Tight binding modeling of halid perovskites
Texte intégral
Figure
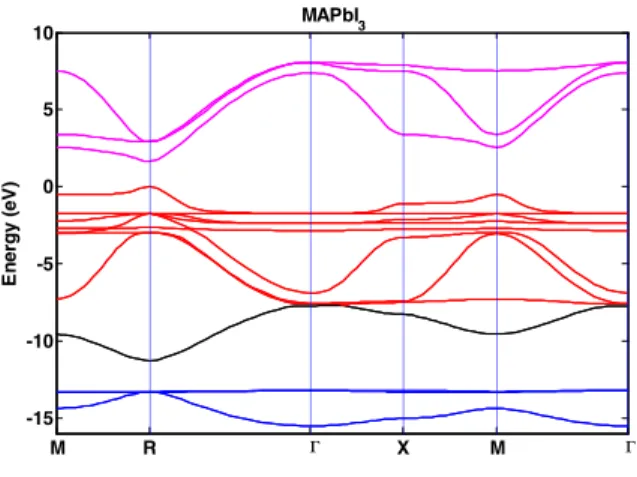
Documents relatifs
The TB parameters are obtained by fitting the LDA+ U bulk band structure to some selected points of the Brillouin zone, with special care in a faithful description of the
5, transferring multi-modal learning on simulated data, as described in that sec- tion, to real data coming from object detection tasks in road traffic, would mean a huge potential
In the previous sections, we showed that a diamond type semiconductor crystal (regular or distorted) can be regarded as a graph ( = (R,£), and described by using
2014 A brief review is given of some basic theorems and methods which give estimates of the spectral limits in systems described within the tight-binding
The objective in this study was to compare the effectiveness of the AP process with that of chemical and Type VII media blasting methods in removing the paint on
The better performance of the SPEEK/PSf-ABIm blend membrane compared to that of the plain SPEEK and Nafion membranes could be attrib- uted, respectively, to the promotion of
Sec22b and Stx1, 3 interact with the lipid transfer proteins E-Syt2 and E-Syt3.. We performed GFP-trap precipitation experiments on lysates
rapport à la déformation. D’autres constantes élastiques telles que le module de cisaillement, le module d'Young, coefficient de Poisson, vitesses du son et la température
