The genetic bases of ecological specialization and the
effects of hybridization in a complex of incipient yeast
species
Thèse
Chris Eberlein
Doctorat en biologie
Philosophiæ doctor (Ph. D.)
Québec, Canada
The genetic bases of ecological
specialization and the effects of
hybridization in a complex of incipient yeast
species
Thèse
Chris Eberlein
Sous la direction de :
Résumé
Il existe des millions d'espèces différentes dans le monde qui ont évolué grâce à des interactions complexes avec leur environnement. La biologie évolutive contemporaine connaît une révolution grâce au séquençage de génomes ainsi qu’au criblage et la manipulation génétique, mais l'objectif reste le même qu'il y a 160 ans: comprendre les mécanismes sous-jacents impliqués dans la spéciation. Cela peut être réalisé en étudiant les mécanismes génétiques impliqués dans l'adaptation locale et la spécialisation écologique lors des premiers événements de spéciation.
L'objectif principal de cette thèse est d'étudier les mécanismes moléculaires qui sous-tendent l'adaptation et la différenciation des populations dans un complexe de jeunes espèces de la levure Saccharomyces paradoxus, naturellement présentes dans les forêts de feuillus d'Amérique du Nord. En utilisant diverses approches, telles que la génomique des populations, la biologie expérimentale, la transcriptomique et le phénotypage à haut débit, nous (1) disséquons les bases génétiques de la spécialisation écologique et (2) étudions les effets de l’hybridation sur la divergence rapide et la spéciation. Nous documentons d’abord que la spécialisation écologique à différentes températures (un phénotype reconnu pour jouer un rôle important dans la divergence de deux principales lignées de S.
paradoxus) est en partie causée par une sélection assouplie avec des compromis.
Les travaux portant sur deux événements d'hybridation inter-espèces démontrent, quant à eux, un croisement entre une espèce hybride et son espèce parentale, ce qui indique que l'hybridation est probablement plus fréquente dans l'évolution des espèces qu'on ne le pensait auparavant.
Nos travaux soulignent l’importance de la différenciation écologique par une sélection relaxée plutôt que par une divergence adaptative de la fixation de mutations bénéfiques. En outre, nos travaux montrent que l'hybridation dans la nature joue probablement un rôle important dans la création d'une nouvelle diversité par le biais de la ségrégation transgressive et que cela peut se répéter par des croisements incluant des espèces hybrides. Des études à venir sur des espèces jeunes et des complexes hybrides permettront de comprendre davantage les bases génétiques de la différenciation des populations, les conséquences de l'hybridation inter-espèces et de sa récurrence dans l'origine des espèces.
Abstract
Millions of different species inhabiting the world have evolved through complex interactions with their environment. Contemporary evolutionary biology is experiencing a revolution in genome sequencing, screening and genetic manipulation technologies. Its aim, however, remains the same as 160 years ago when pioneers like Darwin and Wallace published the first articles about the evolutionary theory: to understand the underlying mechanisms involved in speciation, because such knowledge is key to shed light into species diversification. This can be achieved by studying the genetic mechanisms involved in local adaptation and ecological specialization during early speciation events. The main objective of this work is to investigate the molecular mechanisms underlying adaptation and population differentiation in a young species complex of the budding yeast Saccharomyces paradoxus, naturally found in the North American deciduous forests. Using different approaches, such as population genomics, experimental biology, transcriptomics and high-throughput phenotyping we (1) dissect the genetic bases for ecological specialization and (2) investigate the effect of hybridization in facilitating rapid divergence and speciation. First, we document that the ecological specialization to different temperatures, a phenotype that has been previously shown to play an important role in the divergence of two main S. paradoxus lineages, is partially driven by relaxed selection with trade-offs. Second, with the work on two inter-species hybridization events, we document a back-cross between a hybrid taxa and its parental species, which highlights that hybridization is likely more common in the evolution of species than previously thought.
Our work underlines the importance of ecological differentiation through relaxed selection, rather than adaptive divergence from the fixation of beneficial mutations. Additionally, our findings show that hybridization in nature likely plays an important role in creating new diversity through transgressive segregation, and that this can reiterate through crosses that include hybrid species. Studies on young species and hybrid complexes will enable to further understand the genetic bases of population differentiation and the consequences of inter-species hybridization and its recurrence in the origin of species.
Table of content
RÉSUMÉ ... III ABSTRACT ... IV TABLE OF CONTENT ... V LIST OF TABLES ... VII LIST OF FIGURES ... IX LIST OF ABBREVIATIONS ... XI ACKNOWLEDGEMENTS ... XIII FOREWORD ... XIV
GENERAL INTRODUCTION ... 1
NATURAL SELECTION, POPULATION DIVERGENCE AND SPECIATION ... 2
EVOLUTION AS THE ENGINE FOR DIVERSITY ... 2
NATURAL SELECTION AND THE EVOLUTION OF SPECIES ... 3
POPULATION DIVERGENCE BY THE MEANS OF RANDOM EFFECTS ... 4
NATURAL SELECTION VS. NEUTRAL EVOLUTION AND THEIR DETECTION USING POPULATION DATA ... 5
POPULATION DIFFERENTIATION AT TWO LEVELS:DNA AND RNA ... 8
ECOLOGICAL SPECIATION ... 9
ECOLOGICAL SPECIALIZATION FROM RELAXED SELECTION ... 10
HYBRIDIZATION AND ITS EFFECTS ON LOCAL DIVERSITY ... 11
HYBRIDIZATION IN NATURE ... 11
HYBRIDIZATION FACILITATES SPECIATION ... 14
HYBRID SPECIATION AND ITS RE-OCCURRENCE ... 16
YEASTS IN EVOLUTIONARY BIOLOGY ... 16
THE INCIPIENT SPECIES COMPLEX OF SACCHAROMYCES PARADOXUS ... 18
OVERALL OBJECTIVES OF THIS THESIS ... 20
CHAPTER 1: THE RAPID EVOLUTION OF AN OHNOLOG CONTRIBUTES TO THE ECOLOGICAL SPECIALIZATION OF INCIPIENT YEAST SPECIES ... 22 1.1 RESUMÉ ... 23 1.2 ABSTRACT ... 24 1.3 INTRODUCTION ... 25 1.4 RESULTS ... 28 1.5 DISCUSSION ... 38
1.6 MATERIAL AND METHODS ... 42
1.7 ACKNOWLEDGEMENTS ... 52
1.8 FIGURES ... 53
CHAPTER 2: HYBRIDIZATION IS A RECURRENT EVOLUTIONARY
STIMULUS IN WILD YEAST SPECIATION ... 74
2.1 RESUMÉ ... 75
2.2 ABSTRACT ... 76
2.3 INTRODUCTION ... 77
2.4 RESULTS ... 79
2.5 DISCUSSION ... 89
2.6 MATERIAL AND METHODS ... 91
2.7 ACKNOWLEDGEMENTS ... 109
2.8 FIGURES ... 110
2.9 SUPPLEMENTARY TABLES ... 118
2.10 SUPPLEMENTARY FIGURES ... 128
CONCLUSION ... 153
SUMMARIZING THE PRINCIPAL RESULTS ... 154
GENERAL CONCLUSION ... 154
THE LOSS OF TOLERANCE TO HIGH TEMPERATURE FROM RELAXED SELECTION ... 155
HYBRIDIZATION, HYBRID SPECIATION AND ITS REOCCURRENCE ... 157
PERSPECTIVE ... 159
INFERRING ADAPTIVE EXPRESSION PATTERN FROM THE DETECTION OF CIS-REGULATED GENES ... 160
EVOLUTIONARY-BASED APPROACHES IN COMPARATIVE GENOMICS ... 160
STUDYING ECOLOGICAL SPECIALIZATION USING QTL STUDIES ... 161
OUT OF THE LABORATORY -UNDERSTANDING THE NATURAL HABITAT OF WILD YEAST ... 161
REPEATED HYBRIDIZATION FROM BACK-CROSSES OF HYBRID SPECIES –RARE OR COMMON? ... 162
List of Tables
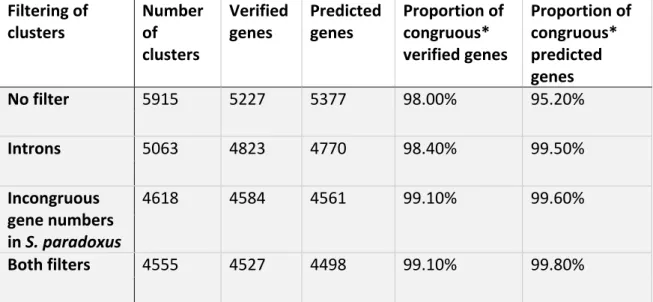
TABLE S 1.9. 1: STRAIN INFORMATION. ... 57 TABLE S 1.9. 2: NUMBER OF CLUSTERS AND SACCHAROMYCES
CEREVISIAE GENES THROUGHOUT THE FILTERING STEPS OF THE ORTHOGROUP INFERENCE PIPELINE. ... 57
TABLE S 1.9. 3: 4555 GENES WITH INFORMATION OF FIXED
SUBSTITUTIONS (USING SPA AS AN OUTGROUP) INCLUDING 76 GENES THAT HAVE SIGNIFICANTLY EVOLVED ASYMMETRICALLY SINCE THE DIVERGENCE FROM SPA (P-VALUE < 0.05). ... 58
TABLE S 1.9. 4: MCDONALD-KREITMAN (MK) TEST OF ALL 4555 GENES
BETWEEN SPB AND SPC. ... 58
TABLE S 1.9. 5: MCDONALD-KREITMAN (MK) TEST BETWEEN SPB AND
SPC REVEALED 124 SIGNIFICANTLY DIFFERENT EVOLVING GENES (FISHER'S EXACT TEST P-VALUE < 0.05). ... 58
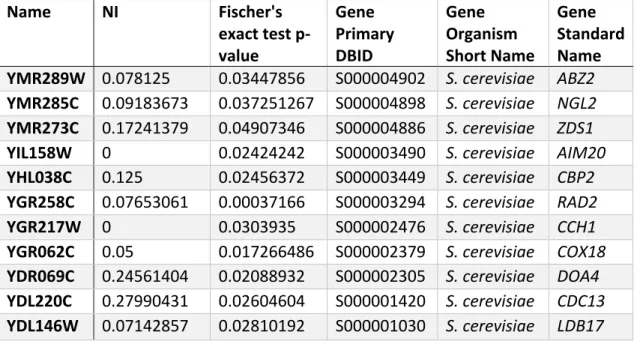
TABLE S 1.9. 6: CANDIDATE GENES UNDER POSITIVE SELECTION WITH
AN NI <1 (FISHER'S EXACT TEST P-VALUE <0.05). ... 59
TABLE S 1.9. 7: GO-TERM ENRICHMENT ANALYSIS USING GORILLA
(EDEN ET AL. 2009) OF THE 76 GENES THAT EVOLVED
SIGNIFICANTLY ASYMMETRIC (FISHER'S EXACT TEST P-VALUE <0.05) DID NOT REVEAL ANY SIGNIFICANT ENRICHMENT AFTER CORRECTING FOR MULTIPLE TESTING. ... 60
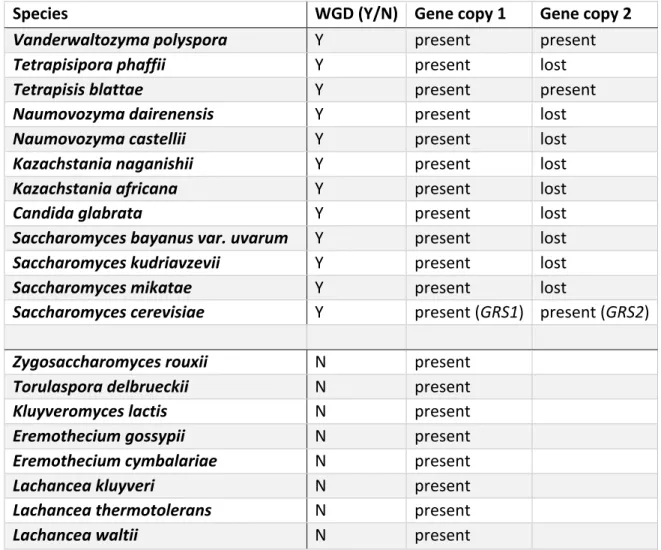
TABLE S 1.9. 8: SYNTENY APPROACH SHOWS THE FREQUENT LOSS OF
THE GRS2-EQUIVALENT PARALOGS IN OTHER POST-WGD SPECIES WITH THE EXCEPTION OF S. CEREVISIAE, VANDERWALTOZYMA AND TETRAPISISPORA BLATTAE... 61
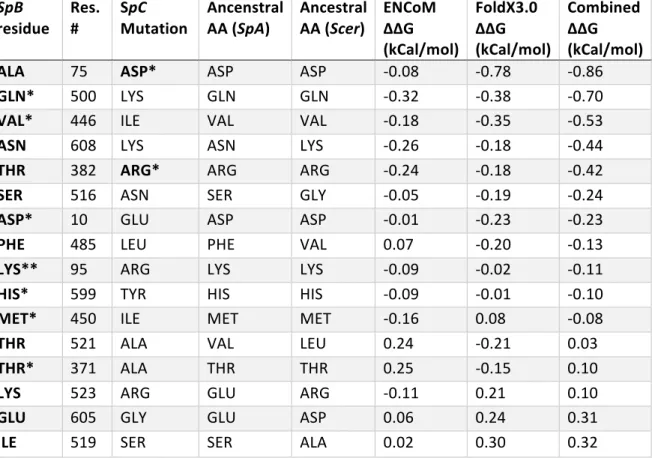
TABLE S 1.9. 9: RESULTS FROM THE PREDICTION OF PROTEIN
STABILITY CHANGE FOR GRS2P FOR RESIDUES THAT HAVE BEEN FIXED BETWEEN SPB AND SPC. ... 62
TABLE S 1.9. 10: COLLECTION OF OLIGONUCLEOTIDES USED. ... 63 TABLE S 1.9. 11: GROWTH MEDIA USED IN THIS STUDY. ... 64 TABLE S 2.9. 1: STRAINS SEQUENCED WITH LONG-READ SEQUENCING
AND DE NOVO ASSEMBLY STATISTICS. ... 118
TABLE S 2.9. 2: FIFTY-ONE FIXED AND INTROGRESSED GENES IN SPC*
WITH SPB ORIGIN. ... 119
TABLE S 2.9. 3: GO ENRICHMENT USING GORILLA (EDEN, ET AL. 2009)
OF THE 51 FIXED AND INTROGRESSED GENES IN SPC* (SPB-LIKE). ... 120
TABLE S 2.9. 4: NINE FIXED AND INTROGRESSED GENES IN SPD THAT
WERE TRANSMITTED FROM SPB TO SPC* TO SPD. THESE GENES OCCUR AT FIXED INTROGRESSED SITES IN SPC* AND WERE USED FOR DATING THE AGE OF THE SPD LINEAGE. ... 121
TABLE S 2.9. 5: SUBSET OF 66 STRAINS FROM THE 5 LINEAGES IN S.
PARADOXUS USED FOR THE ANALYSIS IN BEAST (BOUCKAERT, ET AL. 2014). ... 122
TABLE S 2.9. 6: RESULTS OF DATING THE DIVERGENCE OF S.
PARADOXUS LINEAGES. ... 124
TABLE S 2.9. 7: LIST OF STRAINS AS PART OF THE TRANSCRIPTOMIC
STUDY. ... 125
TABLE S 2.9. 8: ASSEMBLY AND ANNOTATION STATISTICS OF
LINEAGE-SPECIFIC REFERENCE GENOMES FOR THE TRANSCRIPTOMIC
List of Figures
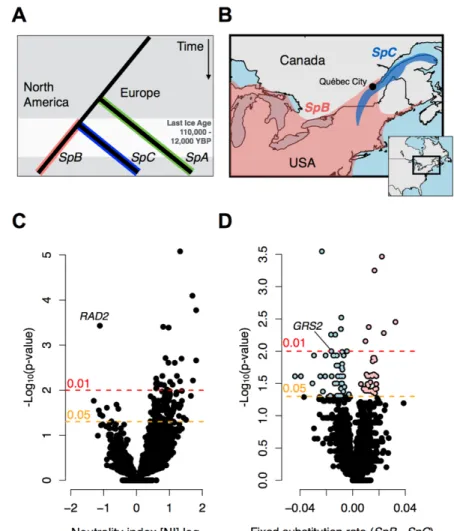
FIGURE 1.8. 1: GEOGRAPHIC DISTRIBUTION AND RAPIDLY EVOLVING
GENES IN THE S. PARADOXUS LINEAGES SPB AND SPC. ... 53
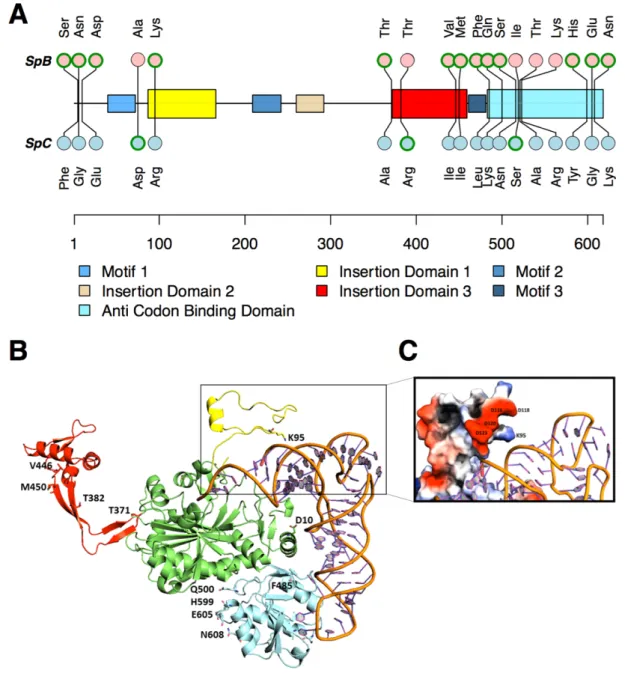
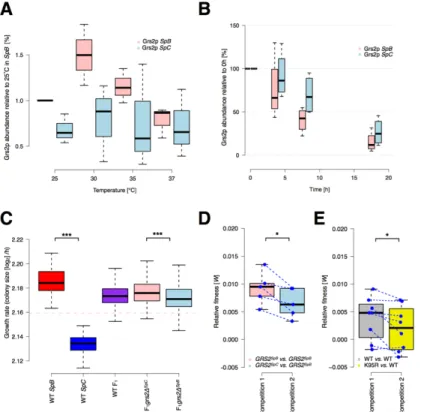
FIGURE 1.8. 2: DOMAINS AND PROTEIN STRUCTURE OF GRS2P. ... 54 FIGURE 1.8. 3: EXPRESSION LEVEL AND CONTRIBUTION TO FITNESS OF
GRS2P IN THE SPB AND SPC LINEAGES. ... 55
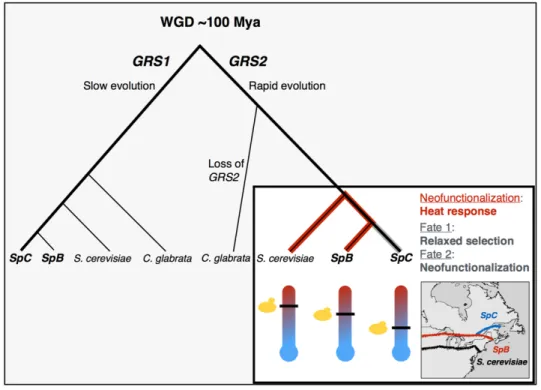
FIGURE 1.8. 4: EVOLUTION OF THE PARALOGS GRS1 AND GRS2 IN S.
PARADOXUS. ... 56
SUPPLEMENTARY FIGURE 1.10. 1: EVOLUTION OF GRS1 AND GRS2 IN S.
PARADOXUS AND S. CEREVISIAE AFTER THE WHOLE-GENOME DUPLICATION (WGD). ... 65
SUPPLEMENTARY FIGURE 1.10. 2: SEQUENCE ALIGNMENTS OF GRS2
OF SPB AND SPC AND THE PREDICTED GRS2PSPB HOMODIMER. .... 66
SUPPLEMENTARY FIGURE 1.10. 3: HIGH GRS1P ABUNDANCE IN SPB
AND SPC. ... 67
SUPPLEMENTARY FIGURE 1.10. 4: LOW ABUNDANCE OF GRS2 IN SPB
AND SPC AS MEASURED BY FLOW CYTOMETRY. ... 68
SUPPLEMENTARY FIGURE 1.10. 5: STRAIN-SPECIFIC GROWTH RATES
ALONG A TEMPERATURE GRADIENT. ... 69
SUPPLEMENTARY FIGURE 1.10. 6: GENE EXPRESSION OF GRS2 AT
30˚C... 70
SUPPLEMENTARY FIGURE 1.10. 7: GRS2P DEGRADATION AT 25˚C IN A
TIME COURSE OF 18H. ... 71
SUPPLEMENTARY FIGURE 1.10. 8: GROWTH RATE OF PARENTAL (SPB
AND SPC) AND HYBRID STRAINS AND THEIR RECIPROCAL
HEMIZYGOTES F1GRS2 SPC AND F1GRS2 SPB. ... 72
SUPPLEMENTARY FIGURE 1.10. 9: COMPETITION ASSAYS AT 30˚C. ... 73 FIGURE 2.8. 1: POPULATION STRUCTURE OF SACCHAROMYCES
PARADOXUS IN NORTH AMERICA. ... 110
FIGURE 2.8. 2: GENOME REARRANGEMENTS SUPPORT THAT SPD
RESULTS FROM THE BACKCROSS OF THE HYBRID SPECIES SPC* WITH ITS PARENT SPB. ... 113
FIGURE 2.8. 3: GENOME-WIDE PATTERN OF INTROGRESSION IN THE
YOUNG AND OLD HYBRIDS. ... 114
FIGURE 2.8. 4: PHENOTYPIC DIVERGENCE AND REPRODUCTIVE
ISOLATION OF SPD. ... 116
SUPPLEMENTARY FIGURE 2.10. 2: PRINCIPAL COMPONENT ANALYSIS
(PCA) FROM GENOME-WIDE SNP DATA DISTINGUISHES THE 5 MAIN S. PARADOXUS GROUPS. ... 129
SUPPLEMENTARY FIGURE 2.10. 3: NUCLEOTIDE DIVERSITY WITHIN AND
SUPPLEMENTARY FIGURE 2.10. 4: SUB-POPULATION STRUCTURE
WITHIN THE SPB LINEAGE. ... 131
SUPPLEMENTARY FIGURE 2.10. 6: THE OBSERVED AND THE FITTED
VALUES OF F4 STATISTICS OBTAINED FOR THE 15 MODELS. ... 133
SUPPLEMENTARY FIGURE 2.10. 7: RANKING OF THE 15 MODELS BASED
ON THEIR FIT TO THE DATA, WHERE ONLY SEQUENCES FROM SPD1 STRAINS (LEFT PLOT) OR SPD2 STRAINS (RIGHT PLOT) WERE
CONSIDERED. ... 134
SUPPLEMENTARY FIGURE 2.10. 9: THE OBSERVED AND THE FITTED
VALUES OF F4 STATISTICS OBTAINED FOR THE 15 MODELS WITH SPD2 STRAINS ONLY. ... 136
SUPPLEMENTARY FIGURE 2.10. 10: SELECTION OF THE BEST TREE
TOPOLOGIES DESCRIBING GENOMIC REARRANGEMENTS USING A MAXIMAL PARSIMONY CRITERION. ... 137
SUPPLEMENTARY FIGURE 2.10. 11: GENES IN SPC* INTROGRESSED
FROM SPB ARE ENRICHMENT FOR THE BIOLOGICAL PROCESS RESPONSE TO AMINO ACID. ... 138
SUPPLEMENTARY FIGURE 2.10. 13: CORRELATIONS BETWEEN COLONY
GROWTH TRAITS AUC, ECS AND MS. ... 140
SUPPLEMENTARY FIGURE 2.10. 14: MULTIPLE FACTOR ANALYSIS (MFA)
PERFORMED ON COMBINED AUC AND MS VALUES. ... 141
SUPPLEMENTARY FIGURE 2.10. 15: PRINCIPAL COMPONENT ANALYSIS
(PCA) PERFORMED ON (A) AUC AND (B) MS VALUES. ... 142
SUPPLEMENTARY FIGURE 2.10. 16: LINEAR DISCRIMINANT ANALYSIS
(LDA) PERFORMED ON (A) AUC AND (B) MS VALUES. ... 143
SUPPLEMENTARY FIGURE 2.10. 17: GROWTH COMPARISON PER
LINEAGE FOR THE (A) AUC AND (B) MS TRAITS. ... 144
SUPPLEMENTARY FIGURE 2.10. 18: DE NOVO ASSEMBLY OF
LINEAGE-SPECIFIC REFERENCE GENOMES FOR MAPPING GENOME-WIDE EXPRESSION DATA. ... 146
SUPPLEMENTARY FIGURE 2.10. 19: AVERAGE GENOME-WIDE
EXPRESSION PROFILES FOR 46 STRAINS FROM ALL MAIN
LINEAGES OF S. PARADOXUS. ... 147
SUPPLEMENTARY FIGURE 2.10. 20: GROUPING ACCORDING TO
PRINCIPAL COMPONENT ANALYSIS OF STRAINS BASED ON
EXPRESSION LEVELS OF 5,160 GENES. ... 148
SUPPLEMENTARY FIGURE 2.10. 21: PRINCIPAL COMPONENT ANALYSIS
SPLITS THE MAIN LINEAGES INTO INDEPENDENT CLUSTERS. ... 149
SUPPLEMENTARY FIGURE 2.10. 22: PAIRWISE COMPARISON OF GENE
EXPRESSION BETWEEN THE LINEAGES SPB, SPC, SPD1 AND SPD2. ... 150
SUPPLEMENTARY FIGURE 2.10. 23: DETECTION OF GENOMIC
List of Abbreviations
AUC Area Under the Curve
bp, kb Base pair, kilo bases
°C Celsius
DMI Dobzhansky-Muller Incompatibilities
ECS Endpoint Colony Size
GWAS Genome-Wide Association Study
h Hour
kCal Kilo calories
log Logarithm
OD Optical density
MK test McDonald-Kreitman test
Mol Unit of amount of substance
MS Mean Slope
Mya Million years ago
PC Principal Component
PCA Principal Component Analysis
QTL Quantitative Trait Locus
SC Synthetic complete (media)
SpA Saccharomyces paradoxus lineage A
SpB Saccharomyces paradoxus lineage B
SpBf Group of SpB that possess a chromosomal
fusion
SpC Saccharomyces paradoxus lineage C
SpC* Saccharomyces paradoxus lineage C*
SpD Saccharomyces paradoxus lineage D
Scer Saccharomyces cerevisiae
WGD Whole Genome Duplication
YBP Years Before Present
‘In a Champagne Supernova in the Sky.’
Acknowledgements
My greatest appreciation goes to Christian Landry, who showed himself as an outstanding group leader, supervisor and friend. I had the chance to participate in exciting research using cutting-edge tools and technologies, while receiving the support of a kind that every Ph.D. student should receive. Thank you Christian!
I am grateful for the help and advice from my committee members Julie Turgeon, Nadia Aubin-Horth, Louis Bernatchez, Juan Carlos Villarreal A. and Jesse Shapiro. Good teachers and advisers show a student where to look and not what to see - Thanks!
In addition, I thank the whole Landry-Lab team! Having so many people with different backgrounds and ideas around has created this outstanding environment of friendship and inspiration, from where I benefited a lot during my last 4 years. Special thanks go to the co-authors and collaborators, who have enabled me to touch different fields of research that were completely new to me.
Special thanks go to my family and close friends. I would not be here finishing my Ph.D. without them. Through many years of studies, I count almost 12 years, and in times of frustration, they have been there supporting me. Thank you TimTim, Bruni, Joerch, uncle, aunt, granny, Maxine, Jonas, Hanna, Nils, Johannes, Françis and Madame Chapeau.
And there is something else. I did not start my Ph.D. in 2014, it started in 2012 in Helsinki, Finland. There are certain people that deserve special thanks for a time that was filled with sadness and endless doubts. Thank you Scott mcCairns, Leena Kaana, Aili Pyhälä and Luka Chevola. And Dominik Begerow; without you I would not be where I am now.
Foreword
This thesis comprises two experimental chapters, chapter 1 and chapter 2. It is rendered by a general introduction and a conclusion. Chapter 1 was published in Molecular Biology and Evolution in 2017. Chapter 2 was first submitted to
Nature Communications in October 2018. It was then re-submitted to Nature Communications with minor modifications in January 2019.
The published chapter 1 can be found under the following reference:
Eberlein C, Nielly-Thibault L, Maaroufi H, Dube AK, Leducq JB, Charron G, Landry CR. 2017. The rapid evolution of an ohnolog contributes to the ecological specialization of incipient yeast species. Molecular Biology and Evolution 34:2173-2186.
C.R.L., L.N.T., J.B.L., C.E., and G.C. planned the experiments. L.N.T. and C.E. performed the bioinformatics analysis on whole-genome data. C.E. performed the cloning and fitness assays. C.E and A.K.D. performed the western blots. H.M. performed the analysis on the protein structure. C.E. wrote the paper with contributions from L.N.T., G.C., J.B.L., A.K.D., H.M. and C.R.L.
The chapter 2, re-submitted to Nature Communications in January 2019, can be traced back with the submission ID: NCOMMS-18-31543-T. The reference of this manuscript is:
Eberlein C., Hénault M., Fijarczyk A., Charron G., Bouvier M., Kohn L.M., Anderson J., Landry C.R. Hybridization is a recurrent evolutionary stimulus in wild yeast speciation.
C.E. and C.R.L. planned this study. C.E., A.F., M.H. and M.B. performed the population genomics analysis. C.E. and M.B. performed the transcriptomic analysis. M.H. performed the phenotypic screen and the long-read sequencing analysis. G.C. performed the reproductive isolation experiment. C.E. wrote the
paper with contributions from M.H., A.F., G.C., M.B., L.K., J.A. and C.R.L. First authorship is shared between C.E. and M.H.
During this thesis, I published (co-authored) four more scientific articles. These articles are not part of this thesis (* first author):
Hénault M*, Eberlein C*, Charron G, Durand É, Nielly-Thibault L, Martin H, Landry CR. 2017. Yeast Population Genomics Goes Wild: The Case of
Saccharomyces paradoxus. In. Cham: Springer International Publishing. p.
1-24.
Leducq J-B*, Nielly-Thibault L*, Charron G*, Eberlein C, Verta J-P, Samani P, Sylvester K, Hittinger CT, Bell G, Landry CR. 2016. Speciation driven by hybridization and chromosomal plasticity in a wild yeast. Nature Microbiology 1:15003.
Bueker B*, Eberlein C*, Gladieux P, Schaefer A, Snirc A, Bennett DJ, Begerow D, Hood ME, Giraud T. 2016. Distribution and population structure of the anther smut Microbotryum silenes-acaulis parasitizing an arctic-alpine plant. Mol Ecol 25:811-824.
Eberlein C*, Leducq JB, Landry CR. 2015. The genomics of wild yeast populations sheds light on the domestication of man's best (micro) friend. Mol Ecol 24:5309-5311.
Natural selection, population divergence and speciation
Evolution as the engine for diversity
Charles Darwin, along with Russel Wallace, were the pioneers in defining the theory of evolution (Darwin and Wallace 1858). In the book ‘The origin of
Species’, Darwin elucidated further that all living organisms are related by
descent and have evolved from earlier forms (Darwin 1859). 160 years later, we are still only at the beginning of fully understanding the underlying evolutionary mechanisms that have driven global diversity and the emergence of 10 to 14 million predicted eukaryotic species (Mora, et al. 2011). The number is even higher for microbial (prokaryotic) species (Locey and Lennon 2016). To understand the emergence of a new species, we need to study the impact and contribution of the evolutionary mechanisms. We distinguish between mutations and migration, two mechanisms that are introducing variation, and genetic drift and natural selection, both acting on variation. In this respect, studying species formation is among the most challenging and important processes in ecology and evolution, where complex interactions between genes and the environment have been shown to play an important role in the origin of biodiversity (Schluter 2001; Rundle and Nosil 2005; Seehausen, et al. 2014). Many question regarding the underlying mechanisms of species formation, such as genomic differentiation, adaptive phenotypic divergence and reproductive isolation remain unanswered and represent some of the biggest challenges of
evolutionary biology in the 21st century (Rice, et al. 2011; Butlin, et al. 2012;
Seehausen, et al. 2014). By focusing on the genetics and genomic underpinnings of diverging populations and their hybrid zones, as well as the nature of adaptation and ‘speciation genes’ (Orr, et al. 2004; Abbott, et al. 2013), we aim to fully understanding the proximate and ultimate causes of speciation. This will eventually enable us to understand and predict changes in biodiversity in the past, present and potentially the future (Papp, et al. 2011; Savolainen, et al. 2013).
Natural selection and the evolution of species
It is debated to what extent speciation is driven by natural selection, reproductive isolation, or both. Charles Darwin himself stated that the key element in the evolution of species is preservation (adaptation) through natural selection (Darwin 1859). Within this thesis, I define a species as a group of living organisms that are able to interbreed and are reproductively isolated from other groups, thus forming stable, independent clusters (Biological species concept; Mayr (1942)). However, defining a species sometimes bears difficulties, which will be further discussed below in the section about hybridization. Natural selection is defined as the process by which individuals of a population that possess the highest survival and reproductive success will be the ones most likely to contribute to successive generations. Over time, their genotype will steadily increase in frequency within the population. Natural selection can be classified into three types, (1) directional selection (when single extreme values of a trait are favored), (2) stabilizing selection (narrowing the phenotypes seen in a population for a non-extreme value) and (3) divergent selection. Under divergent selection, a single population would subdivide into two subpopulations. The subpopulations are selected for the two extreme values of a trait to accommodate for different pressures such as from the environment or mating competition (Gulick 1888). This scenario can be used to demonstrate that speciation happens along a continuum from successful interbreeding individuals in first place, to genetically diverged and reproductively isolated entities that can potentially evolve as new species (Schluter 2001; Rundle and Nosil 2005).
Different forms of speciation have been described. A traditionally more common form of speciation is allopatric speciation, which is speciation by geographic isolation (Mayr 1942) and induced for example by continental drift, glacial cycles or migration events. However, this view has changed a lot during the last 60 years (Mallet 2001; Via 2001), with other forms being similarly plausible such as sympatric speciation, implying the evolution of two species from a single ancestral species in the same geographic region (Coyne 2007), and parapatric
speciation, where geographically adjacent populations evolve into distinct species (Gavrilets, et al. 2000).
It is important to mention that natural selection has a subtype, sexual selection. Sexual selection is defined as the selection through mate-choice, where individuals of one biological sex choose to mate with partners of the opposite sex with specific traits (inter-sexual selection) and compete with individuals of the same sex (intra-sexual selection) (Andersson and Simmons 2006). Reproductive success is therefore limited to the choice of choosing a mating partner or being chosen. And, although there are many well documented cases of sexual selection in different taxa such as birds (Petrie, et al. 1991), mammals (Holekamp, et al. 1996), plants (Moore and Pannell 2011) and fungi (Nieuwenhuis and Aanen 2012), the following introduction will not further cover the topic of sexual selection.
Population divergence by the means of random effects
Genetic differentiation does not necessarily come solely from natural selection. Another key player in the evolution of species is random genetic drift (also known as the Sewall Wright effect) (Wright 1929). While natural selection is the mechanism by which allele frequencies in a population change due to differential survival and reproductive success, random genetic drift is a mechanism that leads to genetic differentiation without selection (Kimura and Crow 1964). Genetic drift describes random changes in allele frequencies in a population, and is highly influenced by population size. In large populations with many individuals contributing to future generations, allele frequencies are more stable and tend to not drift over time.
However, genetic drift will play a larger role in small populations (or when only a few individuals of a population contribute to future generations) and increases the chance that for example low frequency alleles increase in future generations (Wright 1931). Genetic drift is a strong mechanism that can drive the genetic divergence of geographically isolated populations and can therefore play an important role in the evolution of species (Lanfear, et al. 2014). Genetic
drift has especially been associated with bottleneck events or range expansion (Nei, et al. 1975; Barton 1996).
Examples from experimental studies and models that compared the strength of natural selection and genetic drift have shown that in small populations, which experience weak natural selection, genetic drift has a stronger effect in changing and fixing allele frequencies (Lanfear, et al. 2014). In human evolution, facial features were shown to have partially evolved through genetic drift (Ackermann and Cheverud 2004), while the evolution to extreme environments such as Arctic conditions for Greenland Inuit (Fumagalli, et al. 2015) or to high altitudes of Tibetans (Yi, et al. 2010) have resulted from selection.
Natural selection vs. neutral evolution and their detection using population data Presumably most of the species on earth have experienced, at least temporarily, evolution by any type of selection (Orr and Smith 1998; Via 2009). Acquired adaptive phenotypic changes commonly have a polygenic background where many genes are contributing to a phenotypic trait. This is due to the successive fixation of different mutations or variants (variants here refer to standing genetic variation, which, by definition, is the presence of more than one allele at a locus in a population). Each of these single genetic modifications often have small effect sizes with minor contribution to phenotypic changes. Genetic modifications that imply single changes comprising a large effect size are rather rare (Eyre-Walker and Keightley 2007).
The reason why phenotypic changes are often caused by many genetic modifications throughout the genome, can be theoretically explained by the ‘nearly-neutral model of molecular evolution’, proposed by Ohta (1973). The ‘nearly-neutral model of molecular evolution’ is a based on earlier work done by Kimura (1968), who stated in his model (the ‘neutral theory of molecular
evolution’) that the majority of molecular differences within and between species
having strong deleterious effects are abundant, but not retained within populations because of their negative impact on fitness. On the other hand, only a small fraction of mutations is strongly beneficial. The difference in the model by Ohta (1973) is that she proposed that most of the appearing variants are not exactly neutral, but rather slightly deleterious, with some that possess minor beneficial effects, too. That would imply that especially adaptive phenotypic changes between populations or species can sometimes have a genetic basis of large numbers of variants contribution with small effects, which have accumulated and been fixed over time. These characteristics (many variants with very small effect sizes) make studies that aim to link genetic variants to phenotypic traits statistically speaking difficult, as commonly seen in many genome-wide association studies (Ehrenreich, et al. 2010; Kiezun, et al. 2012). These studies often lack of large sampling sizes to actually detect variants with very small contribution to phenotypic traits. Further, complex pleiotropic (genes that influences two or more unrelated phenotypic traits) and epistatic interactions (when the effect of one gene depends on the presence of another) (Arnegard, et al. 2014) often underlie phenotypic changes and contribute to the complexity of the genetic bases of many phenotypic traits.
To distinguish further genetic changes of diverging populations that were driven by selection rather than the fixation of randomly (selectively neutral) occurring mutations, different methods and techniques have been developed (Booker, et al. 2017). With the onset of genetic sequencing in the 1980s, detecting positive selection was mostly based on single gene approaches. A very common method was developed by McDonald and Kreitman (1991), which bases on the assumption that beneficial mutations are more likely preserved in large populations compared to neutral mutations. That would cause a higher number of non-synonymous changes (a nucleotide change that implies the change of an amino acid) over synonymous changes (a nucleotide change that does not imply the change of an amino acid) in regions that bear advantages in a given environment. By comparing two different populations with large effective population sizes (allele frequency changes over consecutive generations are
not influenced by genetic drift), the population experiencing selection would constitute of genes with a higher ratio of fixed variants causing non-synonymous substitution changes over variants causing non-synonymous substitution changes. On the contrary, a high ratio of synonymous substitutions over non-synonymous substitutions could be caused by purifying (negative) selection, which causes the removal of deleterious variants and can lead to stabilizing selection.
Current advances in sequencing technologies lead to single gene approaches being progressively replaced by whole-genome sequence approaches, with the aim to detect genome-wide signatures of positive selection (Turner, et al. 2010; Booker, et al. 2017). A common approach here is the detection of selective sweeps. Selective sweeps are characterized by genetic regions that have a reduction in variation due to strong positive selection of one or more variants in that region. A region that has experienced a selective sweep comprise sites that are generally in high linkage disequilibrium (Smith and Haigh 1974). Linkage disequilibrium is when alleles in proximity are more associated with each other than expected by chance. Selective sweeps are characterized by lower recombination rates since the fitness advantage associated with a particular beneficial mutation causes neighboring alleles to be under selection, too (Smith and Haigh 2009). By detecting these regions in genomes of diverging populations we can define the landscape of adaptation and how selection has contributed to population differentiation (Sabeti, et al. 2007). Salojärvi, et al. (2017) for example conducted a seminal study on this subject, presenting the adaptive landscape of the silver birch. The detected sweeps were enriched for genes associated with environmental responses, encoding for key functions in tree development and physiology.
Interestingly, it has also been shown that selective sweeps can contain higher proportions of slightly deleterious mutations than expected by chance, which spread in a population through the phenomenon called ‘hitchhiking’ (Smith and Haigh 1974). These slightly deleterious sites are genetically linked to highly
beneficial variants and thus are protected from selection. In humans for example, Chun and Fay (2011) identified sites of deleterious mutations that spread through human populations by this effect. Concluding, although positive selection has the effect of increasing the frequency of beneficial alleles in a population, the accumulation and often fixation of deleterious polymorphisms appearing near selective sweeps have, in case of humans, significantly contributed to the high frequency of human disease alleles (Chun and Fay 2011).
Population differentiation at two levels: DNA and RNA
Natural selection takes place on different levels in the organization of the cell. It is broadly accepted that population and species divergence is accompanied by sequence and gene expression level changes (King and Wilson 1975; Bamshad and Wooding 2003; Fraser, et al. 2010; Koenig, et al. 2013; Artieri and Fraser 2014). While studies on protein sequences have a long history dating back to the 60s, gene expression profiles of diverging populations or species have only recently started to accumulate during the last two decades (Wittkopp, et al. 2004; Tirosh, et al. 2009; Fraser 2011; Naranjo, et al. 2015; Kenkel and Matz 2016). Comparative surveys that investigate the evolutionary importance of gene expression differences genome-wide are essential to fully understand the evolutionary processes that contribute to divergence. These studies defined adaptive genome-wide expression levels and allow us to understand the dynamics of genomes during environmental changes within and between species (Bullard, et al. 2010; Fraser 2011; Martin, et al. 2012; Kenkel and Matz 2016; Xu, et al. 2016) complementing genome sequencing approaches.
Divergence in expression levels can be either caused by mutations in the regulatory sequence of the gene itself (cis effect) or by trans-regulatory elements such as transcription factors (trans effects). As discussed above adaptive changes are often polygenic and driven by multiple variants rather than a few or single genes (Bullard, et al. 2010). Using expression data,
identifying coordinated patterns of cis-regulated genes are good indications of positive selection (Bullard, et al. 2010; Martin, et al. 2012). By focusing solely on
cis-regulated genes, their up- or down-regulation can be directly linked to a
gene-specific response to e.g. changing environmental conditions. When such genes are part of the same regulatory system, coordinated up- or down-regulation in expression can indicate an adaptive response, since such coordinated expression changes are less likely to appear under random processes.
Ecological speciation
The genetic mechanisms that underlie phenotypic modifications as a response to natural selection in a given environment, are best studied in populations undergoing ecological speciation. By definition, ecological speciation is the process that results from divergent selection between populations encountering different environments (Van Valen 1976). Ultimately, this will cause the emergence of reproductive barriers as a consequence of divergent selection (Schluter 2001; Rundle and Nosil 2005; Hendry, et al. 2007). With selection acting on traits that are genetically correlated to reproductive isolation, speciation can appear as a by-product of adaptive divergence (Mayr 1947).
There are several good study cases for ecological speciation in nature, such as in plants (Lowry, et al. 2008) or animals (Langerhans, et al. 2007; Nosil, et al. 2008), which have identified the genome-wide pattern of divergence and adaptive radiation that led to reproductive isolation between populations. Ecological speciation is also thought to be the common model underlying diversification in bacteria (Kopac, et al. 2014), which emerge through the adaptation to small ecological niches and persist there while constantly differentiation to other cell lines (strains). Among these study cases, the three-spined stickleback (Gasterosteus aculeatus) system in North America has established itself as a good model to study ecological speciation, with its marine and freshwater populations that correspond to two distinct ecotypes (McKinnon and Rundle 2002; Jones, et al. 2012; Arnegard, et al. 2014). The study by
Colosimo, et al. (2005) showed that the phenotypic transition of a full-plate marine ecotype to a low-plate ecotype was repeatedly driven by selection on a single gene during the occupation of new freshwater environments. Here, genetic changes in the gene Ectodysplasin (Eda) led to the evolution of a plate ecotype, induced by low predation. Even the reverse evolution from a low-plate to a full-low-plate ecotype was demonstrated upon increasing predation (Kitano, et al. 2008). The underlying genetic mechanism for the phenotypic change was selection on standing genetic variation (selection on existing genetic variation), and not the accumulation and fixation of beneficial mutations in the population. These different ecotypes, full- and low-plated, are considered incipient species. Incipient species here means that the different ecotypes of G.
aculeatus represent young species, which persistent in different habitats and
where selection on different traits lead to adaptive divergence that retains them reproductively isolated. At the same time, the evolutionary trade-offs of the low-plate ecotype in freshwater (an environment with low predation) prevents these individuals from re-entering ancestral marine habitats, where they would suffer from low fitness due to higher predation. Since they can still successfully hybridize upon secondary contact, the different ecotypes of G. aculeatus are not fully recognized species (according to the biological species concept).
Ecological specialization from relaxed selection
Being ecologically specialized does not necessarily need to come solely from the effects of natural selection. There are examples where populations and species have become specialists without natural selection being involved in the fixation of alleles. Studies on the eyeless fish Phreatichthys andruzzii, the Mexican cave fish (Calderoni, et al. 2016), has shown its ecological specialization to an environment without light, partially driven by relaxed selection. Because of loss of constraints on the melanopsin and rhodopsin photoreceptors during the occupation of light-less caves, this species has accumulated random mutations in genes linked to vision. However, while the cave fish is now specialized in an environment without light, surviving in habitats outside of caves, where vision is necessary for feeding and coping with
predation, is highly unlikely. Another example of ecological specialization from relaxed selection is the giant panda (Zhao, et al. 2010) feeding on solely eucalyptus leaves. Relaxed selection has caused the accumulation of mutation in the Tas1r1 gene, which encodes the umami taste receptor. In the giant panda, this gene has evolved as a pseudogene (loss of function) and contributed to its specialization in diet that consequently resolves in the trade-off of being restricted to eucalyptus forests. Other examples concern obligate endosymbionts such as Buchnera, which have lost hundreds of genes and become finally dependent on their insect hosts (Moran and Mira 2001).
Hybridization and its effects on local diversity
Hybridization plays a well-recognized role in evolution (Mallet 2005; Seehausen, et al. 2008). It constitutes a potential engine for speciation as well as a unique opportunity for researchers to study reproductive isolation. The following paragraphs will introduce the different aspects of hybridization, how it affects local diversity, enables the appearance of novel phenotypes and promotes speciation.
Hybridization in nature
Hybridization is the process of crossing different varieties or species. It has a long history of application in human agriculture and is still commonly used in breeding programs to enhance or acquire certain combinations of phenotypic traits from different individuals (Konig, et al. 2009; Tester and Langridge 2010). Success or failure of hybridization is strongly anchored with the definition of species. Charles Darwin, who discriminated species by gaps in morphology (= morphological species concept), believed that successful hybridization happens within species, while failure discriminates species (‘sterility within species and
fertility between them’, (Darwin 1859)). However, his concept consequently
failed to explain why dimorphic species (sex-linked morphological characters) could successfully mate and be grouped as one species. On the other hand, Darwin could not explain how morphologically similar forms (two species lacking morphological differences) could fail to hybridize. It took more than half a
century with the work by Ernst Mayr to establish a broadly accepted species concept that describes species as reproductively isolated groups rather than morphologically distinguished entities. Although hybrid sterility represents a key part of his biological species concept (Mayr 1942), his proposal also stated that hybridization between species is possible and cannot be the sole criterion to defining species barriers. Theodosuis Dobzhansky termed the lack of gene exchange between groups of individuals as ‘isolation mechanisms’. He empirically documented that reproductive isolation is a gradual process in the development of two species (Dobzhansky 1936). Successful hybridization was therefore demonstrated to decline with the gradual increase in genetic differentiation from varieties of a species to different species, promoting the appearance of amplifying reproductive isolation mechanisms. In the simplest form, his model of hybrid incompatibilities describes that two single incompatible mutations at different loci, independently fixed in two different and isolated populations, can cause the decrease in fertility upon hybridization (Dobzhansky 1934). This explains why especially young, incipient species, which represent the gray zone along the continuum from populations to species, are often still able to hybridize to some extent, as empirically shown by Roux, et al. (2016).
In contemporary evolutionary biology, the advances in sequencing technology have dramatically improved the detection of ancient, recent and ongoing hybridization, through newly sequenced genomes (e.g. from fossils), thus changing the way we see hybridization (McCoy, et al. 2017; Cahill, et al. 2018; Slon, et al. 2018). Inter-species hybridization, especially between young species, is far more common than most scientists had imagined decades ago. Many of newly sequenced genomes carry imprints from previously unrecognized hybridization events (Mallet 2005; Schumer, et al. 2014). Humans are a good example, where many Ethnic groups possess ancient Neanderthal
variants. These come from hybridization events between Homo
neanderthalensis and Homo sapiens thousands of years ago (McCoy, et al.
2017). In Heliconius butterflies, studies have shown the appearance of repeated inter-species hybridization events that have led to the evolution of several
stable hybrid lineages possessing combined phenotypic traits of parental species (Mavarez, et al. 2006; Heliconius Genome 2012). Inter-species hybridization has also been shown in microorganisms such as the baker’s yeast
Saccharomyces cerevisiae (Marcet-Houben and Gabaldon 2015). And finally,
hybridization has facilitated adaptive radiation for cichlids in the great lakes in Africa after being colonized by a few founder species undergoing hybridization (Seehausen 2004; Meier, et al. 2017).
The extent to which hybridization, ancient or ongoing between species, has impacted evolution has still to be determined. With studies reporting at least 10% of animal species to hybridize, and 25% of plants (Mallet 2005), the field of evolutionary biology and speciation has only just begun to identify the extent of hybridization in nature. Further research will help us understanding the consequences of hybridization from an evolutionary perspective (Mallet 2007; Meier, et al. 2017).
But what makes hybridization so frequent? There are three different fates after hybridization. Firstly, hybridization can accelerate adaptation to new environmental conditions through adaptive introgression (Seehausen 2013; Stukenbrock 2016; Figueiro, et al. 2017). Hybridization enables populations or
species to acquire genetic variation similar as through mutations. In F1 hybrids,
the genetic contribution of both parents is 50:50. In sexually reproducing organisms (2n), the gametes (1n) would harbor admixed parental information from potential crossing-over events that occur during meiosis. During subsequent back-crosses with one parental species, the proportion of the parental genome, with which the hybrid crosses back, will increase over successive generations. Chances that small introgressed fragments from the other parental species (minor parent) will be kept depends on different factors, for example if the introgressed regions possess any adaptive advantage. This can lead to an increase of frequency of such introgressions in subsequent generations. Examples of hybridization affecting adaptive evolution are widespread. Figueiro, et al. (2017) documented that post-speciation admixture
between different cat species has facilitated the adaptive evolution of big cat lineages through the contribution of exchanged genetic material. Here, the authors were able make the link between introgressed regions and adaptive advantages. Other studies showed that the genomes of brown bears possess up to 8.8% of ancient genomic relics from the admixture with polar bears, raising the question of its potential adaptive consequences (Cahill, et al. 2015; Cahill, et al. 2018). However here, introgressions appear only unidirectional from polar into brown bear. The other direction (brown bear into polar bear) is prevented by selection, since brownish-coated bears suffer from low fitness in arctic regions. Furthermore, hybridization has been documented for pathogens, enabling rapid adaptation during the arms race with their host species (Stukenbrock 2016).
The second fate of hybridization is the formation of new, hybrid species (Barton 2001; Mallet 2007; Abbott, et al. 2013; Blanckaert and Bank 2018), which benefits from combined parental or transgressive (extreme) phenotypes. These new features enable the occupancy of potentially novel ecological niches and thus to spatial isolation from the parental species. Geographical isolation can further allow the amplification of reproductive isolation mechanisms to become a new species (Nice, et al. 2013).
Lastly, hybridization can cause the breakdown of species barriers, causing two species to merge back into one (Taylor, et al. 2006; Behm, et al. 2010; Kleindorfer, et al. 2014). The means of hybridization, especially as documented with the first two fates, make such events progressively recognized in the adaptation and evolution of species.
Hybridization facilitates speciation
The possibility that inter-species hybridization enables the emergence of new hybrid species has recently received a lot of attention. However, how the mechanisms of inter-species hybridization can ultimately lead to a new hybrid species are still poorly understood (Schumer, et al. 2015; Blanckaert and Bank 2018; Schumer, Xu, et al. 2018). In addition, there are conflicts in how to define a hybrid species, particularly when determining the minimum proportion of
parental genomes in hybrid species, the degree of reproductive isolation with parental species and their ecological independence (Schumer, et al. 2014; Nieto Feliner, et al. 2017; Schumer, Rosenthal, et al. 2018).
Homoploid hybrid speciation, as defined by Schumer, Rosenthal, et al. (2018), is a type of hybrid speciation that requires the following three conditions to be fulfilled: (1) The new hybrid species must contain the same number of chromosomes as the parental species (no genome duplication). This stands in contrast to many polyploid plant species that have emerged through hybridization (Hegarty and Hiscock 2005). These polyploid hybrid species have doubled their genome as acquired from the two different parental species and subsequently gained an effective reproductive isolation mechanism through the difference in ploidy (Mallet 2007). (2) The hybrid species needs to show reproductive isolation to its parental species. This criterion prevents the persistence of the new hybrid lineage especially in sympatry. (3) The recombination of parental genomes in the hybrid species has created new phenotypic traits that enables it to occupy new ecological niches to ensure long-term persistence.
A detailed view of the genetic mechanisms that enable hybrid speciation has been given by Blanckaert and Bank (2018). In their article, they document that hybrid speciation is possible when several conditions are fulfilled. That is, under a four loci model, which is located on the same chromosome and comprise two
DMI’s in adjacent order, recombination in the F2 breakdown (second offspring
generations after the parental generation) can generate a combination of alleles that cause incompatibilities to both parental species, while the hybrids are compatible and show high fertility among themselves. However, in their model it becomes clear that many factors are important to achieve the opportunity for hybrid speciation, such as the degree of selection, the inheritance pattern of loci (if dominant, codominant or recessive) and population size, while ignoring any ecological factor. Such theoretical models therefore require empirical data for validation.
Hybrid speciation and its re-occurrence
While there have been many discussions about the frequency and mechanisms of hybrid speciation, one might ask instead how frequent recurrent hybrid speciation within the same species complex is. As Blanckaert and Bank (2018) proposed, hybrid speciation can happen for small populations of hybrid individuals within a few hundred generations due to the reciprocal sorting of incompatibilities (Schumer, et al. 2015; Blanckaert and Bank 2018). That was for example shown for ‘Darwin’-finches, in which hybridization caused the emergence of a hybrid species within few generations (Lamichhaney, et al. 2018). However, what are the odds that these species can be involved in a subsequent hybridization event with the possibility to form new hybrid species? What happens when recently evolved incipient hybrid species, cross back with their parents and form a new hybrid population?
Some insights into the mechanism of recurrent hybridization between fully evolved hybrid species have been gained through studies in African cichlids. Here, the colonization of new lakes by at least two young species initiated adaptive radiation. These young species were themselves the result of previous hybridization events and gave rise to new hybrid lineages that took advantage of transgressive phenotypes from recombined parental genomes. These scenarios were continuously repeated in several independent lakes (Joyce, et al. 2011; Keller, et al. 2013; Brawand, et al. 2014; Selz, et al. 2014; Meier, et al. 2017; Meier, et al. 2018). Recurrent hybridization events in these cases were likely possible because of two factors. First, they involved young, relatively recent diverged species, which still had the ability to produce fertile hybrids. Second, the new and unexploited lakes represented a vast of ecological opportunities upon colonization.
Yeasts in evolutionary biology
Our knowledge of population differentiation, speciation and hybridization is biased towards model organisms. Most of the insights into the genetics of early
speciation come from studies with either plants or animals (Meinke, et al. 1998; Schluter 2001; Rundle and Nosil 2005; Seehausen, et al. 2014). For many other taxa such as microbes, very little is known about the genomic modifications as a consequence from changing environments or inter-species hybridization events. For example, eukaryotic microorganisms make a significant part of the world-wide diversity, with 1.3 million of the total 9 million predicted eukaryotic species on earth (Mora, et al. 2011). Prokaryotic species such as bacteria might go into the trillions (Locey and Lennon 2016). Further, microorganisms have an essential role in all ecosystems and are very important in basically all biogeochemical cycling processes (Prosser, et al. 2007). They also offer an outstanding platform for studies on adaptive divergence, facilitated by their fast life cycle, the extensive toolbox of genetic manipulations and controlled monitoring under laboratory conditions, enabling to avoid any effects from varying environmental conditions (Elena and Lenski 2003).
In contrast to well-studied naturally occurring species complexes in animals and plants, experimental evolution studies are dominated by experiments on microorganisms. These have contributed in different ways to the understanding of population divergence and speciation (Barrick, et al. 2009; Barrick and Lenski 2013). Yeast for example play an undisputed role in evolutionary biology and genetics. Saccharomyces cerevisiae entered the light of evolutionary studies in 1996, with being the first eukaryote genome sequenced (Goffeau, et al. 1996), five years before the first genome sequence of humans (Lander, et al. 2001). Now, roughly two decades later, the Saccharomyces sensu stricto species have arguably become the most powerful eukaryotes to study different aspect of evolution, with recent studies on intra-species phenotypic variation and accessibility to more than thousands of sequenced genomes (Peter, et al. 2018), intense resequencing efforts (Yue, et al. 2017) and high-throughput genetic manipulations (Guo, et al. 2018).
Saccharomyces can be split into different groups of species in which some
occurring. S. cerevisiae possesses both, natural populations and others that have undergone massive domestication efforts to fulfill the needs in fermentation processes, responsible for the high variety of alcoholic beverages, or medical and economical purpose. Mankind makes further use of it for the production of biofuel (Buijs, et al. 2013) and secondary metabolites (Siddiqui, et al. 2012). Natural and domesticated strains also coexist in certain environments (Sniegowski, et al. 2002; Almeida, et al. 2015). Almeida, et al. (2015) for example showed that natural and domesticated wine strains coexist in Mediterranean wine yards. Here, genomic data documented asymmetric gene flow from the natural populations into the wine strains, which, suggested by Almeida, et al. (2015), underlines that these natural populations harbor the wild genetic stock of the domesticated wine strains. The domestication of many
Saccharomyces strains world-wide has been driven by different industrial
purposes and created a collection of strains that are genetically diverged (Peter, et al. 2018). Here, the hybridization of such domesticated yeasts is a common procedure in the beer industry, since hybrids can subsequently express new phenotypic variation from admixed parental genotypes and can cause new characteristics during fermentation causing e.g. the change in flavor of beer (Mertens, et al. 2015).
The incipient species complex of Saccharomyces paradoxus
For studying the mechanisms underlying ecological divergence and inter-species hybridization in a natural system, this thesis makes use of the S.
paradoxus system, with some great advantages to study a variety of questions
in evolutionary biology (Hénault, et al. 2017). S. paradoxus, which is genetically the closest sister species to S. cerevisiae, is a naturally occurring yeast that has partially overlapping niches with S. cerevisiae (Charron, Leducq, Bertin, Dubé, et al. 2014). S. paradoxus has never been a target of domestication, which makes it a great system to study evolutionary questions in a natural context. Population genomic approaches have shown substantial inter-species hybridization with S. cerevisiae, where S. cerevisiae has retained sequence fragments from such old hybridisation events (Almeida, et al. 2017; Peter, et al.
2018). Efforts to use S. paradoxus in evolutionary studies have mainly been focused on the European clade. In North America, however, different clades of
S. paradoxus have been used to study different aspects of evolution:
reproductive isolation (Charron, Leducq and Landry 2014), hybrid speciation (Leducq, et al. 2016) and ecological specialization (Eberlein, et al. 2017).
In North American forests, S. paradoxus is mainly associated with maple and oak trees (Sniegowski, et al. 2002; Charron, Leducq, Bertin, Dubé, et al. 2014). Population genomics of North American S. paradoxus lineages has only emerged recently with the study by Leducq, et al. (2016). Here, whole-genome sequencing revealed a separation of the species into two main lineages (respectively lineage SpB and lineage SpC), with its initial division initiated by the Pleistocene glacier ~100,000 years ago (Leducq, et al. 2016). Both lineages have presumably persisted the glacial period in two different refuges (Mississippian and Atlantic refuge), which are known to have supplied shelter for many species during the glacial period (de Lafontaine, et al. 2010; April, et al. 2013). After the glacial decline ~18,000 years ago, the secondary contact zone between SpB and SpC was established in Eastern Québec (Saint Lawrence Valley), Canada. The lineage SpB has its current distribution all over the southern part of the North American continent, while SpC persists in the eastern part of Canada.
The nucleotide divergence based on the study of ~15,000 polymorphisms is 2% between the southern lineage SpB and the eastern lineage SpC (Leducq, et al. 2016). Phenotypic studies have suggested the occurrence of local adaptation, respectively to higher temperature tolerance and freeze-thaw cycle resistance for the SpB lineage (Leducq, et al. 2014) or differences between lineages in the utilization of specific carbon and nitrogen sources (Samani, et al. 2015; Leducq, et al. 2016). In the contact zone of these lineages exists another lineage (SpC*), which was documented as a hybrid between the ancestral lineages SpB and SpC (Leducq, et al. 2016). This hybrid has a mosaic of genomic elements
from both parents and its appearance was traced back to the secondary contact of SpB and SpC after the glacial decline ~18,000 years ago.
Until the publication by Xia, et al. (2017) and following studies by Hénault, et al. (2017), there had been no record of natural hybrids among these three lineages (SpB, SpC and SpC*) in the wild. However, inter-species crosses can be performed under laboratory conditions (Charron, Leducq and Landry 2014). These laboratory hybrids show reduced fertility (measured from spore lethality of inter-lineage crosses) compared to the wild type crosses (crosses of individuals from the same lineage), which is presumably the result of incompatibilities, e.g. chromosomal rearrangements (Charron, Leducq and Landry 2014).
With evidence for reproductive isolation and phenotypic differentiation, the SpB and SpC lineages were described as incipient species (Charron, Leducq and Landry 2014; Leducq, et al. 2016). They persist as stable genetic clusters and thus represent a complex of early speciation. In addition, the hybrid lineage
SpC* was described as a hybrid species (Leducq, et al. 2016), which follows the
definition by Schumer, Rosenthal, et al. (2018).
Apart from SpB, SpC and SpC*, the study of Xia, et al. (2017) identified an unknown lineage (‘Clade d’; later termed SpD), the ancestry of which had not yet been determined. Here, SpD was suggested to be another hybrid lineage. With the likely presence of another hybrid lineage SpD, the North American S.
paradoxus system enables us to study the dynamics of hybridization and its
chances for reoccurrence between incipient species.
Overall objectives of this thesis
The overall objective of this thesis is to study the early divergence of a young (incipient) species complex, to contribute to the underlying genetic mechanisms of ecological specialization and phenotypic divergence. In addition, we elucidate the effects of inter-species hybridization in contributing to genetically new
lineages that take advantage of rapid phenotypic and reproductive isolation from reshuffled parental haplotypes (transgressive segregation).
The thesis is divided into two experimental chapters. The first chapter aimed to study the genetic mechanisms underlying the difference in tolerance to high temperature between two species (SpB, SpC). With their currently lineage-specific distributions in North America, we hypothesized that the tolerance to high temperature has a strong contribution to their lineage-specific distributions (Leducq, et al. 2014). The study was published in 2017 in the Journal Molecular
Biology and Evolution (Eberlein, et al. 2017).
Chapter 2 of this thesis (Eberlein, et al., submitted) documents the effects of inter-species hybridization on local diversity. Here, the aim is to understand the role of inter-species hybridization as a potentially important contributor to global diversity through the evolution of new hybrid species. The thesis uses a previously described hybrid species SpC* (Leducq, et al. 2016) to study the evolutionary dynamic of a hybrid genome that has established itself as an independent species in sympatry with its parental species. Further, this project targets to dissect the role of reoccurring hybridization that involves back-crosses of the hybrid species with its parental species, by using a newly described and largely uncharacterized lineage (SpD) (Xia, et al. 2017). Here, this thesis will give insights into the genomic, transcriptomic and phenotypic fingerprint of these newly emerged hybrid genomes from inter-species hybridization.
Chapter 1: The rapid evolution of an ohnolog contributes to
the ecological specialization of incipient yeast species
1.1 Resumé
L'identification des changements moléculaires qui conduisent à la spécialisation écologique lors de la spéciation est l'un des principaux objectifs de l'évolution moléculaire. Il reste une question à examiner : la spécialisation écologique découle-t-elle strictement des changements adaptatifs et des compromis associés, ou bien découle-t-elle des mutations conditionnellement neutres qui s'accumulent suite au relâchement de la sélection? Nous avons utilisé le séquençage du génome entier, l'annotation du génome et les analyses informatiques pour identifier les gènes qui ont rapidement divergé entre deux espèces naissantes de Saccharomyces paradoxus qui occupent des régions climatiques différentes le long d'un gradient sud-ouest à nord-est. En tant que locus candidats de la spécialisation écologique, nous avons identifié des gènes qui montrent des signatures d'adaptation et des taux accélérés de substitutions d'acides aminés, entraînant une évolution asymétrique entre les lignées. Le gène candidat dans cette étude était GRS2, un gène qui code pour une glycyl-ARNt-synthétase et qui est connue pour être induite de manière transcriptionnelle lors d’un stress thermique chez l'espèce sœur modèle S.
cerevisiae. La modélisation moléculaire, l'analyse de l'expression et la mesure
du taux de croissance suggèrent que l'évolution accélérée de ce gène dans la lignée nordique pourrait être causée par le relâchement de la sélection. GRS2 est apparu pendant la duplication du génome entier survenue il y a 100 millions d'années dans la lignée de levure. Bien que son ohnologue GRS1 ait été préservé dans toutes les espèces formées après la duplication, GRS2 a souvent été perdu et évolue rapidement, ce qui suggère que le sort de cet ohnologue est incertain. Nos résultats suggèrent que l'évolution asymétrique de
GRS2 entre les deux espèces naissantes de S. paradoxus contribue à leur
répartition géographique restreinte et que leur spécialisation écologique découle au moins en partie d'une sélection relâchée plutôt que d'un compromis moléculaire résultant de l'adaptation.