, the 2072 SPARC polymorphism had the clearest association with cancer (see Table 2 and Additional file 1). This SNP is localized in a 44 bp- long conserved sequence between rodents and primates, suggesting that it might belong to a functionally constrained region.
Detection of SPARC variants in tumour samples
Because testing every prediction in our collection would be very labour intensive, we sought to validate experimen-tally the predictions that were made computationally for one of the candidate transcripts. The rationale for SPARC selection was based on the following criteria: (i) multiple hits over a wide range of p-values; (ii) best score for one of the hit (p-value for 2072 c→t = 5 10-17); (iii) multifunc-tional protein; (iv) candidate for tumours with a highly invasive phenotype (i.e., with poor prognosis). A group of 18 acute myeloblastic leukemias (AML) was explored for seeking the four SPARC variants predicted by computa-tional analysis (primers are listed in Addicomputa-tional file 2). Three of them (1551, 1922 and 2072) were detected in some of the samples while the 1474 mutation could not be detected (Table 3). In addition, a 2168 g→a change and a triple base substitution at position 2218 were iden-tified. Allelic frequencies for each SNP in AMLs were com-pared with those in normal controls (n = 20): SNP 2072 and 2168 frequencies were increased in patients versus controls, although the differences were statistically
signif-Table 2: Summary of cancer-associated UTR-SNPs.
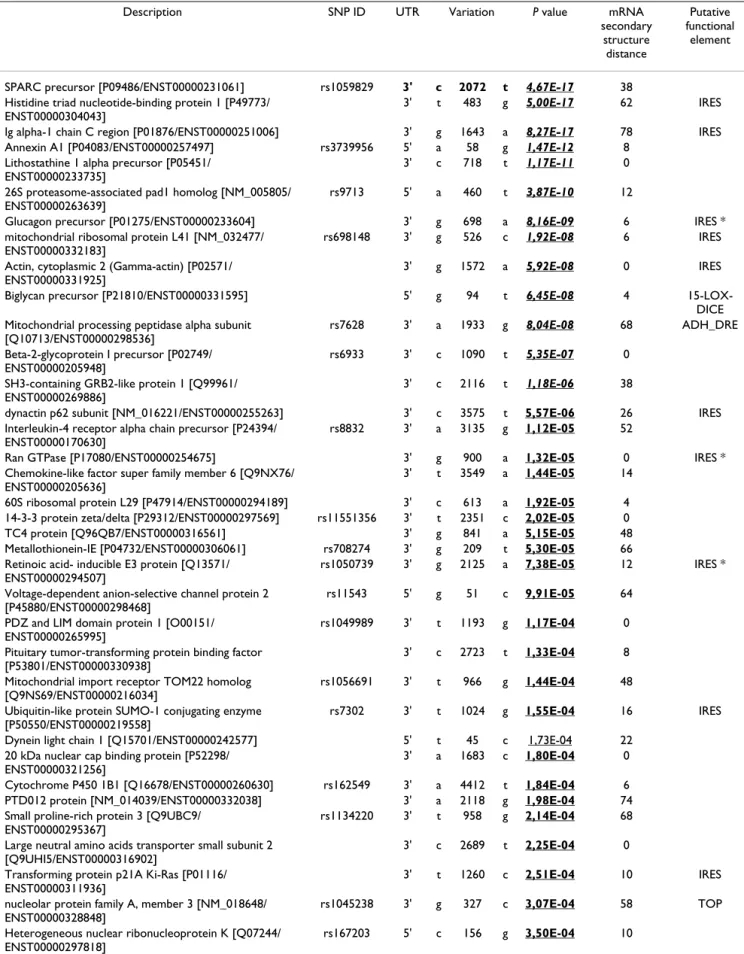
Description SNP ID UTR Variation P value mRNA secondary structure distance Putative functional element
SPARC precursor [P09486/ENST00000231061] rs1059829 3' c 2072 t 4,67E-17 38 Histidine triad nucleotide-binding protein 1 [P49773/
ENST00000304043]
3' t 483 g 5,00E-17 62 IRES Ig alpha-1 chain C region [P01876/ENST00000251006] 3' g 1643 a 8,27E-17 78 IRES Annexin A1 [P04083/ENST00000257497] rs3739956 5' a 58 g 1,47E-12 8
Lithostathine 1 alpha precursor [P05451/ ENST00000233735]
3' c 718 t 1,17E-11 0 26S proteasome-associated pad1 homolog [NM_005805/
ENST00000263639]
rs9713 5' a 460 t 3,87E-10 12
Glucagon precursor [P01275/ENST00000233604] 3' g 698 a 8,16E-09 6 IRES * mitochondrial ribosomal protein L41 [NM_032477/
ENST00000332183]
rs698148 3' g 526 c 1,92E-08 6 IRES Actin, cytoplasmic 2 (Gamma-actin) [P02571/
ENST00000331925]
3' g 1572 a 5,92E-08 0 IRES Biglycan precursor [P21810/ENST00000331595] 5' g 94 t 6,45E-08 4
15-LOX-DICE Mitochondrial processing peptidase alpha subunit
[Q10713/ENST00000298536]
rs7628 3' a 1933 g 8,04E-08 68 ADH_DRE Beta-2-glycoprotein I precursor [P02749/
ENST00000205948]
rs6933 3' c 1090 t 5,35E-07 0 SH3-containing GRB2-like protein 1 [Q99961/
ENST00000269886]
3' c 2116 t 1,18E-06 38
dynactin p62 subunit [NM_016221/ENST00000255263] 3' c 3575 t 5,57E-06 26 IRES Interleukin-4 receptor alpha chain precursor [P24394/
ENST00000170630]
rs8832 3' a 3135 g 1,12E-05 52
Ran GTPase [P17080/ENST00000254675] 3' g 900 a 1,32E-05 0 IRES * Chemokine-like factor super family member 6 [Q9NX76/
ENST00000205636]
3' t 3549 a 1,44E-05 14
60S ribosomal protein L29 [P47914/ENST00000294189] 3' c 613 a 1,92E-05 4 14-3-3 protein zeta/delta [P29312/ENST00000297569] rs11551356 3' t 2351 c 2,02E-05 0 TC4 protein [Q96QB7/ENST00000316561] 3' g 841 a 5,15E-05 48 Metallothionein-IE [P04732/ENST00000306061] rs708274 3' g 209 t 5,30E-05 66 Retinoic acid- inducible E3 protein [Q13571/
ENST00000294507]
rs1050739 3' g 2125 a 7,38E-05 12 IRES * Voltage-dependent anion-selective channel protein 2
[P45880/ENST00000298468]
rs11543 5' g 51 c 9,91E-05 64 PDZ and LIM domain protein 1 [O00151/
ENST00000265995]
rs1049989 3' t 1193 g 1,17E-04 0 Pituitary tumor-transforming protein binding factor
[P53801/ENST00000330938]
3' c 2723 t 1,33E-04 8
Mitochondrial import receptor TOM22 homolog [Q9NS69/ENST00000216034]
rs1056691 3' t 966 g 1,44E-04 48 Ubiquitin-like protein SUMO-1 conjugating enzyme
[P50550/ENST00000219558]
rs7302 3' t 1024 g 1,55E-04 16 IRES Dynein light chain 1 [Q15701/ENST00000242577] 5' t 45 c 1,73E-04 22
20 kDa nuclear cap binding protein [P52298/ ENST00000321256]
3' a 1683 c 1,80E-04 0
Cytochrome P450 1B1 [Q16678/ENST00000260630] rs162549 3' a 4412 t 1,84E-04 6 PTD012 protein [NM_014039/ENST00000332038] 3' a 2118 g 1,98E-04 74 Small proline-rich protein 3 [Q9UBC9/
ENST00000295367]
rs1134220 3' t 958 g 2,14E-04 68 Large neutral amino acids transporter small subunit 2
[Q9UHI5/ENST00000316902]
3' c 2689 t 2,25E-04 0
Transforming protein p21A Ki-Ras [P01116/ ENST00000311936]
3' t 1260 c 2,51E-04 10 IRES
nucleolar protein family A, member 3 [NM_018648/ ENST00000328848]
rs1045238 3' g 327 c 3,07E-04 58 TOP Heterogeneous nuclear ribonucleoprotein K [Q07244/
ENST00000297818]
RECS1 protein homolog [Q969X1/ENST00000258412] 3' g 2920 a 3,70E-04 0 G1/S-specific cyclin D2 [P30279/ENST00000261254] 3' a 6471 c 3,78E-04 20 Zinc finger protein 384 [Q8TF68/ENST00000319770] rs6786 3' t 3145 c 4,32E-04 0 Myosin regulatory light chain 2 [P19105/
ENST00000217652]
rs7811 3' a 1107 g 4,68E-04 22 Neuron specific protein family member 2 [Q9Y328/
ENST00000303177]
rs4457100 3' a 1030 g 5,46E-04 8
Sorting nexin 4 [O95219/ENST00000251775] 3' t 2178 a 5,63E-04 ND IRES * Paired amphipathic helix protein Sin3b [O75182/
ENST00000248054]
rs1044880 3' c 4888 t 6,27E-04 ND 15-LOX-DICE FK506-binding protein 1A [P20071/ENST00000262925] 3' a 518 g 6,30E-04 ND
Epsin 4 [Q14677/ENST00000296951] rs254682 3' t 3136 c 6,86E-04 ND 40S ribosomal protein S5 [P46782/ENST00000196551] 5' c 27 t 6,94E-04 ND Rho-related GTP-binding protein RhoH [Q15669/
ENST00000303700]
rs2245466 5' g 175 c 6,99E-04 ND Death-associated protein. [Q9BUC9/ENST00000230895] rs267927 5' t 163 c 1,69E-03 ND Inhibitor of apoptosis protein 1 [Q13489/
ENST00000263464]
5' t 539 g 4,88E-03 ND Kruppel-like factor 6 [Q99612/ENST00000173785] 3' c 1206 t 9,50E-03 ND
The Table shows a selection of 50 UTR-SNPs (out of 358) with significantly different allele frequency in normal versus tumoral tissues (exact Fisher's test; p < 0.01). UTR-SNPs are ranked by decreasing p-value. Swissprot protein accession references and Ensembl transcript accession references are indicated between brackets. Candidate positive after the multiple testing corrections are set in italics (Bonferroni), in bold (Benjamini and Hochberg) or underlined (candidate positive after a resampling procedure). Predictive effect of the polymorphisms on RNA secondary structure and putative UTR functional elements is indicated. Asterisk means that the reference allele sequence is modified by the cancer-associated UTR-SNP. For full data access, see Additional file 1. Accession numbers and SNP rs numbers are indicated in column 1 (description) and column 2 (SNP ID), respectively.
Table 2: Summary of cancer-associated UTR-SNPs. (Continued)
icant only for the last one. Of note, the computer-based procedure failed to identify the 2168 g→a substitution because the reference SPARC RNA available from Ensembl (release 16.3) was only 2104-bp- long. Moreover, since our algorithm is exclusively devoted to the detection of substitutions and not of indels, the three base insertion at position 2218 also was not identified through the in silico screen. In any case, for the four UTR-SNPs predicted through the computer-based procedure, results from experimental validation correlated with the p-values obtained from the EST scanning. Moreover, this analysis indicates that the in silico approach presented here can help to select candidate genomic regions within which mutations can be sought.
Patterns of substitution
In addition to a gene-centric view, SNPs can be character-ized by type of nucleotide change and putative functional effect. The objective of this section was to examine the substitution patterns among the cancer-associated UTR-SNPs identified by our computer-based procedure. We explored the distribution of the various types of sim-ple substitution SNPs in the different sets of candidate UTR-SNPs, i.e. the complete dataset of UTR-SNPs (n = 20,304), the total pool of cancer-associated UTR-SNPs (n = 358), and the subset of UTR-SNPs which were positive after the resampling procedure (n = 92) and that are less likely to correspond to false positives. The transition rates were around 70 % and the transversion rates were ~30%
in the different categories, in accordance with previous genome-wide estimates [33,34]. In all cases, the most common substitution was C→T (see Additional file 3 and Additional file 4 for a graphical representation); however, this type of change was 1.5 times less frequent in the pool of UTR-SNPs positive after the resampling procedure as in the total dataset (18.5 % versus 27.8 %, respectively). At the same time, the T→C transition accounted for 16.3 % of all single nucleotide substitutions within this pool
ver-sus 9.6 % within the total dataset. The couple of
comple-mentary substitutions A↔T followed a similar distribution in the total and cancer-associated datasets. Similarly, G→T and A→C frequencies were of similar magnitude in the three datasets; however, one can see that the frequencies for the complementary substitutions T→G and C→A behave in opposite manner: T→G substitutions were over-represented in the pool of UTR-SNPs positive after the resampling procedure (8.7 % versus 4.2 % in the total sampling) whereas only ~2 % of UTR-SNPs were of type C→A in the cancer-associated datasets (versus 4.9 % in the total pool of UTR-SNPs). Last, while the global fre-quencies of C↔G did not differ significantly between the different datasets (see Additional file 4, panel A), when the SNPs are reported respective of the direction of change, the frequencies of the pairs C→G and G→C showed a pattern reversal in the pool of UTR-SNPs posi-tive after the resampling procedure compared to the total dataset (1.1 % versus 6.3 % for C→G, and 6.5 % versus 2.6 % for G→C, respectively). Together, these results show that the ratios of several types of substitutions differ
between the entire dataset of UTR-SNPs and cancer-asso-ciated alleles.
Possible impact of cancer-associated UTR-SNPs on mRNA secondary structure and UTR regulatory elements
Although many of the UTR-SNPs identified in our experi-ment are not expected to be functional, but rather to act as markers for functional variants yet to be discovered else-where in the gene or even possibly in a nearby gene, it is possible that at least a fraction represent functional SNPs. Therefore, we decided to assess the putative structural and functional consequences of the tumor-associated UTR-SNPs on mRNA metabolism (mRNA secondary structure and putative regulatory sites).
Sequence changes in the UTR regions can affect mRNA folding, that in turn may impact transcript stability, mRNA processing or translational control [35-40]. To assess the possible effects of our set of cancer-associated UTR-SNPs on mRNA secondary structures, we checked with computer subroutines available in the RNAMute tool [41] that are based on energy minimization methods (Vienna and MFold) [42,43] whether these changes would be predicted to induce conformational rearrange-ments. This program was used to compute predicted sec-ondary structures, differences in secsec-ondary structures and corresponding free energy changes (ΔG) for a 100-nt win-dow around the UTR-SNP site. 'Variant' inputs of length 100-nt were extracted from two groups of sequences: (i) sequences that displayed the cancer-associated UTR-SNPs identified through the computer-based procedure; (ii)
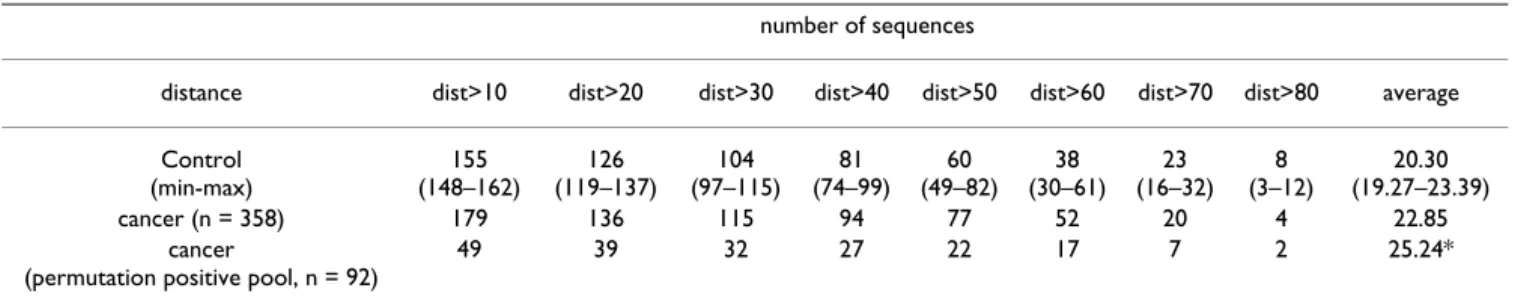
randomly chosen sequences displaying UTR-SNPs that were not associated with the tumoral state. For each group, 'Reference' inputs were also generated from the corresponding normal allele sequences. Table 4 gives the results of variant to reference comparisons (n = 358) for the cancer-associated pool and for 10 different control datasets. Our data reveals a slight trend for cancer-associ-ated SNPs to be found in higher distances than control SNPs. Notably, this trend becomes statistically significant (Two Sample T-test; p < 0.05) when only the cancer-asso-ciated SNPs positive after the permutation test (n = 92) are being considered. Among these cancer-associated UTR-SNPs, 41 (44.6%) were predicted to have no or a minor effect on RNA secondary structure (dist < 10), 29 (31.5%) were predicted to induce significant conformational changes in the folding (distance values between 10 and 50) and 22 (23.9%) were predicted to lead to high dis-tance values with respect to their reference alleles (dist > 50) (see Table 2 and Additional file 1). In only 31.5 % of the cases (29/92) the reference allele displayed the highest negative energy value, suggesting that the majority of can-cer-associated UTR-SNPs lead to more stable transcripts. However, this result should be balanced by the fact that UTR-SNPs associated with mRNA stabilizing structures have higher chance to be detected than those associated with degrading elements. The cancer-associated mutation which was predicted to cause the greatest change on mRNA structure is a c→t polymorphism on ENST00000206380 (distance = -84 using Vienna's RNADistance), a transcript that shares no similarity with any sequence in public databases. The 1551 and 2072
Table 3: Results of SPARC genotyping analysis in AML samples.
AML patients Healthy donors OR (95% CI)
SNP 1474 GG 13 20 GA 0 0 AA 0 0 Allelic Frequency A (%) SNP 1551 GG 2 7 GC 8 7 CC 3 6 Allelic Frequency C (%) 53 47 2.9 (0.5–17.3) SNP 1922 TT 13 5 TG 2 1 GG 0 0 Allelic Frequency G (%) 13 0.8 (0.05–10.5) SNP 2072 CC 4 10 CT 7 3 TT 4 4 Allelic Frequency T (%) 50 32 3.9 (0.9–17.5) SNP 2168 GG 3 11 GA 6 1 AA 4 5 Allelic Frequency A (%) 54 32 6.1 (1.2–31.2) AML: acute myeloblastic leukaemia; OR: odds ratio; CI: confidence interval.
SNPs on SPARC were predicted to have a positive effect on mRNA stability (with distances of + 56 and + 38, respec-tively) while the 1922 polymorphism had only a mild predicted impact (distance = + 4).
Next, putative UTR functional elements potentially affected by cancer-associated SNPs were searched for using UTRscan [44]. Most of the cancer-associated poly-morphisms did not lie within or at the immediate vicinity
of cis-regulatory elements (see Table 5 and Additional file 1). A fraction of 153 UTR-SNPs out of 358 (42.7%) had an assignment to known UTR regulatory regions. When only the 92 hits positive after the permutation test are considered, the percentage of polymorphisms predicted to impact UTR functional elements remains relatively constant (37/92, i.e., 40.2 %). As shown in Table 5, a total of 9 regulatory elements out of the 31 included in the UTRSite database were located near or at
cancer-associ-Table 5: Putative UTR regulatory elements affected by cancer-associated UTR-SNPs.
UTR-SNPs Ref (n = 20,304) UTR-SNPs Var (n = 20,304) CANCER Ref (n = 358) CANCER Var (n = 358) CANCER Ref (permutation pool, n = 92) CANCER Var (permutation pool, n = 92) Total IRES 5766 5802 92 94 20 20 Gained 625 11 3 Lost 589 9 3 Total 15-LOX-DICE 1788 1744 37 36 8 8 Gained 90 2 Lost 134 3 Total TOP 491 490 11 10 5 5 Gained 1 Lost 2 1 Total K-Box 287 287 3 3 0 0 Gained 16 Lost 16 Total GY-Box 174 168 5 5 0 0 Gained 9 Lost 15 Total Brd-Box 111 116 2 1 1 0 Gained 13 Lost 8 1 1 Total ADH-DRE 44 50 1 1 1 1 Gained 6 Lost 0 Total CPE 26 30 1 1 0 0 Gained 12 Lost 1 Total SECIS-2 12 10 1 0 1 0 Gained 1 Lost 3 1 1
The results identify cis-regulatory elements located in the immediate vicinity of or at the UTR-SNP sites. UTR regulatory elements can be 'gained' or 'lost' when reference allele sequences are modified by cancer-associated SNPs.
Table 4: Prediction of UTR-SNPs affecting mRNA folding structures.
number of sequences
distance dist>10 dist>20 dist>30 dist>40 dist>50 dist>60 dist>70 dist>80 average Control (min-max) 155 (148–162) 126 (119–137) 104 (97–115) 81 (74–99) 60 (49–82) 38 (30–61) 23 (16–32) 8 (3–12) 20.30 (19.27–23.39) cancer (n = 358) 179 136 115 94 77 52 20 4 22.85 cancer
(permutation positive pool, n = 92)
49 39 32 27 22 17 7 2 25.24*
'Control': average of experiments with UTR-SNPs not associated with cancer phenotype (n = 358, 10 independent control trials). Results were statistically analyzed using the two sample t-test ('*', p < 0.05).
ated SNP sites. Based on the UTRScan analysis, sequences close to or containing Internal Ribosomal Entry Site (IRES) elements were identified as preferential targets for cancer-associated polymorphisms, which is expected since this class of elements is the most abundant in our UTR-SNP dataset (first column of Table 5). Interestingly, a number of cancer-associated variant sequences dis-played potential regulatory elements (IRES, 15-LOX-DICE) that were not apparent in the reference allele sequences. Inversely, some UTR functional elements (IRES, 15-LOX-DICE, TOP, Brd-Box and SECIS-2) were detected only in reference allele sequences but not in var-iant ones. Thus, cis-acting regulatory elements may be gained or lost when reference allele sequences are modi-fied by cancer-associated SNPs. Loss of a SECIS-2 (for selenocysteine insertion sequence) regulatory element in the 3'-UTR of ENST00000288332 may be particularly rel-evant. Indeed, out of the 20,304 UTR-SNPs included in our dataset, only 12 were mapped to untranslated regions containing SECIS-2 elements. ENST00000288332 encodes a putative glutathione peroxidase, i.e., a seleno-protein, and SECIS elements are required for the transla-tional incorporation of the unusual amino acid selenocysteine in these enzymes [45,46]. Last, two physi-cally close cancer-associated SNPs (3726 a→g and 3743 c→t) resulted in supplementary regulatory elements (IRES and LOX-15-DICE, respectively) in the 3'-UTR of
brain-type glycogen phosphorylase, a proposed biomarker of
gas-trointestinal tumours [47,48].
Altogether, these results provide evidence that at least a subset of cancer-associated SNPs might have functional consequences on mRNA stability and/or expression.
Discussion
Owing to advances in biotechnology and bioinformatics progress, researchers can now capture "molecular por-traits" of various particular cancers using gene chips or SAGE data. These methods provide information on tens of thousands of genes simultaneously, and some variations in genes might be directly related to the cancer phenotype. Transcriptome analysis not only gives information about gene expression levels in normal versus cancer cells, but also about genetic variations. In that respect, large-scale scanning of EST databases have previously been used for identification of SNPs in genes involved in a various number of disorders [49-51]. As noted elsewhere [8,9,15,52], EST-based strategies have inherent limita-tions, including poor sequencing depth, variations in library sizes, poor quality annotation and differences in transcript sampling. Moreover, large-scale computational studies may be hampered by artifacts produced during EST library preparation, e.g. uncertainty concerning the origin of the samples or use of pools of different cell types. With these caveats in mind, in this study, we made the
assumption that UTR-SNP profiles may help to propose novel molecular signatures in cancer. Using a novel com-putational strategy, a set of ~350 UTR-SNPs presumably associated with the cancer phenotype was identified, and then characterized using bioinformatics tools. This list contains novel markers as well as candidate SNPs that could alter both mRNA stability, i.e., transcript abun-dance, and translational regulation of cancer-associated genes, i.e., protein levels. Because some UTR-SNPs may affect transcript and protein abundance, their knowledge could somehow bridge a gap between differential gene expression studies and cancer phenotype evidences. Hence, a prolongation of our study is the determination of UTR-SNPs that correlate with aberrant gene expression in cancer cells. As novel UTR regulatory sites are identified and more methods are developed to analyze mRNA sec-ondary structure, future plans may include development of integrated and large-scale computational tools to pre-dict UTR-SNPs with potential phenotypic consequences. Once these computational tools will be made available, it will be of interest to determine if the proportion of UTR-SNPs predicted as deleterious increases at low allelic fre-quencies, mirroring previous studies that were focused on nsSNPs [50,53,54]. While out of the scope of this cancer-oriented study, other genome-centric approaches may be useful such as examination of base composition around the UTR-SNP position, exploration of neighbouring-nucleotide effects, or functional annotation of the variant transcripts.
Determination of the allele frequencies for several UTR-SNPs and study of the haplotype structure of some of the loci would also likely constitute profitable avenues of research. In that respect, one of the testable hypotheses of our work is related to DNCL1. This gene encodes a highly conserved multifunctional protein known to play impor-tant roles in a variety of processes including cell prolifera-tion, apoptosis and cytoskeleton organizaprolifera-tion, and whose deregulation could influence tumour progression [55-61]. We have recently identified and experimentally character-ized a DNCL1 tumour variant (corresponding to a Gly to Cys substitution at amino acid position 79) [7], and we report here an UTR-SNP located in the 5'-UTR of the DNCL1 transcript (introducing a t→c change at position 45, see Additional file 1). The G79C mutation was shown to induce a clear conformational change to DNCL1 and to reduce substantially the in vitro target binding capabilities compared to the wild-type version [7]. As the possibility exists that the 5'-UTR polymorphim may be in linkage dis-equilibrium with the G79C mutation, it will be interesting to investigate both polymorphisms in samples from healthy and diseased donors.
Although potential UTR-SNPs relevant for cancer associa-tion studies could be successfully identified through
inno-vative computer-based procedures, it is worth stressing that the candidate SNPs should be verified through exper-imental methods such as RT-PCR, microarrays and geno-typing experiments, as described here for the polymorphisms located on SPARC. SPARC is a gene involved in a number of diseases including rheumatoid arthritis, scleroderma, tumor development and metastasis [62-67]. Our computer-based screen revealed four UTR-SNPs located in the 3'-UTR of SPARC (1474 g→a, 1551 g→c, 1922 t→g and 2072 c→t) that were significantly associated with tumor libraries. Out of these four UTR-SNPs, three were confirmed in tissue samples (1551, 1922 and 2072) and one was experimentally validated as can-cer-associated in AML samples (2072). During the course of the study, two additional cancer-associated polymor-phisms were discovered through the genotyping experi-ments (2168 g→a and a 3-bp insertion at position 2218). Interestingly, a distinct polymorphism within the SPARC gene, namely 998 c→g, has been associated with suscepti-bility to and clinical manifestations of scleroderma [68]. Therefore, SPARC genetic polymorphisms may represent useful candidate SNPs for screening either susceptibility to cancer (2072 c→t and 2198 g→a) or scleroderma pathogenesis (998 c→g). Moreover, recent studies have reported increased risk of cancer in patients with sclero-derma [69]. Although underlying explanations are still lacking, one possibility is that alterations in SPARC could represent a common risk factor. In this hypothesis, it is noteworthy that the 1922 t→g UTR-SNP present on SPARC has been associated with scleroderma [68], in addition to cancer (our screen). In conclusion, knowledge of SPARC polymorphisms could provide potential candi-date UTR-SNPs for both diseases, either separately or in combination. Last, it will be worth testing experimentally whether the identified UTR-SNPs affect gene expression. In addition to relative quantification of allelic expression by quantitative RT-PCR or Western blotting on human samples with different genotypes, functional evaluation will require demonstration of allele-specific effects on mRNA expression or stability. This can be addressed through nuclear run-on experiments and mRNA half-life studies, and construction of chimeric genes encoding the luciferase reporter sequence with the wild type or the mutated alleles. Information derived from post-genomic bioinformatics when combined to laboratory observa-tions has the potential to greatly increase our understand-ing of the role of polymorphisms involvunderstand-ing untranslated regions in disease pathogenesis.
Conclusion
In the search for non-coding genetic variation associated with cancer, no systematic attention has been paid so far to the changes that occur in untranslated regions of mRNA. This work is a first, genome-wide attempt to iden-tify UTR-SNPs (and flanking sequences) to prioritize for
further studies in the field of cancer biomarker research. Computational analysis suggests that a proportion of can-cer-associated UTR-SNPs may have the potential to signif-icantly affect mRNA secondary structure and/or functionally important RNA regions. The in silico approach described here therefore sets the stage for the next phase of characterization of UTR-located functional variants in human cancer.
Methods
Data preparation
We have used an EST-based pipeline to scan for UTR-located polymorphisms associated with libraries of can-cerous origin. Human ESTs from dbEST [70] (October 2004 release) were first extracted using the ACNUC sequence retrieval system [71]. ESTs were classified according to their UNIGENE library features [72] as previ-ously described [6]. The eVOC ontology [73] (October 2004) for anatomical sites and pathology types was then used to classify the libraries through a number of criteria such as tissue origin and pathological context including tumor state. This well-accepted hierarchical vocabulary provided us with a mean to determine when a specific tis-sue was part of an organ and when a specific label was part of the 'tumoral' state. A total of 5135 'tumor' and 2503 'normal' (i.e. non-pathological) libraries were catalogued. Our approach to EST clustering used the human genome as a reliable guide. ENSEMBL RNAs [74] annotated on human genome assembly (release 16.3) were used as a backbone for the clustering of dbEST sequences using MEGABLAST (alignment length ≥ 100 bp and similarity ≥ 95%) [75]. In order to avoid paralogous false positive assignation, only best EST hit matches were subsequently selected. RNA clustering of ESTs in both normal and tumoral tissues was the starting point for in silico mining of UTR-SNPs associated with tumoral phenotype.
SNP detection
We have developed an algorithm to identify exonic SNPs in multiple alignments of various ESTs associated to a par-ticular annotated transcript. This algorithm takes advan-tage of EST library redundancy and performs four filters to reduce the effect of sequencing inaccuracies at each posi-tion. The first filter required that each position within a multiple alignment of ESTs should have an exact match with the reference RNA (windows length = 10 bp around each variant position). The second filter considered a position as informative if the number of libraries in the multiple alignment was superior to a fixed minimum threshold (library number ≥ 5). The third filter of the algo-rithm required the variant to be found at least two times independently i.e., in two different libraries. A last inde-pendent filter that required a minimum of two variant ESTs in one of the libraries was subsequently added in order to increase further the stringency of the cancer
asso-ciation mining strategy. We then combined detection method information (library and EST depth coverage) and nucleotide substitution features (e.g., transition/ transversion, position in 5'- or 3'-UTR) for the UTR-SNPs that have been filtered out. Statistical analysis was per-formed using R [76]. Genomic data were stored in a local PostgreSQL database (GeMCore) [77] using PERL and Java script.
Cancer association
Finally for each informative SNP that has both normal and tumoral EST coverage, an exact Fisher's test was per-formed in order to statistically evaluate the association of a particular variant with the tumoral state. We privileged the counting of ESTs rather than a count per library because of the frequent lack of precision concerning the origin of the source tissues and the use of pooled samples. To adjust p-values produced by the Fisher's exact test, three approaches were used: (a) Bonferroni, and (b) Benjamini and Hochberg corrections, which are very conservative methods for controlling the false discovery rate, and (c) a resampling procedure. The standard Bonferroni correc-tion multiplies the uncorrected p-value by the number of statistical tests. The Benjamini and Hochberg correction consists of ranking all p-values and adjusting each by mul-tiplying by the total number of tests and dividing by the rank of that p-value. The resampling procedure simulates the distribution of the minimum p-value that we would expect if there was no association with cancer. To do this, reference and variant margins were fixed at each SNP; Fisher's exact test was then performed for 1,000 resampled datasets, and the smallest p-value was recorded. This resa-mpling procedure was repeated for n = 10,514 SNPs, from which an empirical distribution of the minimum p-value was obtained. From this distribution, we estimated the p-value that corresponded to the conventional 5% thresh-old. The intensity of the bias of tumoral versus normal allele frequency was calculated according to the following formula:
Ib = (a/V) - [(T - a)/R], where 'a' is the number of tumoral
variants, 'V' the total number of variants, 'T' the sum of tumoral counts (variant plus reference) and R the total number of reference alleles (Ib being close to 1 in case of strong association).
In silico characterization of UTR-located SNPs
UTRScan [78] was used to identify putative cis acting ele-ments patterns in the regions containing cancer-associ-ated SNPs. UTRScan looks for UTR functional elements by searching through user submitted query sequences for the patterns defined in the UTRsite collection [44]. To test the potential effects of the detected polymorphisms on mRNA folding, we took advantage of the RNAMute appli-cation [41].
In vitro detection of SPARC variants
Genomic DNA was extracted from bone marrow in patients with acute myeloblastic leukemia (AML) and peripheral blood in healthy donors. The phenol/chloro-form method was used for DNA extraction according to standard procedures. Primers to explore SPARC polymor-phisms detected by computational procedures were designed based on the DNA sequence from GenBank (entry: BC072457). The sequences of the primers are listed in Additional file 2. Amplification was carried out in an iCycler Thermal Cycler (Bio-Rad, Hercules, CA, USA): 1 μg of DNA was amplified in a 25 μl of final reaction vol-ume containing 10 × buffer II, 2 mM MgCl2, 0.2 μl of 25 mM dNTP's mixture, 0.4 μl of 20 pmol/μl forward and reverse primers and 0.2 μl of Fast-Taq polymerase (5 u/μl) (Roche Diagnostics, Indianapolis, IN, USA). PCR proce-dure consisted of 35 cycles of denaturation at 94°C for 15 sec, annealing at 60°C for 60 sec, with an initial denatur-ation of the DNA at 94°C for 5 min before PCR, and a final extension at 72°C for 5 minutes. The PCR products were sequenced in an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
Authors' contributions
AA designed the study, analyzed the data and drafted the manuscript. VN developed the algorithm for the differen-tial display procedure, participated in the data analyses and reviewed the manuscript. NG and RLP performed the genotyping experiments. DB and AC carried out the RNA secondary structure mutation predictions and reviewed the manuscript. DM and CG provided funding and super-vision for the work. All authors read and approved the final manuscript.
Additional material
Additional File 1
Complete list of cancer-associated UTR-SNPs. UTR-SNPs with signifi-cantly different allele frequency in normal versus tumoral tissues (exact Fisher's test; p < 0.01). Hits are ranked by decreasing p value (see Method section). Sequences for which no annotation is available are referred to as 'NULL'. Bias intensity is given for information. Candidate positive after the multiple testing corrections are underlined. Information concerning the SNPs present on SPARC appears in bold. References appear for vali-dated SNPs from dbSNP (last column). Ambiguity code: R = G↔A; M = C↔A; K = G↔T; Y = C↔T; S = G↔C; W = A↔T. Nucleotide sequence of the reference RNA is shown within a 10-bp interval around each SNP site. Putative cis acting elements located in the regions containing cancer-associated SNPs were identified with UTRScan (see text for details). RNAMute was used to compute distances between variant and reference alleles. Gene symbols were from HGNC (HUGO Gene Nomenclature Committee). UTR-SNPs lying on transcripts for which non-synonymous SNPs were previously identified [7] are marked in light grey (first col-umn).
Click here for file
[http://www.biomedcentral.com/content/supplementary/1471-2164-8-2-S1.xls]
Acknowledgements
V. N is supported by a grant from INRA. A. A is recipient of a fellowship from the CNRS.
RLP was supported by a grant from the AECC. The authors wish to thank Audrey Barthelaix for critical reading of the manuscript and Sandy Jacquier for helpful comments.
References
1. Rajkovic A, Yan MSC, Klysik M, Matzuk M: Discovery of germ
cell-specific transcripts by expressed sequence tag database anal-ysis. Fertil Steril 2001, 76(3):550-554.
2. Wang J, Liang P: DigiNorthern, digital expression analysis of
query genes based on ESTs. Bioinformatics 2003, 19(5):653-654.
3. Scheurle D, DeYoung MP, Binninger DM, Page H, Jahanzeb M, Naray-anan R: Cancer gene discovery using digital differential
dis-play. Cancer Res 2000, 60(15):4037-4043.
4. Baranova AV, Lobashev AV, Ivanov DV, Krukovskaya LL, Yankovsky NK, Kozlov AP: In silico screening for tumour-specific
expressed sequences in human genome. FEBS Lett 2001, 508(1):143-148.
5. Brentani H, Caballero OL, Camargo AA, da Silva AM, da Silva WA Jr, Dias Neto E, Grivet M, Gruber A, Guimaraes PE, Hide W, Iseli C, Jon-geneel CV, Kelso J, Nagai MA, Ojopi EP, Osorio EC, Reis EM, Riggins GJ, Simpson AJ, de Souza S, Stevenson BJ, Strausberg RL, Tajara EH, Verjovski-Almeida S, Acencio ML, Bengtson MH, Bettoni F, Bodmer WF, Briones MR, Camargo LP, Cavenee W, Cerutti JM, Coelho Andrade LE, Costa dos Santos PC, Ramos Costa MC, da Silva IT, Este-cio MR, Sa Ferreira K, Furnari FB, Faria M Jr, Galante PA, Guimaraes GS, Holanda AJ, Kimura ET, Leerkes MR, Lu X, Maciel RM, Martins EA, Massirer KB, Melo AS, Mestriner CA, Miracca EC, Miranda LL, Nobrega FG, Oliveira PS, Paquola AC, Pandolfi JR, Campos Pardini MI, Passetti F, Quackenbush J, Schnabel B, Sogayar MC, Souza JE, Valentini SR, Zaiats AC, Amaral EJ, Arnaldi LA, de Araujo AG, de Bessa SA, Bicknell DC, Ribeiro de Camaro ME, Carraro DM, Carrer H, Car-valho AF, Colin C, Costa F, Curcio C, Guerreiro da Silva ID, Pereira
da Silva N, Dellamano M, El-Dorry H, Espreafico EM, Scattone Fer-reira AJ, Ayres FerFer-reira C, Fortes MA, Gama AH, Giannella-Neto D, Giannella ML, Giorgi RR, Goldman GH, Goldman MH, Hackel C, Ho PL, Kimura EM, Kowalski LP, Krieger JE, Leite LC, Lopes A, Luna AM, Mackay A, Mari SK, Marques AA, Martins WK, Montagnini A, Mourao Neto M, Nascimento AL, Neville AM, Nobrega MP, O'Hare MJ, Otsuka AY, Ruas de Melo AI, Paco-Larson ML, Guimaraes Pereira G, Pesquero JB, Pessoa JG, Rahal P, Rainho CA, Rodrigues V, Rogatto SR, Romano CM, Romeiro JG, Rossi BM, Rusticci M, Guerra de Sa R, Sant' Anna SC, Sarmazo ML, Silva TC, Soares FA, Sonati Mde F, de Freitas Sousa J, Queiroz D, Valente V, Vettore AL, Villanova FE, Zago MA, Zalcberg H: The generation and utilization of a
cancer-ori-ented representation of the human transcriptome by using expressed sequence tags. Proc Natl Acad Sci USA 2003, 100(23):13418-13423.
6. Aouacheria A, Navratil V, Barthelaix A, Mouchiroud D, Gautier C:
Bioinformatic screening of human ESTs for differentially expressed genes in normal and tumor tissues. BMC Genomics
2006, 7:94.
7. Aouacheria A, Navratil V, Wen W, Jiang M, Mouchiroud D, Gautier C, Gouy M, Zhang M: In silico whole-genome scanning of
can-cer-associated nonsynonymous SNPs and molecular charac-terization of a dynein light chain tumour variant. Oncogene
2005, 24(40):6133-6142.
8. Qiu P, Wang L, Kostich M, Ding W, Simon JS, Greene JR: Genome
wide in silico SNP-tumor association analysis. BMC Cancer
2004, 4(1):4.
9. Imyanitov EN, Togo AV, Hanson KP: Searching for
cancer-associ-ated gene polymorphisms: promises and obstacles. Cancer
Lett 2004, 204(1):3-14.
10. Strausberg RL, Simpson AJ, Wooster R: Sequence-based cancer
genomics: progress, lessons and opportunities. Nat Rev Genet
2003, 4(6):409-418.
11. Chakravarti A: It's raining SNPs, hallelujah? Nat Genet 1998,
19(3):216-217.
12. Collins FS, Guyer MS, Charkravarti A: Variations on a theme:
cat-aloging human DNA sequence variation. Science 1997, 278(5343):1580-1581.
13. Syvanen AC, Landegren U, Isaksson A, Gyllensten U, Brookes A:
First International SNP Meeting at Skokloster, Sweden, August 1998. Enthusiasm mixed with scepticism about sin-gle-nucleotide polymorphism markers for dissecting com-plex disorders. Eur J Hum Genet 1999, 7(1):98-101.
14. Hudson BI, Stickland MH, Futers TS, Grant PJ: Effects of novel
pol-ymorphisms in the RAGE gene on transcriptional regulation and their association with diabetic retinopathy. Diabetes 2001, 50(6):1505-1511.
15. Schmitt AO, Specht T, Beckmann G, Dahl E, Pilarsky CP, Hinzmann B, Rosenthal A: Exhaustive mining of EST libraries for genes
differentially expressed in normal and tumour tissues. Nucleic
Acids Res 1999, 27(21):4251-4260.
16. Peppel K, Vinci JM, Baglioni C: The AU-rich sequences in the 3'
untranslated region mediate the increased turnover of inter-feron mRNA induced by glucocorticoids. J Exp Med 1991, 173(2):349-355.
17. Duan J, Sanders AR, Molen JE, Martinolich L, Mowry BJ, Levinson DF, Crowe RR, Silverman JM, Gejman PV: Polymorphisms in the
5'-untranslated region of the human serotonin receptor 1B (HTR1B) gene affect gene expression. Mol Psychiatry 2003, 8(11):901-910.
18. Miller GM, Madras BK: Polymorphisms in the 3'-untranslated
region of human and monkey dopamine transporter genes affect reporter gene expression. Mol Psychiatry 2002, 7(1):44-55.
19. Goto Y, Yue L, Yokoi A, Nishimura R, Uehara T, Koizumi S, Saikawa Y: A novel single-nucleotide polymorphism in the
3'-untrans-lated region of the human dihydrofolate reductase gene with enhanced expression. Clin Cancer Res 2001, 7(7):1952-1956.
20. van der Velden AW, Thomas AA: The role of the 5' untranslated
region of an mRNA in translation regulation during develop-ment. Int J Biochem Cell Biol 1999, 31(1):87-106.
21. Gray NK: Translational control by repressor proteins binding
to the 5'UTR of mRNAs. Methods Mol Biol 1998, 77:379-397.
22. Bartel DP, Chen CZ: Micromanagers of gene expression: the
potentially widespread influence of metazoan microRNAs.
Nat Rev Genet 2004, 5(5):396-400.
Additional File 2
Polymerase chain reaction primers for detecting SPARC UTR-SNPs.
Click here for file
[http://www.biomedcentral.com/content/supplementary/1471-2164-8-2-S2.xls]
Additional File 3
Percentages of the different types of simple substitution SNPs.
Click here for file
[http://www.biomedcentral.com/content/supplementary/1471-2164-8-2-S3.xls]
Additional File 4
Distribution of the different types of simple substitution SNPs (graphical representation). (A) Substitutional patterns observed among UTR-SNPs. Transition rates were 67.2 % in the complete dataset of UTR-SNPs, 72.6 % in the total pool of cancer-associated UTR-SNPs, and 66.3 % in the subset of UTR-SNPs which were positive after the resampling procedure. Of the 358 cancer-associated UTR-SNPs, 260 were transition events while 298 were transversion events. When considering the 92 UTR-SNPs positive after the resampling procedure, 61 were transition events and 31 were transversion events. (B) The proportions for each pair of complemen-tary substitutions are graphed next to each other for ease of comparison. Student t-test not significant (p > 0.05).
Click here for file
[http://www.biomedcentral.com/content/supplementary/1471-2164-8-2-S4.pdf]
23. Audic Y, Hartley RS: Post-transcriptional regulation in cancer.
Biol Cell 2004, 96(7):479-498.
24. Motamed K: SPARC (osteonectin/BM-40). Int J Biochem Cell Biol 1999, 31(12):1363-1366.
25. Bornstein P, Sage EH: Matricellular proteins: extracellular
mod-ulators of cell function. Curr Opin Cell Biol 2002, 14(5):608-616.
26. Bradshaw AD, Sage EH: SPARC, a matricellular protein that
functions in cellular differentiation and tissue response to injury. J Clin Invest 2001, 107(9):1049-1054.
27. Brekken RA, Sage EH: SPARC, a matricellular protein: at the
crossroads of cell-matrix. Matrix Biol 2000, 19(7):569-580.
28. Hanahan D, Weinberg RA: The hallmarks of cancer. Cell 2000,
100(1):57-70.
29. Zhou Y, Luoh SM, Zhang Y, Watanabe C, Wu TD, Ostland M, Wood WI, Zhang Z: Genome-wide identification of chromosomal
regions of increased tumor expression by transcriptome analysis. Cancer Res 2003, 63(18):5781-5784.
30. Pasqualucci L, Neumeister P, Goossens T, Nanjangud G, Chaganti RS, Kuppers R, Dalla-Favera R: Hypermutation of multiple
proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature
2001, 412(6844):341-346.
31. Koivisto PA, Hyytinen ER, Matikainen M, Tammela TL, Ikonen T, Sch-leutker J: Kruppel-like factor 6 germ-line mutations are
infre-quent in Finnish hereditary prostate cancer. J Urol 2004, 172(2):506-507.
32. Vax VV, Gueorguiev M, Dedov II, Grossman AB, Korbonits M: The
Kruppel-like transcription factor 6 gene in sporadic pituitary tumours. Endocr Relat Cancer 2003, 10(3):397-402.
33. Stephens JC, Schneider JA, Tanguay DA, Choi J, Acharya T, Stanley SE, Jiang R, Messer CJ, Chew A, Han JH, Duan J, Carr JL, Lee MS, Koshy B, Kumar AM, Zhang G, Newell WR, Windemuth A, Xu C, Kalbfleisch TS, Shaner SL, Arnold K, Schulz V, Drysdale CM, Nandabalan K, Jud-son RS, Ruano G, Vovis GF: Haplotype variation and linkage
dis-equilibrium in 313 human genes. Science 2001,
293(5529):489-493.
34. Zhao Z, Boerwinkle E: Neighboring-nucleotide effects on single
nucleotide polymorphisms: a study of 2.6 million polymor-phisms across the human genome. Genome Res 2002, 12(11):1679-1686.
35. Decker CJ, Parker R: Mechanisms of mRNA degradation in
eukaryotes. Trends Biochem Sci 1994, 19(8):336-340.
36. Darzacq X, Singer RH, Shav-Tal Y: Dynamics of transcription and
mRNA export. Curr Opin Cell Biol 2005, 17(3):332-339.
37. Mitchell P, Tollervey D: mRNA turnover. Curr Opin Cell Biol 2001,
13:320-325.
38. Jansen RP: mRNA localization: message on the move. Nat Rev
Mol Cell Biol 2001, 2(4):247-256.
39. Macdonald P: Diversity in translational regulation. Curr Opin Cell
Biol 2001, 13(3):326-331.
40. Bashirullah A, Cooperstock RL, Lipshitz HD: RNA localization in
development. Annu Rev Biochem 1998, 67:335-394.
41. Churkin A, Barash D: RNAmute: RNA secondary structure
mutation analysis tool. BMC Bioinformatics 2006, 7(1):221.
42. Hofacker IL: Vienna RNA secondary structure server. Nucleic
Acids Res 2003, 31(13):3429-3431.
43. Zuker M: Mfold web server for nucleic acid folding and
hybrid-ization prediction. Nucleic Acids Res 2003, 31(13):3406-3415.
44. Pesole G, Liuni S: Internet resources for the functional analysis
of 5' and 3' untranslated regions of eukaryotic mRNAs.
Trends Genet 1999, 15(9):378.
45. Muller C, Wingler K, Brigelius-Flohe R: 3'UTRs of glutathione
peroxidases differentially affect selenium-dependent mRNA stability and selenocysteine incorporation efficiency. Biol
Chem 2003, 384(1):11-18.
46. Wingler K, Bocher M, Flohe L, Kollmus H, Brigelius-Flohe R: mRNA
stability and selenocysteine insertion sequence efficiency rank gastrointestinal glutathione peroxidase high in the hier-archy of selenoproteins. Eur J Biochem 1999, 259(1–2):149-157.
47. Tashima S, Shimada S, Yamaguchi K, Tsuruta J, Ogawa M: Expression
of brain-type glycogen phosphorylase is a potentially novel early biomarker in the carcinogenesis of human colorectal carcinomas. Am J Gastroenterol 2000, 95(1):255-263.
48. Shimada S, Tashima S, Yamaguchi K, Matsuzaki H, Ogawa M:
Car-cinogenesis of intestinal-type gastric cancer and colorectal cancer is commonly accompanied by expression of brain
(fetal)-type glycogen phosphorylase. J Exp Clin Cancer Res 1999, 18(1):111-118.
49. Emahazion T, Jobs M, Howell WM, Siegfried M, Wyoni PI, Prince JA, Brookes AJ: Identification of 167 polymorphisms in 88 genes
from candidate neurodegeneration pathways. Gene 1999, 238(2):315-324.
50. Halushka MK, Fan JB, Bentley K, Hsie L, Shen N, Weder A, Cooper R, Lipshutz R, Chakravarti A: Patterns of single-nucleotide
poly-morphisms in candidate genes for blood-pressure homeosta-sis. Nat Genet 1999, 22(3):239-247.
51. Bennet AM, Naslund TI, Morgenstern R, de Faire U: Bioinformatic
and experimental tools for identification of single-nucleotide polymorphisms in genes with a potential role for the devel-opment of the insulin resistance syndrome. J Intern Med 2001, 249(2):127-136.
52. Liu D, Graber JH: Quantitative comparison of EST libraries
requires compensation for systematic biases in cDNA gener-ation. BMC Bioinformatics 2006, 7:77.
53. Sunyaev S, Ramensky V, Koch I, Lathe W 3rd, Kondrashov AS, Bork P: Prediction of deleterious human alleles. Hum Mol Genet 2001, 10(6):591-597.
54. Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, Shaw N, Lane CR, Lim EP, Kalyanaraman N, Nemesh J, Ziaugra L, Friedland L, Rolfe A, Warrington J, Lipshutz R, Daley GQ, Lander ES:
Character-ization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet 1999, 22(3):231-238.
55. Vadlamudi RK, Kumar R: p21-activated kinase 1: an emerging
therapeutic target. Cancer Treat Res 2004, 119:77-88.
56. Fuhrmann JC, Kins S, Rostaing P, El Far O, Kirsch J, Sheng M, Triller A, Betz H, Kneussel M: Gephyrin interacts with Dynein light
chains 1 and 2, components of motor protein complexes. J
Neurosci 2002, 22(13):5393-5402.
57. Naisbitt S, Valtschanoff J, Allison DW, Sala C, Kim E, Craig AM, Wein-berg RJ, Sheng M: Interaction of the postsynaptic density-95/
guanylate kinase domain-associated protein complex with a light chain of myosin-V and dynein. J Neurosci 2000, 20(12):4524-4534.
58. Puthalakath H, Villunger A, O'Reilly LA, Beaumont JG, Coultas L, Cheney RE, Huang DC, Strasser A: Bmf: a proapoptotic
BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science 2001, 293(5536):1829-1832.
59. Puthalakath H, Huang DC, O'Reilly LA, King SM, Strasser A: The
proapoptotic activity of the Bcl-2 family member Bim is reg-ulated by interaction with the dynein motor complex. Mol
Cell 1999, 3(3):287-296.
60. Schnorrer F, Bohmann K, Nusslein-Volhard C: The molecular
motor dynein is involved in targeting swallow and bicoid RNA to the anterior pole of Drosophila oocytes. Nat Cell Biol
2000, 2(4):185-190.
61. Fan J, Zhang Q, Tochio H, Li M, Zhang M: Structural basis of
diverse sequence-dependent target recognition by the 8 kDa dynein light chain. J Mol Biol 2001, 306(1):97-108.
62. Nakamura S, Kamihagi K, Satakeda H, Katayama M, Pan H, Okamoto H, Noshiro M, Takahashi K, Yoshihara Y, Shimmei M, Okada Y, Kato Y: Enhancement of SPARC (osteonectin) synthesis in
arthritic cartilage. Increased levels in synovial fluids from patients with rheumatoid arthritis and regulation by growth factors and cytokines in chondrocyte cultures. Arthritis Rheum
1996, 39(4):539-551.
63. Vuorio T, Kahari VM, Black C, Vuorio E: Expression of
osteonec-tin, decorin, and transforming growth factor-beta 1 genes in fibroblasts cultured from patients with systemic sclerosis and morphea. J Rheumatol 1991, 18(2):247-251.
64. Ledda MF, Adris S, Bravo AI, Kairiyama C, Bover L, Chernajovsky Y, Mordoh J, Podhajcer OL: Suppression of SPARC expression by
antisense RNA abrogates the tumorigenicity of human melanoma cells. Nat Med 1997, 3(2):171-176.
65. Ledda F, Bravo AI, Adris S, Bover L, Mordoh J, Podhajcer OL: The
expression of the secreted protein acidic and rich in cysteine (SPARC) is associated with the neoplastic progression of human melanoma. J Invest Dermatol 1997, 108(2):210-214.
66. Thomas R, True LD, Bassuk JA, Lange PH, Vessella RL: Differential
expression of osteonectin/SPARC during human prostate cancer progression. Clin Cancer Res 2000, 6(3):1140-1149.
Publish with BioMed Central and every scientist can read your work free of charge "BioMed Central will be the most significant development for disseminating the results of biomedical researc h in our lifetime."
Sir Paul Nurse, Cancer Research UK
Your research papers will be:
available free of charge to the entire biomedical community peer reviewed and published immediately upon acceptance cited in PubMed and archived on PubMed Central yours — you keep the copyright
Submit your manuscript here:
http://www.biomedcentral.com/info/publishing_adv.asp
BioMedcentral 67. Sage EH: Terms of attachment: SPARC and tumorigenesis.
Nat Med 1997, 3(2):144-146.
68. Zhou X, Tan FK, Reveille JD, Wallis D, Milewicz DM, Ahn C, Wang A, Arnett FC: Association of novel polymorphisms with the
expression of SPARC in normal fibroblasts and with suscep-tibility to scleroderma. Arthritis Rheum 2002, 46(11):2990-2999.
69. Pearson JE, Silman AJ: Risk of cancer in patients with
sclero-derma. Ann Rheum Dis 2003, 62(8):697-699.
70. DbEST: Expressed Sequence Tags database [http://
www.ncbi.nlm.nih.gov/dbEST/]
71. Gouy M, Gautier C, Attimonelli M, Lanave C, di Paola G: ACNUC –
a portable retrieval system for nucleic acid sequence data-bases: logical and physical designs and usage. Comput Appl
Bio-sci 1985, 1(3):167-172.
72. Unigene: organized view of the transcriptome [ftp://
ftp.ncbi.nih.gov/repository/UniGene/]
73. Evoke: expression ontology toolkit [http://www.evocontol
ogy.org/]
74. Ensembl database [http://www.ensembl.org/]
75. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lip-man DJ: Gapped BLAST and PSI-BLAST: a new generation of
protein database search programs. Nucleic Acids Res 1997, 25(17):3389-3402.
76. The Comprehensive R Archive Network [http://stat.cmu.edu/
R/CRAN/]
77. GeM (Genomic Mapping) Website [http://pbil.univ-lyon1.fr/
gem/gem_home.php]