Association entre les variants rares du gène Abraxas et
la susceptibilité au cancer du sein:
une étude cas-témoins
Mémoire
Anne-Laure Renault
Maîtrise en physiologie-endocrinologie
Maître ès sciences (M.Sc.)
Québec, Canada
© Anne-Laure Renault, 2014
III
Résumé du mémoire
Ce mémoire est le fruit d’un travail de deux ans dans le laboratoire de Génomique des Cancer du Dr Simard. La première partie du mémoire est une mise en contexte sur le cancer du sein et sur les deux principales voies de réparation des cassures doubles brins de l’ADN, puis se poursuit par la problématique ayant menée à l’élaboration de mon projet. Cette partie s’achève par le descriptif des techniques de laboratoire et des outils d’analyse utilisés pour ce projet.
En deuxième partie se trouve l’article scientifique rédigé à partir des résultats obtenus pendant mes recherches, avant de se terminer par une conclusion.
V
Table des matières
RESUME DU MEMOIRE ... III
TABLE DES MATIERES ... V
LISTE DES ILLUSTRATIONS ... VII
LISTE DES ABREVIATIONS... IX
REMERCIEMENTS ... XI
AVANT-PROPOS ... XIII
INTRODUCTION ... 1
1. LE CANCER DU SEIN ... 2
2. LA SUSCEPTIBILITE AU CANCER DU SEIN ... 6
2.1 Gènes de susceptibilité à forte pénétrance ... 6
2.3 Gènes de susceptibilité à faible pénétrance ... 7
2.4 La susceptibilité au cancer du sein : un modèle polygénique. ... 8
2.5 Identification de nouveaux gènes de susceptibilité : quels enjeux ? ... 9
3. LES VOIES DE REPARATION DE L’ADN ... 10
3.2.1 Activation et recrutement de BRCA1 ... 11
3.2.2 Réparation des cassures doubles brins ... 11
3.3 Régulation des voies de réparation de l’ADN ... 13
4. BRCA1 PROTÉINE CENTRALE DE LA RECOMBINAISON HOMOLOGUE ... 15
4.1 BRCA1 ... 15 4.2 Le complexe BRCA1/BARD1 ... 16 4.3 Le complexe A ... 17 4.3.1 La protéine Rap80 ... 17 4.3.2 La protéine BRCC36 ... 17 4.3.3 La protéine BRE ... 18 4.3.4 La protéine MERIT40 ... 18 4.3.5 La protéine Abraxas ... 18 4.3.6 Assemblage du complexe A ... 19
4.4 Le complexe B ... 20 4.5 Le complexe C ... 21
5. PROBLÉMATIQUE ... 23
CHAPITRE 1 ... 30
1. RÉSUMÉ DE L’ARTICLE ... 32
DISCUSSION ... 71
PERSPECTIVES ... 75
CONCLUSION ... 79
FIGURES ANNEXES ... 83
BIBLIOGRAPHIE ... 89
VII
Liste des illustrations
Figure 1 : Schéma de l'histologie de la glande mammaire. ... 2
Figure 2: Répartition des cas de cancer du sein dans la population générale ... 4
Figure 3 : Gènes de susceptibilité au cancer du sein connus à ce jour. ... 6
Figure 4 : Fréquence de l’allèle à risque des gènes de susceptibilité au cancer du sein et leur risque relatif dans la population générale ... 8
Figure 5 : Mécanismes potentiellement impliqués dans le risque de cancer du sein et les gènes connus qui y sont associés. ... 9
Figure 6 : Les deux voies majeures de réparation des cassures doubles brins de l'ADN ... 13
Figure 7 : BRCA1 et ses partenaires d'interaction lors de la réparation par recombinaison homologue. ... 16
Figure 8 : Composition du complexe A (adapté de Wang 2012) ... 17
Figure 9 : Les protéines du complexe A et leurs domaines d’interaction. ... 19
Figure 10 : Composition du complexe B (adapté de Wang 2012) ... 21
Figure 11 : Composition du complexe C (adapté de Wang 2012) ... 22
Figure 12 : Positionnement des acides aminés dans le plan de Grantham. ... 25
Figure 13 : Calcul du GD et du GV ... 26
Figure 14 : Echelle de risque du modèle AlignGVGD. ... 27
Figure 15 : Séquence nucléotidique des exons où des variants ont été identifiés et séquence protéique correspondante. ... 85
Figure 16 : Schéma du vecteur pcDNA3.1 ... 87
IX
Liste des abréviations
ABR: Abraxas Binding Rap80 domain ADN : Acide DésoxyriboNucléique ANKRD16 : Ankyrin Repeat domain 16 ATM : AtaxiaTelengiectasia Mutated
BRCA1 : Breast Cancer Susceptibility Gene 1 BRCA2 : Breast Cancer Susceptibility Gene 2 BRCA3 : Breast Cancer Susceptibility Gene 3 BRCT : BRCA1 C-Terminal domain
BRIP : BRCA1 Interacting Protein c-terminal helicase 1 BRCC36 : BRCA1/BRCA2 Containing Complex 3 BRE : Brain and Reproductive organ-Expressed BARD1 : BRCA Associated Ring Domain 1
BABAM1 : BRISC And BRCA1 A-complex Member 1 CASP8 : Caspase 8
CHEK2 : Checkpoint Kinase 2 CtIP : C-terminal Interacting Protein
COX11 : Cytochrome c Oxidase assembly homologue 11 DDR : DNA Damage Response
DSB : Double Strand Breaks DUB : DeUbiquination
RE : Récepteur des Estrogènes
FGFR2 : Fibroblast Growth Factor Receptor 2 G2 : Growth cell phase 2
GD : Grantham Deviation GV : Grantham Variation
GWAS : Genome Wide Association Study HR : Homologous Recombination IR : Ionizing Radiations
JAMM/MPN+: JAB1/MPN/Mov34 metalloenzyme M : Mitosis phase
MAP3K1 : Mitogen Activated Protein Kinase Kinase Kinase 1 NEK10 : NIMA-related Kinase 10
NGS : New Generation Sequencing NHEJ : Non-Homologous End Joining PALB2 : Partner and Localizer BRCA2 PTEN : Phosphatase and Tensin Homologue RR : Risque Relatif
RT-QPCR : ReTro-Quantitative Polymerase Chain Reaction S-P-x-F : Serine-Proline-amino acid-Phenylalanine
STK11 : Serine/Threonine kinase 11 TP53 : Tumor Protein 53
TNRC9 : TOX high mobility group box family member 3 THS : Traitements Hormonaux Substitutifs
TOPB : Topoisomerase Binding
UEV : Ubiquitin-conjugating Enzyme Variant UIM : Ubiquitin Interactor Motif
ZMIZ1: Zinc finger, MIZ-type Containing 1 ZNF: Zinc Finger protein
XI
Remerciements
Mes recherches, la rédaction de ce mémoire et de l’article qui sera soumis prochainement ont été possibles grâce à toute une équipe. Je tiens à remercier tout particulièrement le Pr Jacques Simard de m’avoir accordé sa confiance pour mes deux années de maîtrise dans son laboratoire.
Je remercie également les assistants de recherche Martine Tranchant, Stéphane Dubois et Guy Reimnitz d’avoir pris de leur temps pour me former à la paillasse, répondre à mes questions et de m’avoir aidée dans l’acquisition de mes données ; les Dres Penny Soucy et Martine Dumont pour leur aide dans l’élaboration des affiches et dans la rédaction de l’article, et Marie-Cécile Symons pour son soutien et ses encouragements.
Une pensée pour Natalie Paquet de la plateforme de biopuces du CRCHUQ et Sylvie Desjardins de la plateforme de séquençage et de génotypage du CRCHUQ pour leur aide dans la mise en place technique du projet.
Je souhaite également remercier le Dre Fabienne Lesueur et toute l’équipe du Centre International de Recherche sur le Cancer de Lyon pour son accueil et pour m’avoir formée à la technique de HRM.
Je remercie le Dr Sean Tavtigian et les membres du Breast Cancer Family Registry, John L. Hopper, Irene L. Andrulis, Melissa C. Southey et Esther John pour avoir mis à notre disposition les ADNs de 2453 femmes.
Je remercie nos collaborateurs sur le projet pour leur aide et leur expertise lors des essais fonctionnels : les Drs Jean-Yves Masson et Stéphane Gobeil ainsi que leurs assistants de recherche Yan Coulombe et Louis-Jean Bordeleau.
D’un point de vue personnel je remercie ma famille et mes amis de m’avoir soutenue, suivie et encouragée dans mon projet, même à 5000km de distance !
XIII
Avant-propos
Ce mémoire est le fruit d’un an et demi de recherche sur l’identification d’un nouveau gène de susceptibilité au cancer du sein. Il commence par une mise en contexte sur la maladie et sur les voies de réparation de l’ADN pour mieux appréhender la problématique de mon projet qui était de cribler tous les exons du gène Abraxas chez 2453 individus par la technique de « High Resolution Melting curve » (HRM). Après avoir reçu une formation à cette technique par la Dre Fabienne Lesueur (Centre International de Recherche sur le Cancer, Lyon, France), il m’a fallu la mettre au point au CRCHUQ. Suite à l’analyse par HRM j’ai validé la présence de variants par séquençage de Sanger puis déterminé l’impact de ces variants sur la protéine à l’aide d’outils in
silico. Finalement, j’ai réalisé des essais fonctionnels en collaboration avec le Dr Stéphane Gobeil (CRCHUQ)
pour obtenir une lignée cellulaire déficiente en Abraxas, et avec le Dr Jean-Yves Masson et son assistant de recherche Yan Coulombe (Centre de Recherche en Cancérologie du CRCHUQ) pour les essais d’immunofluorescence.
L’article inséré dans la deuxième partie de ce mémoire résume les résultats obtenus pour ce projet par moi-même. M’ont également aidée lors de la rédaction du papier : l’étudiante au doctorat Yosr Hamdi et la Dre Penny Soucy.
Le Breast Cancer Family Registry est également cité pour avoir mis à disposition l’ADN génomique de 2453 femmes et leurs données personnelles (âge, ethnie, centre de recrutement, cas ou contrôle).
XV
« La vie de l’homme dépend de sa volonté ; sans volonté, elle serait
abandonnée au hasard »
2
1. Le cancer du sein
Le cancer du sein est l’une des formes de cancer qui affecte le plus les femmes dans le monde avec 1 000 000 de personnes diagnostiquées positives à la maladie et 400 000 décès par an 1. En 2013, la Société Canadienne du
Cancer estime que 23 800 canadiennes apprendront qu’elles sont atteintes d’un cancer du sein et que 5 000 décèderont des suites de leur maladie 2. Bien que la majorité des personnes affectées par la maladie soient des
femmes, les hommes peuvent eux aussi être touchés. Généralement les hommes qui développent un cancer du sein appartiennent à une famille où le gène BRCA2 est muté.
Il existe différents types de cancer que l’on peut diviser en deux grandes catégories :
- les non invasifs (in situ), localisés uniquement dans la glande mammaire et parmi lesquels on retrouve : le carcinome canalaire : type de cancer du sein le plus fréquent. Il s’agit d’une forme précoce de cancer qui se développe à l’intérieur des canaux de lactation et qui ne se propage pas aux ganglions et à d’autres organes.
le carcinome lobulaire (ou néoplasie lobulaire) : les cellules cancéreuses sont retrouvées dans la partie lobulaire du sein.
- les invasifs (infiltrant) sont une forme de cancer où les cellules cancéreuses ont traversé la paroi du canal de lactation et vont s’infiltrer dans les ganglions ou d’autres organes sous forme de métastases. Dans cette catégorie il est possible d’observer :
un carcinome lobulaire un carcinome canalaire
A B
Figure 1 : Schéma de l'histologie de la glande mammaire.
A. Localisation du carcinome canalaire. B. Localisation du carcinome lobulaire. Sur les schémas : A : Canal galactophore ; B : Lobules ; C : Sinus galactophore ; D : Mamelon ; E : Gras ; F : Muscle pectoral : G : Côte
Le cancer du sein est une maladie complexe car son développement est influencé par un ensemble de facteurs endogènes et exogènes qui interagissent entre eux. Certains facteurs augmentent le risque de développer la maladie :
- l’âge,
- les antécédents personnels de cancer, - les antécédents familiaux de cancer du sein, - la densité mammaire,
- une hyperplasie atypique,
- des mutations dans les gènes BRCA,
- des troubles génétiques rares : syndromes de Li-Fraumeni, de Cowden, de Peutz-Jeghers ou l’Ataxie-télengiectasie,
- être de descendance Ashkénaze,
- les facteurs hormonaux endogènes : apparition des premières menstruations avant 12 ans, ménopause après 50 ans,
- les facteurs hormonaux exogènes : prise tardive de contraceptifs oraux, traitements hormonaux substitutifs (THS) lors de la ménopause,
- les facteurs environnementaux : exposition aux radiations ionisantes (IR) et aux produits chimiques,
- avoir un statut économique élevé,
- les facteurs liés aux habitudes de vie : habitudes alimentaires, sédentarité, cigarette, alcool. Inversement, l’allaitement, la multiparité et une première maternité précoce diminuent le risque de développer la maladie 3,4.
L’âge et l’histoire familiale de cancer du sein sont les premiers facteurs de risque (RR). En effet une femme de 50 ans dont la sœur a développé un cancer du sein possède un risque relatif de 1.8, alors qu’une femme du même âge dont la mère et la sœur ont développé la maladie possède un RR de 17.1. Le risque relatif qu’une femme développe un cancer du sein augmente avec l’âge et avec le nombre d’apparenté qui ont déjà développé la maladie.
Les femmes les plus à risque, c’est-à-dire celles qui ont un risque de 10% ou plus de développer la maladie, sont retrouvées dans 12% de la population 5. D’autre part, la moitié de la population générale a un risque de cancer du
sein inférieur ou égal à 3%. Cette sous-population représente 12% des cas de cancer du sein. Les personnes ayant un risque élevé de cancer du sein sont donc présentes dans une toute petite partie de la population (Figure1). Il est donc important d’identifier les femmes ayant un risque accru de cancer du sein pour améliorer leur diagnostic et leur suivi.
4
Figure 2: Répartition des cas de cancer du sein dans la population générale
Les risques sont définis en valeur logarithmique ; la moyenne du risque pour l’ensemble de la population est déterminée à 1.0. La distribution des individus qui développent un cancer du sein est déplacée vers la droite. La déviation standard décrit l’étendue du risque entre les valeurs hautes et basses dans la population, et donc le potentiel de discrimination des différents niveaux de risque entre individus. (Pharoah et al, Polygenic susceptibility to breast cancer and implications for prevention, Nature Genetics, 2002)
6
2. La susceptibilité au cancer du sein
On sait aujourd’hui que pour expliquer la susceptibilité au cancer les variations génétiques importent plus que le style de vie ou les facteurs environnementaux 6,7. Dans certains gènes la présence de mutations augmente le risque de
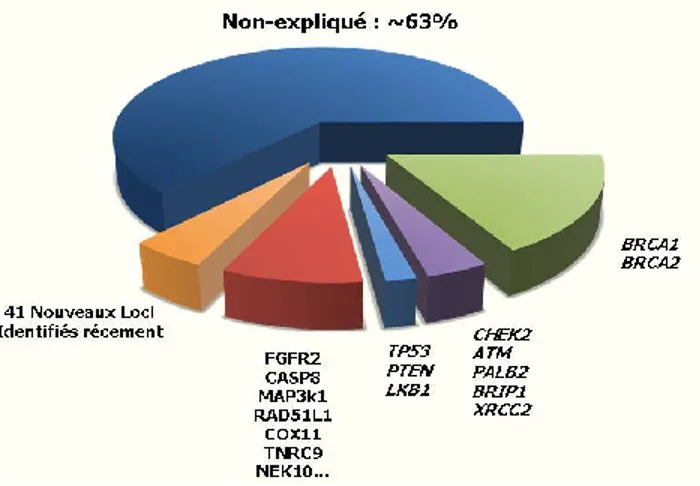
développer un cancer du sein. Ces gènes que l’on nomme « gènes de susceptibilité » expliquent à ce jour environ 35% du risque génétique de développer la maladie 1 (Figure 2).
Figure 3 : Gènes de susceptibilité au cancer du sein connus à ce jour.
Parmi les gènes de susceptibilité au cancer du sein actuellement connus, les gènes de susceptibilité à forte pénétrance (BRCA1, BRCA2, TP53, STK11) permettent d’expliquer 10 à 15% du risque de développer la maladie, les gènes de susceptibilité de pénétrance intermédiaire (CHECK2, ATM, PALB2, BRIP1, XRCC2) représentent environ 3% du risque, les gènes de pénétrance faible (FGFR2, CASP8, MAP3k1, RAD51L1, COX11, TNRC9, NEK10…) expliquent environ 14% du risque et les 41 nouveaux loci identifiés par GWAS permettraient d’expliquer environ 5% du risque. Il reste encore environ 63% du risque de développer la maladie à expliquer.
2.1 Gènes de susceptibilité à forte pénétrance
Les premiers gènes de susceptibilité mis en évidence, Breast Cancer susceptibility gene 1 (BRCA1), en 1994 8, et
Breast Cancer susceptibility gene 2 (BRCA2) en 1995 9, on été identifiés lors d’études de liaison dans des familles à
haut risque de cancer du sein. BRCA1 et BRCA2 sont des gènes à forte pénétrance, ce qui signifie qu’une forte proportion des porteurs de mutations exprime la maladie. La fréquence de l’allèle délétère de ces gènes est très rare (1/800 pour BRCA1 et 1/500 pour BRCA2), mais confère un risque relatif de cancer du sein de 10 à 30 fois celui de la population générale 10(Figure 3).
Tumor Protein p53 (TP53)11 et Serine/Threonine kinase 11 (STK11 ou LKB1) 12 sont aussi des gènes de susceptibilité
à forte pénétrance identifiés par d’autres études de liaison.
2.2 Gènes de susceptibilité à pénétrance modérée
Les études de liaison n’ont jamais révélé l’existence d’un gène de type « BRCA3 » qui aurait pu expliquer une proportion significative des cas de cancer du sein familiaux inexpliqués. Les études d’association ont permis d’identifier de nouveaux gènes de susceptibilité grâce à la mise en évidence de variants rares présents majoritairement chez les cas de cancer du sein13. C’est de cette manière que les gènes à pénétrance modérée ont
été découverts : Phosphatase and Tensin homologue (PTEN) 14,15, Checkpoint kinase 2 (CHEK2) 16, Ataxia
telangiectasia mutated (ATM) 14,17, BRCA1 interacting protein C-terminal helicase 1 (BRIP1) 18 et Partner and
Localizer of BRCA2 (PALB2) 19. La fréquence de l’allèle à risque de chacun de ces gènes est plutôt rare, moins de
1%, sauf dans certaines populations. Par exemple, chez les allemands et les finlandais, la délétion 1100delC dans
CHEK2 a une fréquence de 1% 16.
Les gènes de susceptibilité à pénétrance modérée confèrent un risque relatif pour les porteurs 2 à 4 fois supérieur à celui de la population générale 20 (Figure 3) et expliquent environ 3% du risque de développer un cancer du sein.
2.3 Gènes de susceptibilité à faible pénétrance
Malgré les études de liaison et d’association, les trois-quarts des cas de cancer du sein familiaux restaient inexpliqués. Avec les progrès technologiques et l’augmentation du nombre d’individus dans les cohortes, les Genome
Wide-Association Studies (GWAS) ont permis de cartographier des gènes de susceptibilité à faible pénétrance 21,22
telsque FGFR2, CASP8, TNRC9, MAP3k1, 2q35, 6q25, 5q12, Cox11, NEK10, RAD51L1, 11q13, ZMIZ1, CDKN2A/B,
ZNF35, ANKRD16, ainsi que 72 nouveaux loci 10. Les variants associés au cancer du sein sont des variants
communs dont la fréquence est de plus de 5% et confèrent un risque relatif faible inférieur à 1.5 fois celui de non porteurs 10. Cette sous catégorie de gène de susceptibilité explique environ 14% du risque de développer un cancer
8
Figure 4 : Fréquence de l’allèle à risque des gènes de susceptibilité au cancer du sein et leur risque relatif dans la population générale
(M. Ghoussaini, P. Pharoah, D. Easton, Inherited Genetics Susceptibility to Breast Cancer The Beginning of the End or the End of the
Beginning, The American Journal of Pathology, 2013)
2.4 La susceptibilité au cancer du sein : un modèle polygénique.
Malgré les progrès techniques et l’accès à des cohortes de plus en plus importantes, environ 65% du risque génétique lié au cancer du sein reste à ce jour inexpliqué 1. L’hypothèse actuelle est que le cancer est transmis selon
un model polygénique 6 : chaque variant pris individuellement confère un risque minime, mais l’effet cumulé de ces
variants augmente fortement le risque de développer la maladie 21.
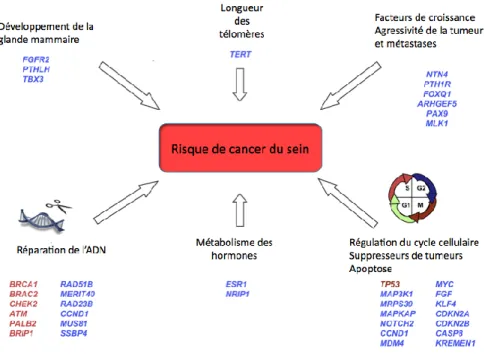
L’une des démarches actuelles pour identifier de nouveaux gènes de susceptibilité est de re-séquencer des gènes candidats sélectionnés selon leur rôle biologique :
- régulation du cycle cellulaire, - voie de réparation de l’ADN, - apoptose,
- carcinogenèse,
- interaction avec BRCA1 ou BRCA2, - stréroïdogenèse,
Figure 5 : Mécanismes potentiellement impliqués dans le risque de cancer du sein et les gènes connus qui y sont associés.
Les gènes en rouge correspondent aux gènes de susceptibilité à forte et moyenne pénétrances, alors que les gènes en bleu correspondent aux gènes à faible pénétrance. (Adapté de Ghoussaini M, Pharoah PDP, Easton DF, Pharoah PD. Inherited Genetic Susceptibility to Breast Cancer. The American Journal of Pathology, 2013)
2.5 Identification de nouveaux gènes de susceptibilité : quels enjeux ?
Le re-séquençage de gènes candidats permettrait d’identifier des variants rares de risque intermédiaire (2 à 4 fois celui de la population générale) qui ne sont ni détectables par études de liaison (risque trop faible) ni par études pangénomiques (fréquence trop faible). Ces variants permettraient d’expliquer une partie du risque génétique lié au cancer du sein actuellement inexpliquée.
L’identification de nouveaux gènes de susceptibilité a pour but d’enrichir l’information clinique et de comprendre les mécanismes cellulaires de carcinogenèse afin de trouver de nouvelles cibles thérapeutiques pour améliorer les soins des patientes. Un autre objectif est la stratification des femmes à risque pour mieux adapter leur dépistage et leur suivi. Les femmes à risque élevé seraient suivies plus précocement et de façon plus fréquente ce qui à long terme réduirait les effets secondaires des traitements et augmenterait la qualité de vie des patientes. Inversement, une stratification des femmes à faible risque diminuerait les coûts de santé publique en réduisant le nombre de mammographies.
10
3. Les voies de réparation de l’ADN
La réparation de l’ADN étant un mécanisme important dans le maintien de l’intégrité du génome et dans la non-prolifération des cellules tumorales nous avons choisi de nous y intéresser plus particulièrement.
Les cellules sont continuellement soumises à des facteurs génotoxiques internes et externes qui entraînent soit des modifications chimiques de l’ADN soit des lésions. Parmi les agents génotoxiques endogènes on retrouve :
- les métabolismes oxydatifs qui produisent des radicaux libres qui se fixent à l’ADN créant alors des cassures,
- les stress à l’ADN qui engendrent des lésions au niveau des télomères, - la recombinaison V(D)J du système immunitaire,
- la recombinaison chromosomique lors de la méiose.
Les facteurs exogènes auxquels nous sommes exposés sont principalement les agents chimiques et les rayonnements infra-rouges et radioactifs 23,24.
Les dommages les plus importants qui peuvent survenir sont les cassures doubles brins de l’ADN (Double Strand
Breaks = DSBs) car elles mettent en péril l’intégrité et la stabilité du génome. Pour répondre à ces altérations et
limiter la prolifération des cellules endommagées, les cellules ont mis en place des mécanismes de réponse aux dommages à l’ADN (DNA Damage Response = DDR) 24. Différents signaux se propagent dans les cellules pour
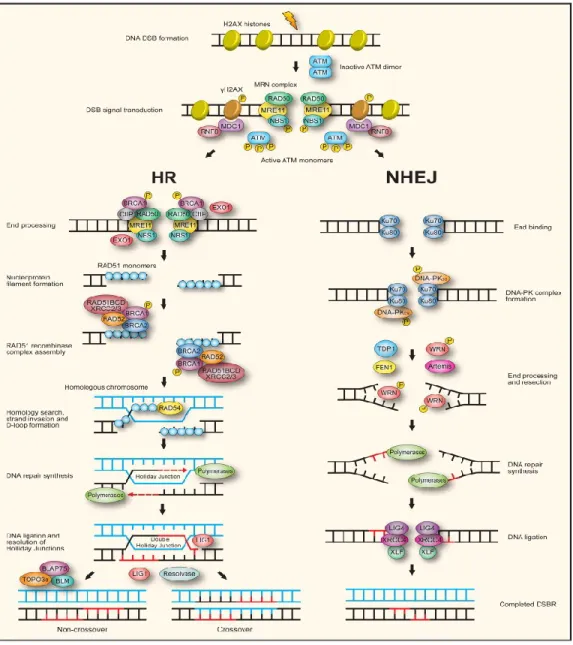
induire soit l’activation des voies de l’apoptose, soit un arrêt du cycle cellulaire pour permettre la réparation des cassures de l’ADN 25. Il existe deux voies majeures de réparation : la recombinaison homologue (Homologous
Recombination = HR) et la réparation par jonction des extrémités non-homologue (Non Homologous End Joining =
NHEJ) 26. Cependant il est important de noter qu’il existe d’autres mécanismes de réparation moins importants,
comme la jonction alternative des extrémités et l’appariement simple brin de l’ADN 26.
3.1 La réparation par Jonction des Extrémités Non Homologues (NHEJ)
Le mécanisme de réparation par NHEJ peut intervenir lors de toutes les phases du cycle cellulaire 23,25,26. C’est un
mécanisme considéré comme simple car peu de protéines sont requises : l’hélicase Ku70/Ku80, l’endonucléase Artemis/DNA-PKcs, les polymérases λ et μ, et le complexe de ligation XRCC4/ADN ligaseIV/XLF 24,26.
L’hélicase Ku70/80, qui ne s’associe qu’aux extrémités de l’ADN, est la première protéine recrutée de part et d’autre du site de cassure. Une fois appariée à l’ADN elle forme un point d’ancrage pour les autres protéines et va renforcer leur liaison à l’ADN. Le complexe Artemis/DNA-PKcs impliqué dans la résection des brins possède des fonctions 5’ et 3’ endonucléases qui lui permettent de couper les extrémités non compatibles de chaque côté de la cassure 23-24.
Enfin, la jonction des brins se fait grâce au complexe de ligation, composé des protéines XRCC4, ADN ligase IV et XLF 25, qui peut liguer des brins compatibles ou incompatibles 23 (Figure 5). Il est important de noter que l’ordre de
liaison des nucléases, des polymérases et de la ligase est variable selon les cassures et selon les extrémités d’une même cassure. Cette variabilité, ainsi que la capacité des protéines à lier l’ADN indépendamment de sa taille et de sa structure, fait de la réparation par NHEJ un mécanisme flexible capable de réparer un large éventail de cassures
23. L’inconvénient majeur de ce modèle de réparation est qu’il crée des erreurs car les bases éliminées lors de la
résection ne seront pas remplacées, provoquant alors une perte d’information génétique. Dans le cas extrême où il y a deux sites de cassures, la ligation peut se faire entre ces deux sites, ce qui entraîne une translocation chromosomique 25,26.
La réparation par NHEJ est donc rapide, simple et flexible mais n’assure totalement pas le maintien de l’intégrité du génome.
3.2 La réparation par Recombinaison Homologue (HR)
La recombinaison homologue est un mécanisme basé sur l’utilisation de séquences complémentaires qui vont servir de modèle à la réplication des brins cassés 25. L’ADN étant répliqué lors des phases S et G2 du cycle cellulaire, la HR
ne peut avoir lieu qu’à ces stades du cycle 25,26. L’activation de cette voie de réparation et la localisation des sites de
cassures sont assurées par un ensemble de protéines de signalisation 26 (Figure 5).
3.2.1 Activation et recrutement de BRCA1
La réparation homologue commence par l’activation et le recrutement de BRCA1 aux sites de cassures suite à une cascade de phosphorylations et de recrutements protéiques 27.
Le complexe MRN formé par les protéines Mre11, Rad50 et Nsb1 est le premier complexe recruté aux sites de cassures. Il est suivi par le recrutement d’ATM qui a pour rôle de phosphoryler les histones H2Ax (dont le nom devient alors γH2Ax), ce qui entraîne un premier signal de recrutement des protéines nécessaires au DDR 28 et un
relâchement de la chromatine 25-27. Parallèlement, ATM va permettre l’activation de BRCA1 de façon directe ou
indirecte 27:
- de façon directe en phosphorylant BRCA1,
- de façon indirecte soit en phosphorylant CHEK2 qui va à son tour phosphoryler BRCA1, soit en phosphorylant CHEK1 qui va fixer le domaine BRCT de BRCA1.
La protéine BRCA1 activée est alors recrutée par différents complexes protéiques dépendamment des fonctions qu’elle doit effectuer.
3.2.2 Réparation des cassures doubles brins
La résection des brins par le complexe MRN/CtIP/BRCA1 permet d’obtenir de l’ADN simple brin qui pourra infiltrer la chromatide sœur 25,29. Pour éviter la dégradation de l’ADN simple brin avant sa réparation, les protéines de réplication
12 A (RPA) vont venir s’y attacher pour former un manteau protecteur. BRCA2 va ensuite acheminer les protéines Rad51 qui vont remplacer les protéines RPA pour permettre la recherche de la séquence complémentaire du brin cassé par invasion de la chromatide sœur. Ce phénomène d’invasion se traduit par la formation de jonction de Holliday 25,26,29. Après réplication des brins cassés on obtient des produits issus soit d’un crossing-over soit d’un non
crossing-over 25.
La réparation par recombinaison homologue permet de réparer les cassures doubles brins d’ADN complexes tout en maintenant l’intégrité du génome. Contrairement à la réparation par NHEJ, la HR basée sur un système de copie n’insère généralement pas d’erreurs dans le génome 25,26. Un autre avantage de cette voie de réparation est sa
capacité à repartir une fourche de réplication à partir d’un simple brin. Néanmoins, la recombinaison homologue peut entraîner une perte d’hétérozygotie si le chromosome paternel remplace le chromosome maternel, ou inversement. On peut également observer des insertions ou délétions si les séquences répétées ne sont pas alignées correctement lors de l’invasion des brins 25.
Figure 6 : Les deux voies majeures de réparation des cassures doubles brins de l'ADN
Les deux voies majeures de réparation des cassures doubles brin de l’ADN sont représentées : à gauche la
recombinaison homologue, possible quand la chromatide sœur est disponible, c'est-à-dire en phase S et G2 ; à droite la réparation par NHEJ réalisable à toutes les étapes du cycle cellulaire. (Dennis Kjølhede Jeppesen et al, DNA repair deficiency in neurodegeneration, Progress in neurobiology,2011)
3.3 Régulation des voies de réparation de l’ADN
Les cellules ont mis en place des mécanismes de régulation à différents niveaux pour favoriser la réparation de l’ADN soit par recombinaison homologue soit par jonction des extrémités non homologues.
Tout d’abord lors du cycle cellulaire, les protéines impliquées dans la recombinaison homologue vont être activées par des protéines kinases S/G2 dépendantes, limitant ainsi leur action aux stades S et G2 du cycle cellulaire 25,30.
14 Par exemple, CtIP qui intervient à la fois dans la NHEJ et la HR ne peut favoriser la résection des brins qu’une fois phosphorylée 31.
Les phénomènes d’ubiquitination et de sumoylation constituent un autre mécanisme de régulation 25. Ils jouent un rôle
important dans le recrutement des protéines de réparation mais également dans leur détachement au niveau des lésions. Par exemple, l’ubiquitination de la protéine Ku70/Ku80 par RNF8 provoque son détachement de la chromatine, ce qui favorise la résection des brins 25.
Un troisième mécanisme de régulation est observé au niveau des chromosomes. Les cohésines, les condensines et les protéines SMC5/6 maintiennent la structure des chromosomes, ce qui confine la réparation aux sites de cassures et limite ainsi la recombinaison entre des séquences différentes 25.
Enfin, un dernier niveau de régulation se produit par les interactions entre BRCA1 et différents complexes protéiques que nous allons détailler plus loin.
Le fonctionnement des ces deux voies majeures de réparation des cassures doubles brins de l’ADN dépend donc d’un ensemble de facteurs cellulaires, protéiques et structuraux.
4. BRCA1 protéine centrale de la recombinaison homologue
Les mutations localisées dans les gènes impliqués dans la stabilité du génome étant responsables de la moitié des cas de cancer du sein 32, nous avons choisi de nous intéresser aux gènes qui interviennent dans la voie de réparation
des cassures doubles brins de l’ADN par le biais de BRCA1.
4.1 BRCA1
BRCA1 est une phosphoprotéine nucléaire appartenant à la famille des suppresseurs de tumeurs et impliquée dans différentes voies de signalisation. Elle contrôle la prolifération cellulaire des tissus hormono-dépendants en se liant au récepteur d’oestrogènes REα, inhibant par exemple la transciption du gène VEGF (Vascular Endothelial Growth
Factor) responsable de l’angiogenèse des endothélium 33. Une autre fonction de BRCA1 est de contrôler le passage
des cellules des phases G2 à M : en cas de dommage à l’ADN, BRCA1 s’associe à CHEK1 pour inhiber l’action de cycline B qui permet le passage des cellules en mitose 34,35. BRCA1 est également essentielle à la réparation des
cassures doubles brins de l’ADN par recombinaison homologue. Elle propage le signal de DDR lorsqu’elle est associée à divers complexes impliqués dans la stabilité du génome.
La capacité de BRCA1 à se lier à REα ainsi que ses fonctions dans le contrôle de la prolifération cellulaire et le maintien de l’intégrité du génome en font une protéine centrale pour comprendre le développement et la propagation du cancer du sein.
D’un point de vue structural, BRCA1 est une protéine de 1863 acides aminés et composée de trois domaines : - un domaine coiled-coil,
- un domaine RING en N-terminal, que l’on retrouve également chez des protéines de régulation du cycle cellulaire, chez les suppresseurs de tumeur et chez les proto-oncogènes.
- deux domaines BRCT en C-terminal, qui permettent l’interaction avec d’autres protéines de façon phospho-dépendante.
BRCA1 se lie à divers complexes indépendants par le biais de ses différents domaines ce qui lui permet d’assurer des rôles spécifiques (Figure 6).
16
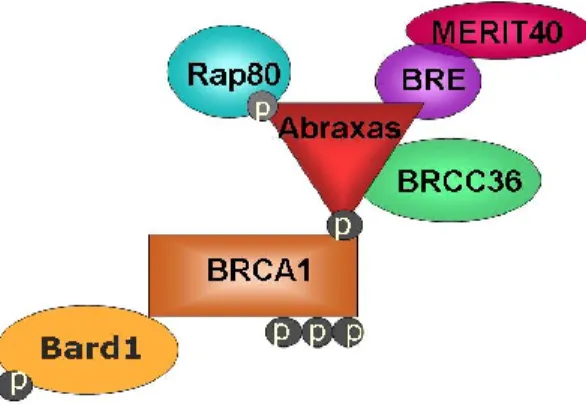
Figure 7 : BRCA1 et ses partenaires d'interaction lors de la réparation par recombinaison homologue.
BRCA1 interargit avec diverses protéines dont le complexe A (CCDC98, Rap80, BRCC36, BRE, MERIT40), le complexe B (BRIP1), le complexe C (CtIP, MRE11, RAD50 et NSB1) ainsi qu’avec la protéine BARD1.
4.2 Le complexe BRCA1/BARD1
Le domaine RING de BRCA1 est connu pour avoir une activité d’ubiquitine ligase (E3) qui facilite le transfert des ubiquitines aux protéines cibles 36. BRCA1 et BARD 1 (BRCA1 Associated RING Domain 1) forment un complexe
stable en interagissant avec leur domaine RING respectif 36-40. BARD1 stimule l’activité d’ubiquitine ligase de BRCA1
au niveau :
- de BRCA1 elle-même, - de CtIP,
- des histones H2AX, ce qui sert de signal à la translocation de protéines aux sites de cassures doubles brin de l’ADN 39.
Il a été démontré que des mutations présentent dans le domaine RING de BRCA1 nuisent à l’activité d’ubiquitine ligase de la protéine et prédisposent au cancer du sein 36.
4.3 Le complexe A
Le complexe A est composé de cinq protéines : Abraxas, Rap80, BRCC36, BRE et MERIT40, toutes nécessaires au bon fonctionnement du complexe (Figure 7). En effet, chacune d’entre elles possède différents domaines protéiques qui leur permettent de former un complexe coordonné et fonctionnel.
Figure 8 : Composition du complexe A (adapté de Wang 2012)
4.3.1 La protéine Rap80
Rap80 ou Ubiquitin Interactor Motif Containing 1 (UIMC1), est une protéine nucléaire qui possède des Ubiquitin-Interacting Motifs (UIMs) permettant la fixation spécifique des chaînes d’ubiquitines de type lysine 63 (K63). Ce type d’ubiquitination est un mécanisme que l’on retrouve au cours de la régulation de la transcription, dans la signalisation des protéines 41 et dans le recrutement des facteurs nécessaires au DDR 30,42,43.
4.3.2 La protéine BRCC36
BRCC36 (BRCA1-BRCA2-Containing Complex subunit 3) est une enzyme de la famille des protéines de dé-ubiquitination (DUBs) qui possède des domaines JAMM/MPN+ activés de façon Zn²+ dépendante 43,44. Ces domaines
possèdent une spécificité de liaison aux chaînes de poly-ubiquitination de type K63, et sont essentiels au rôle de dé-ubiquitination de BRCC36 pour réguler l’accumulation des poly-ubiquitines sur la chromatine 41. BRCC36 possède
18
4.3.3 La protéine BRE
La protéine BRE (Brain and Reproductive organ-Expressed), également nommée BRCC45, est une protéine nucléaire et cytoplasmique exprimée fortement dans le cerveau, les ovaires et les testicules 43. Son expression et ses
fonctions dépendent du contexte dans lequel elle se trouve : modulation des signaux de transduction et réponse au stress en se liant au TNFα, survie et voie apoptotiques en réponse aux dommages à l’ADN. BRE est caractérisée par une sensibilité aux radiations ionisantes qui se traduit par une faible expression de celle-ci. D’un point de vue structural, BRE possède deux Variants Enzymatiques de conjugaison d’Ubiquitine (UEV) qui servent de médiateur de fonction au sein du complexe A 45. L’UEV localisé en C-terminal va permettre la liaison avec MERIT40 alors que les
deux motifs semblent importants dans la liaison avec Abraxas et BRCC36 44 (Figure 8)
4.3.4 La protéine MERIT40
La protéine MERIT40 également appelée BABAM1 (BRISC And BRCA1-A Complex Member 1) ou NBA1, intervient dans la résistance des cellules aux IR et dans la régulation du cycle cellulaire 44. D’un point de vue structural
BABAM1 possède en C-terminal un motif proline-x-x-arginine (PXXR) qui lui permet d’interagir de façon directe avec BRE 43. Le gène BABAM1 est déjà identifié comme gène de susceptibilité au cancer du sein à faible pénétrance et
comme gène de modification du risque chez les porteuses de mutations dans BRCA1.
4.3.5 La protéine Abraxas
Abraxas également appelée CCDC98 (Coiled-Coil Domain Containing 98) ou FAM175A (FAMily with sequence similarity 175, member A) est une petite protéine nucléaire composée de trois domaines qui maintiennent la cohésion entre tous les membres du complexe A : en C-terminal, le motif SPxF permet la liaison avec les domaines BRCT de BRCA1 43, au centre de la protéine le domaine coiled-coil interagit avec celui de BRCC36, en N-terminal le domaine
MNP- fixe BRE, et les domaines coiled-coil et MNP- permettent la liaison avec Rap80 43 (Figure 8). Situé sur le
chromosome 4 au niveau du locus q.21, Abraxas est un gène de 24 kb composé de 9 exons codants.
Abraxas possède un homologue nommé Abro1 (Abraxas Brother 1) ou FAM175B (FAMily with sequence similarity 175, member B) situé sur le chromosome 8. Les séquences protéiques d’Abraxas et d’Abro1 sont semblables sauf
que la dernière ne présente pas le domaine SPxF en C-terminal. Cette caractéristique empêche la protéine Abro1 de se lier à BRCA1. De plus, une étude à montrer qu’Abraxas est principalement localisée dans le noyau, en vue d’intervenir dans la réparation des cassures doubles brins de l’ADN, alors que Abro1 est retrouvé majoritairement dans le cytoplasme. À ce jour, aucune explication n’a été apporté sur le rôle de Abro1 et sur le fait que ces protéines se ressemblent presque parfaitement mais ne jouent pas le même rôle.
Figure 9 : Les protéines du complexe A et leurs domaines d’interaction.
Chaque protéine est représentée avec ses domaines : Rap80 composée de ses domaines d’Ubiquitin-Interacting Motifs (UIMs), son domaine Abraxas Interacting with Rap80 (AIR) et ses deux doigts de Zinc en C-terminale ; Abraxas formée par son domaine MNP-, son domaine coiled-coil et son motif Serine-Proline-x-Phenylalanine (SPxF) ; BRCC36 avec ses domaines MNP+ et coiled-coiled-coiled-coil ; BRE composée de ses deux domaines d’Ubiquitin Enzymatic Variant (UEV) ; NBA1 (BABAM1) formée de son motif PxxR et la partie C-terminal de BRCA1 contenant ses deux domaines BRCT. Les interactions entre les domaines des différents membres du complexe A sont représentées par les traits horizontaux.
4.3.6 Assemblage du complexe A
Le recrutement des 5 membres du complexe fait suite à la phosphorylation des histones H2AX par ATM et au recrutement de MDC1. Une fois MDC1 phosphorylé par ATM il y a recrutement de RNF8 et d’Ubc13 aux sites de cassures de l’ADN. RNF8 va générer une poly-ubiquitination de type K63 qui va se lier aux histones γH2AX. Rap80 capte le signal de poly-ubiquitination par le biais de son domaine IUM ce qui entraîne le recrutement d’Abraxas, BRE, BRCC36 et MERIT40. Grâce à sa spécificité de liaison aux poly-ubiquitinations de type K63, RAP80 sert de transducteur de signal au complexe A dans la réponse aux dommages à l’ADN 43.
Abraxas est l’autre protéine essentielle au complexe car elle associe les protéines entre-elles et fait le lien avec BRCA1. Elle est l’élément central du complexe et le médiateur de l’information (Figures 6 et 7). MERIT40 stabilise le complexe et maintient son intégrité grâce à son interaction avec la protéine BRE 28. Il a été démontré que la
diminution de l’expression de ces deux protéines entraîne une diminution de la quantité d’Abraxas, de RAP80 et de BRCC36, probablement suite à leur dégradation par le protéasome 43. Le complexe A peut donc être caractérisé
comme une holoenzyme 44 constituée d’une partie catalytique (BRCC36) activée par des cofacteurs (Rap80,
20
4.3.7 Les fonctions du complexe A
Le complexe A est le premier complexe recruté en réponse aux dommages à l’ADN pour acheminer BRCA1 au niveau des histones γH2AX 46. Bien que le recrutement du complexe A soit essentiel à la recombinaison homologue
ses fonctions sont actuellement males définies. D’un coté, l’activité de ligase 3 de BRCA1 permettrait d’ubiquitiner les histones pour relâcher la chromatine et faciliter l’accès aux autres protéines de réparation, alors que BRCC36 dé-ubiquitinerait les chaines de poly-ubiquitination de type K63 pour maintenir l’équilibre de la chromatine. Deux hypothèses ont été émises concernant l’activité de BRCC36 au sein du complexe. La première est que la dé-ubiquitination abolirait le signal de DDR ce qui limiterait le recrutement des nucléases et régulerait la résection des extrémités cassées de l’ADN 46. Le complexe A serait donc l’élément régulateur de la résection des cassures doubles
brins et éviterait une recombinaison homologue incontrôlée. La seconde hypothèse est que la dé-ubiquitination des histones et des protéines impliquées dans le DDR entraînerait le signal de fin de réparation et donc un retour à la normale de la réplication, de la transcription et de la prolifération cellulaire 32.
Le complexe A est également impliqué dans la régulation du cycle cellulaire lors du passage des points de contrôle de la phase G2 à M 29,44.
Chaque protéine est essentielle au bon fonctionnement du complexe. En effet différentes études ont démontré que les mutations qui empêchent une partie du complexe de se former entraînent l’absence ou la diminution du recrutement de BRCA1 aux sites de dommages 28,46. De plus, les cellules déficientes en une protéine du complexe
présentent une augmentation de leur sensibilité aux IR ainsi qu’une incapacité à interrompre le cycle cellulaire en phase G2 en cas de dommages à l’ADN 29,46.
4.4 Le complexe B
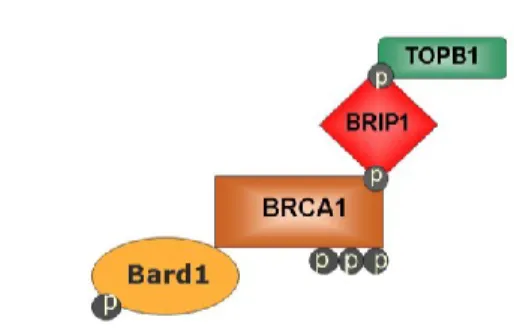
Le complexe B est formé de BRCA1, BRIP1 (BRCA1 Interacting Protein C-terminal helicase 1 également appelé FANCJ ou BACH1) et TOPB1 (Figure 9). BRIP1 est une hélicase appartenant à la famille des RecQ DEAH dont le rôle principal est de fixer et dérouler l’ADN.
Le complexe B permet d’initier la réparation homologue en déroulant l’ADN aux niveaux des cassures doubles brins. Il coordonne également les déplacements de Rad51 sur le nucléofilament 29.
Figure 10 : Composition du complexe B (adapté de Wang 2012)
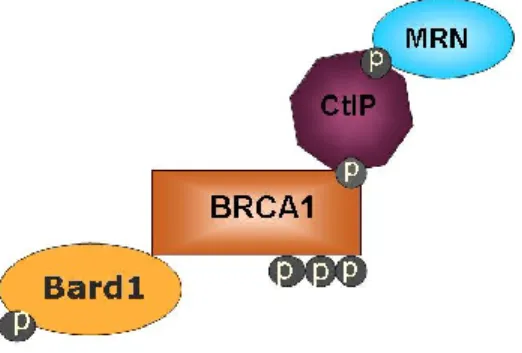
4.5 Le complexe C
Le complexe C est formé par la protéine CtIP (C-terminal Intarcting Protein) et par le complexe MRN, constitué des protéines NBS1, MRE11 (Meiotic REcombination 11) et RAD50 29 (Figure 10).
CtIP est une protéine nucléaire essentielle au déroulement du cycle cellulaire et au maintien de l’intégrité du génome
42. Un des premiers rôles de CtIP est de promouvoir le passage de la phase G1 à S lors du développement
embryonnaire en se liant au promoteur de gènes impliqués dans le passage des points de contrôle du cycle cellulaire
47. Lorsque CtIP interagit avec les protéines de la famille Rb, on observe l’inhibition de certains facteurs de
transcription de manière à limiter la prolifération cellulaire 47.
MRE11 est une protéine nucléaire qui intervient dans la réparation des cassures doubles brins de l’ADN ainsi que dans le maintien de la longueur des télomères. Elle possède une activité 3’-5’ exonucléase mais également une activité 5’-3’ endonucléase ce qui lui permet d’intervenir à la fois dans réparation par NHEJ, lorsqu’elle se complexe avec Rad50, et dans la réparation par HR lorsqu’elle s’associe avec la ligase E3.
NBS1 est associée au syndrome de Nijmegen, caractérisé par une instabilité chromosomique, un retard de la croissance, une déficience immunitaire et une prédisposition au cancer.
Enfin, Rad50 est essentielle à la réparation des cassures doubles brins l’ADN, à l’activation des points de contrôle du cycle cellulaire, à la recombinaison méiotique et au maintien des télomères. Des études ont montré que cette protéine est nécessaire à la viabilité et à la croissance cellulaire 47.
La première étape de formation du complexe C est l’activation de CtIP par phosphorylation 48. Une fois activée, CtIP
se fixe à un domaine BRCT de BRCA1 par reconnaissance du motif SPxF. C’est à ce moment qu’intervient le complexe MRN qui va permettre à BRCA1 d’être acheminée aux sites des cassures doubles brins de l’ADN par le biais d’une interaction CtIP/NSB1 31. CtIP joue donc le rôle de médiateur entre BRCA1 et le complexe MRN,
22 déacétylations de CtIP vont favoriser la résection des extrémités cassées de l’ADN, première étape de la réparation par recombinaison homologue.
Le duo BRCA1/CtIP est également impliqué dans la réparation par jonction des d’extrémités non-homologues lors de la phase G1 27, notamment pendant la translocation chromosomique. La présence de mutations dans les domaines
BRCT de BRCA1 nuit à la liaison avec CtIP, ce qui diminue l’efficacité de la réparation 31.
Figure 11 : Composition du complexe C (adapté de Wang 2012)
Les complexes A, B et C s’associent avec BRCA1 via le domaine BRCT de façon mutuellement exclusive, ce qui permet à chacun de jouer son rôle dans les étapes précédant la réparation des cassures doubles brins de l’ADN par HR. Le but commun de ces trois complexes est de préparer l’ADN pour l’invasion du brin endommagé au niveau de la chromatide sœur.
5. PROBLÉMATIQUE
L’identification de nouveaux gènes de susceptibilité est essentielle pour comprendre les mécanismes cellulaires qui génèrent les tumeurs, pour identifier de nouvelles cibles thérapeutiques et pour améliorer le diagnostic ainsi que le suivi des patientes.
On estime qu’une fraction inexpliquée du risque génétique de cancer du sein est susceptible d'être expliquée par des variants plus rares de risque intermédiaire. Tel que démontré par l’analyse des gènes ATM, CHEK2, BRIP1 et
PALB2, les variants délétères chez ces gènes ne confèrent pas un risque suffisamment élevé, ou sont trop rares,
pour être identifiés par des études de linkage, et sont également trop rares pour être détectés par des GWAS. Très peu de gènes ont été analysés jusqu’à présent dans cette catégorie de variants, et il est donc probable que beaucoup d'autres loci de susceptibilité de ce type sont encore à identifier et pourraient expliquer une proportion importante du risque de cancer du sein.
L'approche la plus efficace pour identifier des gènes de susceptibilité de risque intermédiaire est le criblage de mutations dans le cadre d’études cas-témoins. Cette approche comporte 4 étapes principales:
1. la sélection de gènes candidats en fonction de leur rôle biochimique,
2. le criblage des gènes pour la présence de variants dans la région codante et les jonctions introns-exons dans une série de cas de cancer du sein et de témoins appariés au niveau de l’ethnicité, 3. l’analyse détaillée de la séquence est effectuée selon les variants observés dans le but de les
catégoriser quant à leur probabilité d'interférer avec la fonction de la protéine codée par le gène, 4. la somme des fréquences des variants les plus susceptibles d'altérer la fonction des gènes est
comparée dans les cas de cancer du sein par rapport au contrôle. Si celle-ci est significativement plus élevée dans les cas que chez les contrôles, il est donc probable qu’il s’agisse d’un gène de susceptibilité. Les analyses bio-informatiques qui comparent la distribution gradée des substitutions dans les cas de cancer vs. les contrôles peuvent identifier de nouveaux gènes de susceptibilité en complétant les analyses de mutations délétères.
L’ensemble des gènes codant pour des protéines impliquées dans la réparation des cassures doubles brins de l’ADN sont de bons gènes candidats à la susceptibilité au cancer du sein. C’est pour cette raison que nous avons sélectionné le gène Abraxas codant pour une protéine qui interagit avec BRCA1 et qui est l’élément central du complexe A. De plus, une étude de liaison réalisée dans des familles finlandaises à haut risque de cancer a identifié la mutation R361Q dans 3 familles sur 125 et a été associée au risque de développer la maladie. À noter qu’il a été démontré que l’utilisation de cas familiaux augmente considérablement la puissance de détection de risque conféré par des variants comparativement à une étude de taille similaire mais dont les cas ne pas sélectionnés pour ce critère.
24 L’objectif premier de mon projet de maitrise était de déterminer si Abraxas peut être un gène de susceptibilité au cancer du sein de risque intermédiaire par une étude de type cas/témoins, où les cas ne possèdent pas une histoire familiale de cancer du sein. Pour ce projet l’étude du BCFR s’adaptait parfaitement car elle est composée de 1332 cas sans histoire familiale de cancer du sein, non porteurs de mutations dans les gènes BRCA1/BRCA2, sélectionnés selon l’âge au diagnostic (à ou avant 45 ans), selon l’origine (Caucasien, Asiatique de l’est, Afro-Américain ou Latino) et selon l’origine de leurs grands-parents. Les 1121 témoins ont été choisis de façon à faire correspondre les critères de sélection avec un cas. Le deuxième objectif du projet était de vérifier si la mutation R361Q, identifiée dans les familles finlandaises à haut-risque de cancer du sein, était retrouvée parmi les cas de notre étude.
Approches expérimentales :
Pour identifier les variants, les 9 exons codants d’Abraxas ont été criblés par la méthode de « High Resolution
Melting Curve ». Cette technique qui consiste à identifier des individus porteur d’une variation en fonction de la
température d’association des brins d’ADN d’un amplicon est réalisée en trois étapes : Tour d’abord on réalise une PCR dans laquelle on ajoute du SYBR Green, un agent fluorescent, qui va s’intercaler entre les brins d’ADN à chaque cycle d’amplification. Les PCR ont été mises au point pour les 9 exons d’Abraxas, et l’amplification a été réalisée sur le LightCycler480 de Roche®. Une fois l’amplification terminée, on augmente la température à 95°C pour séparer les brins d’ADN, puis on diminue progressivement la température pour reformer les doubles-brins d’ADN des amplicons. La diminution de la température est progressive pour favoriser la formation des hétéroduplexes, dans le cas où l’individu présenterait une variation dans l’amplicon étudié. C’est dans cette phase que le SYBR Green va jouer son rôle. En effet, pendant la re-formation des doubles brins d’ADN le SYBR Green va s’insérer entre les brins, comme pendant la PCR, et le LightCycler480 va mesurer l’intensité de fluorescence. On observe une diminution drastique de l’intensité de fluorescence au moment de la demi-dénaturation des brins. L’analyse des échantillons permet d’identifier les individus pour lesquels la température d’association des brins est différente, ce qui indique la présence d’un variant chez ces individus pour un amplicon donné. Pour identifier la nature de la variation il faut ensuite séquencer l’amplicon d’intérêt par séquençage de Sanger.
Il s’agit actuellement de l’approche la plus avantageuse en termes de coût-efficacité. En effet, cette méthode simple basée sur la détection de variant en mesurant la température de dissociation des amplicons de PCR d’une région du génome permet d’analyser 384 échantillons en même temps. Les échantillons présentant une température de dissociation différente sont ensuite séquencés par la méthode de Sanger pour identifier le variant. Une fois les conditions de PCR mise au point, il est facile d’analyser 3 plaques de 384 échantillons en une journée. La présence d’un SNP fréquent dans l’amplicon d’intérêt peut représenter un inconvénient en masquant des variants rares qui passeront inaperçu lors de l’analyse. Progressivement cette technique va laisser place au séquençage de nouvelle génération (NGS) dont les coûts baissent année après année.
Les algorithmes Align-GVGD, PolyPhen2 et SIFT nous ont servi pour estimer la pathogénicité des variants faux-sens rares susceptibles d'être délétères. Ces trois outils de prédiction sont basés sur la conservation des protéines au cours de l’évolution. On sait qu’en général plus une protéine, ou un domaine protéique, est conservé au cours de l’évolution plus ses fonctions sont essentielles à la cellule. Partant de ce principe, s’il y a une mutation dans un domaine conservé il y a de forte probabilité qu’elle soit pathogène.
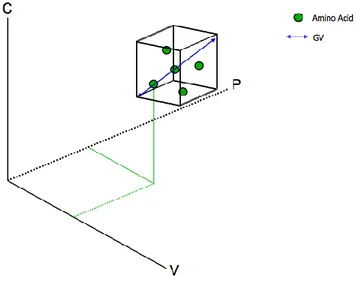
Align GVGD est un outil de prédiction basé sur le modèle mathématique de Grantham qui permet de calculer la distance entre deux acides aminés positionnés dans un plan en fonction de leur composition, de leur polarité et de leur volume (Figure 11). La première étape pour l’utilisation de ce programme a été d’aligner la séquence protéique d’Abraxas provenant de différentes espèces allant de l’Homo sapiens à la Xenopus laevis. Ensuite, pour une position donnée, l’acide aminé de chaque espèce sauf celui de l’Homme est positionné dans le plan de Grantham. On trace alors un cube englobant chaque acide aminé et on calcule la Variation de Grantham (GV) qui correspond à la distance entre les coins opposés du cube (Figure 11).
Figure 12 : Positionnement des acides aminés dans le plan de Grantham.
Dans le plan l’axe C représente la composition, l’axe V le volume et l’axe P la polarité de l’acide aminé.
Puis, dans le même plan, on place l’acide aminé présent dans la séquence de l’Homme à la position d’intérêt et on calcule la Déviation de Grantham (GD) qui est égale à la distance entre l’arête la plus proche du cube et la position de l’acide aminé de l’Homme (Figure 12).
26
Figure 13 : Calcul du GD et du GV
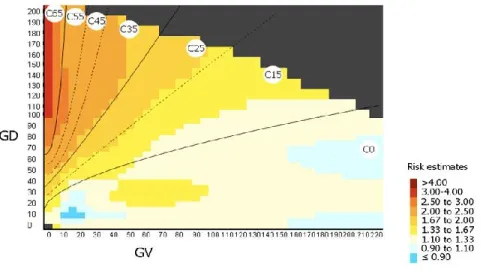
Le rapport GV/GD nous donne une évaluation de l’impact de la variation sur la protéine selon une échelle de risque définie de C0, classe de risque la plus faible, à C65 classe de risque la plus forte (Figure 13) en passant par cinq classes intermédiaires : C15, C25, C35, C45 et C55. Replaçons maintenant ce modèle mathématique dans le contexte de conservation des protéines au cours de l’évolution : si à une position donnée l’acide aminé est conservé chez toutes les espèces utilisées pour l’alignement il n’y aura pas de cube formé dans le plan de Grantham mais uniquement un point, donc GV=0. Si l’acide aminé dans la séquence humaine est le même que pour les autres espèces, alors sa position dans le plan dans Grantham sera la même que celle des autres espèces, et donc GD=0. D’après l’échelle de risque GV/GD=0 correspond à la classe de risque C0, et donc en toute logique l’impact sur la protéine est nul puisque l’acide aminé est conservé. En revanche, si l’acide aminé présent chez l’Homme diffère, le GD variera selon la composition, le volume et la polarité de cet acide aminé. Plus ces trois variables changent plus le GD augmente, et par conséquent la classe de risque aussi. Donc l’impact du variant sur la protéine est de plus en plus pathogène à mesure que les variables diffèrent de l’acide aminé conservé chez les autres espèces.
Pour SIFT j’ai également utilisé l’alignement protéique réalisé pour AlignGVGD. La différence entre ces deux outils de prédiction étant la manière de calculer l’impact du variant sur la protéine et la façon de les classer. En effet, SIFT calcule des probabilités normalisées pour toutes les substitutions possibles à chaque position. Le variant est classé « délétère » si la probabilité est ≤0,05 et « toléré » si la probabilité est 0,05. En ce qui concerne le troisième outil de prédiction, PolyPhen2, l’alignement utilisé est celui proposé par le serveur et les variants sont classés comme « bénin », « possiblement dommageable » et « Probablement dommageable ».
Figure 14 : Echelle de risque du modèle AlignGVGD.
L’échelle est comprise entre C0 (risque le plus faible) et C65 risque le plus élevé, en passant par les risques intermédiaires C15, C25, C35, C45 et C55.
Cette approche devait conduire à l'identification de variants relativement rares qui représentent une catégorie importante de variants à étudier dans le contexte d'une prédisposition génétique car ces variants sont plus susceptibles d'être des variants causals.
Bien que mon projet était la caractérisation des variants rares de la séquence codante d’Abraxas, j’ai approfondie l’étude de certains d’entre eux par des essais fonctionnels.
Pour 2 variants classés dommageables pour la protéine par les outils in silico, j’ai voulu vérifier les prédictions en évaluant leur impact au niveau cellulaire. Nous savons qu’Abraxas joue un rôle central dans la réparation des cassures doubles-brins de l’ADN, donc pour tester l’hypothèse que les variants ont réellement un impact sur la protéine et peuvent influencer la localisation d’Abraxas aux sites de cassures, le laboratoire du Dr Jean-Yves Masson a réalisé des essais d’immunofluorescence. Le but de ces essais est d’induire des cassures doubles-brins à l’ADN par un agent chimique, l’hydroxy-urée, et de regarder si Abraxas est toujours recrutée aux sites de cassures. Les cellules MCF-7 (Michigan Cancer Foundation-7) que nous avons utilisées ont d’abord été « knockées » pour le gène
Abraxas via un shRNA pour que la protéine endogène n’influent pas les résultats observés. Nous avons sélectionné
les cellules qui présentaient un « knock-down » de 85% pour les transfecter avec un vecteur contenant le cDNA complet d’Abraxas et une séquence HA-Flag qui peut être reconnue spécifiquement par un anticorps anti-HA. Les cellules transfectées expriment donc la protéine majoritairement de façon exogène. Grâce à leur séquence HA-Flag nous pouvons marquer ces protéines par un anticorps primaire anti-HA, puis localiser la protéine via un anticorps secondaire anti-anticorps primaire couplé à un fluorochrome. Pour pouvoir observer la localisation d’Abraxas aux sites de cassures doubles-brins de l’ADN, les histones g-H2AX, qui sont un marqueur des sites de cassures, sont
28 révélés en rouges par la même technique que pour Abraxas (Figure 3 de l’article), et Abraxas est marquée en vert. Le DAPI colore le noyaux de façon à vérifier que les protéines sont bien retrouvées dans le noyaux.
J’ai également voulu vérifié si l’un des variant situé au début de l’exon 1 pouvait influencer l’épissage alternatif de l’intron 1. Pour cela j’ai utilisé la technique du mini-gène qui consiste à insérer dans le vecteur pcDNA3.1 les exons et une cinquantaine de paires de bases de l’intron d’intérêt. Le vecteur pcDNA3.1 (Figure Annexe 2) possède un promoteur T7 qui permet d’initier la transcription et une séquence spécifique qui est transcrite en même temps que les exons. Une fois le vecteur construit il a été transfecté dans les cellules HEK293T. Le but est d’ensuite extraire l’ARN total des cellules puis de faire une RT-PCR dirigée sur notre transcrit exogène grâce à la séquence spécifique du vecteur. Enfin, on dépose le produit PCR sur gel pour observer la taille des transcrits en présence ou non du variant d’intérêt.
Ce projet représentait donc une source de variants additionnels, en plus des variants communs et variants fonctionnels de régulation de l’expression génique, à être évalués pour une association avec le cancer du sein dans la population générale.
30
32
1. Résumé de l’article
Environ 35% du risque familial de cancer du sein est expliqué par les allèles de susceptibilité actuellement connus, il reste donc 65% de l’héritabilité inexpliquée ce qui suggère qu’il existe d’autres gènes de susceptibilité à découvrir. Les cassures doubles brins de l’ADN sont l’évènement causant le plus de dommages aux cellules car elles mettent en péril la stabilité et l’intégrité du génome. La majorité des gènes de susceptibilité au cancer du sein déjà identifiés, incluant BRCA1, BRCA2, CHEK2, ATM, BRIP1 et PALB2 sont impliqués dans les voies de réparation des cassures doubles brins de l’ADN. Abraxas est un membre du complexe A qui achemine BRCA1 aux sites de dommages à l’ADN lors de la réparation par recombinaison homologue. Des évidences montrent que des mutations dans la séquence protéique d’Abraxas empêchent le recrutement de BRCA1 aux sites de cassures et augmentent la sensibilité des cellules aux radiations ionisantes. De plus Abraxas a déjà été identifié comme gène de susceptibilité au cancer du sein dans des familles finlandaises à haut risque de cancer du sein. Pour déterminer si Abraxas peut être un gène de susceptibilité à pénétrance intermédiaire, nous avons postulé que des mutations germinales dans ce gène pourraient augmenter le risque de développer un cancer du sein.
Les neufs exons codants d’Abraxas ont été analysés par « High Resolution Melting Curve » chez 1330 cas de cancer du sein précoces et chez 1123 témoins provenant de l’étude du Breast Cancer Family Registry. Seize variants rares on été identifiés : 9 d’entre eux sont synonymes, 4 synonymes et 3 introniques. L’impact des substitutions non-synonymes sur la protéine a été évalué par analyses in silico, et deux d’entre elles ont été classées avec les plus hauts grades de dommages. En dépit de ces résultats, notre étude ne nous permet pas d’associer ces variants avec le risque de cancer du sein.
Notre étude suggère que les mutations rares localisées dans le gène Abraxas n’ont pas d’impact sur la susceptibilité au cancer du sein dans l’étude du Breast Cancer Family Registry.
34
2. Article
L’article actuellement en cours de corrections par les différents collaborateurs sera soumis dans le journal PlosOne
Abraxas (FAM175A) and breast cancer susceptibility: no evidence of association in the Breast Cancer Family Registry
Anne-Laure Renault1, Fabienne Lesueur2, Penny Soucy1, Yosr Hamdi1, Yan Coulombe3, Stéphane
Gobeil4, Florence Le Calvez-Kelm5, Maxime Vallée5, The Breast Cancer Family Registry6,7,8,9,10, John L.
Hopper6, Irene L. Andrulis7, Melissa C. Southey8, Esther M. John9,10, Jean-Yves Masson3, Sean V.
Tavtigian11, Jacques Simard1
1Cancer Genomics Laboratory, CHUQ Research Center, Quebec City, Canada, 2INSERM, U900, Mines
ParisTech, Institut Curie, Paris, France, 3Genome Stability Laboratory, Laval University Cancer
Research Center, Quebec City, Canada, 4CHUQ Research Center, Faculty of Medicine, Laval University,
Quebec City, Canada, 5Genetic Cancer Susceptibility, International Agency for Research on Cancer,
Lyon, France, 6Center for Molecular, Environmental, Genetic and Analytical Epidemiology, School of
Population Health, EGA The University of Melbourne, Victoria, Australia, 7Departement of Molecular
Genetics, Samuel Lunenfeld Research Institute, Mount Sinai Hospital, Toronto, Canada, 8Genetic
Epidemiology Laboratory, The University of Melbourne, Victoria, Australia, 9Cancer Prevention
Institute of California, Fremont, USA, 10Stanford University School of Medicine and Stanford Cancer
Institute, Stanford, USA, 11Department of Oncological Sciences, Huntsman Cancer Institute,
Abstract
Background: Currently, only about 35% of the familial risk of breast cancer can be explained by the
known susceptibility alleles, leaving approximately 65% of the heritability still unexplained and suggesting other susceptibility genes remain to be discovered. DNA double-strand breaks are one of the most damaging events occurring in a cell, as it can disrupt the integrity and stability of the genome. The majority of breast cancer susceptibility genes already identified, including BRCA1,
BRCA2, CHECK2, ATM, BRIP1 and PALB2, are involved in the DNA double-strand breaks repair
pathway. Abraxas is involved in this pathway as a member of the “A complex” which leads BRCA1 to DNA damage sites during homologous recombination repair. Mutations in Abraxas impair BRCA1 recruitment to DNA damage foci and increase cell sensitivity to ionizing radiation. Moreover, a recurrent germline mutation has been reported in Finnish high-risk breast cancer families. To determine if Abraxas could be a breast cancer susceptibility gene in other populations, we conducted a population-based case-control mutation screening study the Breast Cancer Family Registry.
Methodology/Principal finding: The nine coding exons of Abraxas were screened by High
Resolution Melting curve analysis in 1,332 early-onset breast cancer cases and 1,123 controls. Sixteen rare distinct variants were identified: of these one was an in-frame deletion, eight were non-synonymous, four were non-synonymous, two were intronic and one was located in the 5’UTR of the gene. Two variants (p.Thr141Ile and p.Gly39Val) were predicted to affect protein function, and we confirmed experimentally that both of them diminish phosphorylation of h-H2AX, an important DNA damage signaling event.
Conclusion: Overall, likely damaging or neutral variants were evenly represented among cases and
controls suggesting that rare variants in Abraxas may explain only a small proportion of hereditary breast cancer.