9 6 6 f
fijJlfJf
FACULTE DES SCIENCESTHESE
PRESENTEE A L'ECOLE DES GRADUES DE L'UNIVERSITE LAVAL
POUR L'OBTENTION DU GRADE DE MAITRISE ES SCIENCES
PAR
DENIS MAHEUX
Bachelier ès Sciences, Université Laval
THERMODYNAMIQUE DE LA POLYMERISATION
DU TETRAHYDROFURANNE
EN SOLUTION DANS LE BENZENE,
En reconnaissance à mes parents
Ill
REMERCIEMENTS
Je tiens è exprimer, en premier lieu, ma plus pro fonde reconnaissance à mon directeur de thèse, M. le professeur J. Léonard, pour ses précieux conseils, ses encouragements sou tenus et ses critiques constructives. Le Dr. Léonard restera pour moi, non seulement un scientifique accompli, mais un ami sincère et dévoué.
Je profite également de 1'occasion qui m'est donnée pour remercier le Département de Chimie pour son aide financière.
Je m'en voudrais ici d'oublier l'aide que m'ont ap portée mes camarades de laboratoire dont j'ai toujours apprécié
la franche cordialité et la bonne humeur.
Je remercie vivement MM. F. Bouchard, M. Favennek, G. Marier et R. Crépault pour la construction et la réparation de différents appareils, indispensables à la réussite de ce travail.
Finalement, je me dois de faire l'éloge de mon épouse Pierrette, comme secrétaire et de lui exprimer toute ma grati tude pour le zèle, la patience et le dévouement qu'elle a su manifester au cours de ces deux dernières années.
THERMODYNAMIQUE DE LA POLYMERISATION
DU TETRAHYDROFURANNE
EN SOLUTION DANS LE BENZENE,
V
TABLE DES MATIERES
Page
SOMMAIRE... vii
INTRODUCTION ... 1
CHAPITRE I: PARTIE THEORIQUE 1. 1) LA POLYMERISATION...3
1.1.1) L'amorçage... 3
1.1.2) La propagation. . ... 4
1.1.3) La terminaison... 5
1.2) THERMODYNAMIQUE DE POLYMERISATION ... 7
1.2.1) Effets des propriétés thermodynamiques du milieu sur les équilibres monomère-polymère 7 1.2.2) Effet de la température . ...11
CHAPITRE II: PARTIE EXPERIMENTALE 2.1) LE SYSTEME A VIDE... 13
2.2) LES REACTIFS... 15
2.2.1) L'amorceur... .. . 15
2.2.2) Le solvant et le monomère... 16
2.3) LA POLYMERISATION... . 17
2.4) DETERMINATION DES CONCENTRATIONS DE MONOMERE ET DE POLYMERE... 19
VI
CHAPITRE III: LES RESULTATS
3.1) POSITION D'EQUILIBRE ET REVERSIBILITE DU SYS
TEME...21
3.2) LES MESURES DE VOLUMES ET LE CALCUL DES CON CENTRATIONS ...24
3.3) DETERMINATION DES MASSES MOLAIRES... .. . 35
CHAPITRE IV: LES CALCULS ET LA DISCUSSION DES RESULTATS 4.1) LE CALCUL DE AGlc/RT EN UTILISANT LES FRAC TIONS VOLUMIQUES... 48
4.2) LA VARIATION DE [M]£ AVEC LA TEMPERATURE. ... 52
4.3)
L'INFLUENCE DU CHOIX DE LA VALEUR DExmp
SUR LA DETERMINATION DE AHlc ET DE ASlc... 544.4) LE CALCUL DE AGlc/RT EN UTILISANT LES CHA LEURS INTEGRALES DE MELANGE ... 56
CONCLUSION...61
APPENDICE A LISTE DES FIGURES... 63
APPENDICE B LISTE DES TABLEAUX ... 64
APPENDICE C LISTE DES SYMBOLES... »... 65
SOMMAIRE
Au cours de certaines polymérisations anioniques ou cationiques il est possible d'obtenir un équilibre
monomère-polymère. La position d'équilibre è une tempé
rature donnée dépend de la concentration du polymère présent è l'équilibre et du solvant utilisé pour une polymérisation donnée. Ces effets sont étudiés dans le cas de la polymé risation cationique du tétrahydrofuranne (THF) dans le ben zène. L'amorce de la réaction s'opère avec un sel d'oxonium; le trjuéthyloxonium hexafluorophosphate (EtgO+PFg) dans des concentrations au voisinage de 10"" ^ M. Les polymérisations sont effectuées sous vide dans des ampoules de verre calibrées et sont terminées lorsque le système a atteint l'équilibre. La détermination de la concentration du monomère à l'équi libre s'effectue par différence du poids du monomère initial de celui du polymère obtenu par évaporation à sec de la solu tion mère dans une étuve è vide è la température de la pièce. La précision des données è l'équilibre dépend donc de la pré cision des volumes initiaux de monomère et de solvant ainsi que de la détermination du poids du polymère recueilli par séchage.
1
INTRODUCTION
Au cours d'une polymérisation anionique ou catio nique en solution, il est possible d'obtenir des polymères "vivants" possédant une distribution de masses molaires très étroite. Ces réactions sont réversibles et il s'établit
alors un équilibre monomère-polymère. Ce sujet a fait l'objet d'une revue (1) qui nous a permis de constater la faiblesse de nos connaissances sur l'effet des propriétés thermodyna miques du milieu et sur la position d'équilibre d'une poly mérisation. Cependant, des études récentes (2)(3) montrent qu'è une température donnée, la position d'équilibre peut dépendre de la concentration du polymère présent à l'équi
libre. Ces études mettent en lumière l'influence des propriétés thermodynamiques du milieu. Il est montré (3) que le solvant utilisé joue un rèle prédominant et que si l'on connaît les paramètres thermodynamiques du système, il est possible de calculer le rendement d'une polymérisation et de prévoir les principales caractéristiques du produit obtenu.
L'objet des travaux de la présente thèse est l'é tude des équilibres monomère-polymère au cours de la poly mérisation cationique du tétrahydrofuranne (THF). lia été définitivement établi qu'au cours de la polymérisation en masse du THF un équilibre monomère-polymère était atteint
(4). Il existait de sérieuses indications qu'une situation identique prévalait pour la polymérisation en solution (5).
Comme certaines données thermodynamiques du système benzène- THF sont disponibles (6), nous avons procédé à la polymérisa tion cationique du THF dans le benzène.
CHAPITRE I
PARTIE THEORIQUE
De façon générale, les éthers cycliques polymé- risent par voie cationique. Ils peuvent polymériser en masse
(en 11 absence de solvant) ou encore en solution, en présence d'amorceurs tels que les acides forts de Lewis ou les sels dérivés de ces acides. Quoi qu'il en soit, le seul problème est de choisir 11amorceur le plus efficace pour chacune des polymérisations demandées.
1.1) LA POLYMERISATION
1.1.1) L1 amorçage
L'amorçage de la réaction de polymérisation peut se faire de différentes façons (4); tout d'abord,
4
--- ou par formation d'un ion oxonium (in situ) --- ou par addition d'un ion carbonium
--- ou encore par abstraction d'hydrogène.
Dans le cas qui nous intéresse on produit 1'amorçage par réac tion avec un sel trialkyloxonium préalablement formé. Il
s'agit du triéthyloxonium hexafluorophosphate (Et 0+PF~) qui 3 6
réagit avec le tétrahydrofuranne (THF) pour former le premier ion oxonium du THF. La réaction d'amorçage s'écrit comme suit :
+ PF: b + Bt20 (CH3-CH2>3° PF6 CH,-CH,
<
2
I
2
^H2"CH2 + yCH -> ch3-ch2-q( ^ I-CH, ch2-ch2Amorceur THF ion oxonium
du THF
1.1.2) La propagation
Les espèces qui se propagent lors de la polyméri
sation cationique du THF sont donc des ions oxonium tertiaires. La polymérisation a lieu lors de 1'attaque nueléophylique de 1'oxygène du THF (monomère) sur le carbone alpha de 1'oxygène de l'ion oxonium. Nous avons donc:
+ —ch2-ch2-o \:h -ch
t 2
2 2 H H2C-- CH 22
9» .CH -CH —ch9-ch -cr zI
~.H2-iH/
h2cz \:H h2c- ■CH,2
2
Ne considérant que les attaques effectives sur les carbones de la partie cyclique de l'ion oxonium, il en résulte une croissance de la chaîne et un nouvel ion oxonium, semblable au premier, est alors formé avec la seule différence qu'il possède en plus une unité monomérique.
La réaction de propagation s'effectue donc par une ouverture du cycle du THF (de l'ion oxonium) et on obtient un polymère linéaire sans ramification:
ch2-ch2
CH3-CH2—E-0—(-CH2—I
(le polymère "vivant")
xch2-ch2
où "n" peut prendre les valeurs 1,2,3.... Le polymère formé est appelé polymère "vivant". Il peut polymériser ou dépo- lymériser sa chaîne suivant les fluctuations des conditions d'équilibre (les changements de température ou de concen tration du monomère). La "vivacité" du polymère provient essentiellement de la charge positive sur 1'oxygène de l'ion oxonium à l'extrémité de la chaîne polymérique. Cet oxygène chargé porte le nom de "centre actif".
1.1.3) La terminaison
Pour éviter les variations de longueur de chaîne une fois 1'équilibre monomère-polymère atteint, on doit "tuer" le polymère, c'est-à-dire que l'on doit détruire les centres actifs. "L'arme" la plus efficace semble être le méthanol,
tout d'abord par sa miscibilité avec la solution polyméri que et par son apport des ions OH . La réaction de termi naison est vraisemblablement la suivante:
+ PF™ + CHgOH --- ^--- 0—(-CH2-^3-CH2OH mëthanol "polymère mort" polymère
"vivant" —°'
I
CH2~CH2
Le polymère "mort" est un polyéther linéaire comprenant une extrémité hydroxylée.
7
1.2) THERMODYNAMIQUE DE POLYMERISATION
1.2.1) Effets des propriétés thermodynamiques du milieu sur les équilibres monomère-polymère.
Un état d'équilibre entre le monomère et le polymère vivant peut être atteint au cours d'une polymérisation anio nique ou cationique. Dainton et Ivin postulaient en 1958 (7) que la concentration du monomère à 1'équilibre était constante ê. une température donnée et ils présentaient la relation:
ln[M]e = AGp/RT, où AGp est la variation en énergie libre pour la conversion d'une mole de monomère liquide en une mole1 de polymère amorphe en présence d'un solvant. [M] est la concen tration du monomère ê. 1'équilibre exprimée en moles/litre, R est la constante des gaz et T la température en degrés Kelvin.
Ivin et Léonard ont récemment observé (2) que dans le cas d'une polymérisation anionique à 1'équilibre de l'a- méthylstyrène dans le tétrahydrofuranne ù certaines tempéra tures, que la concentration du monomère è 1'équilibre décroît linéairement avec 1'augmentation de la concentration du poly mère.
Prenant en considération l'effet de la concentration du polymère sur la concentration du monomère à 1'équilibre,
ils ont dérivé une expression pour 1'énergie libre de polymé risation (8): -AGm + AG^c + AGp = 0, où les indices m, le, et p représentent respectivement le monomère, la conversion d'une
8
mole de monomère liquide en une mole1 de polymère amorphe li quide, et le polymère. A partir de l'expression de Flory- Huggins (9) pour 1'énergie libre molaire partielle du monomère et du polymère dans un système ternaire, ils ont obtenu:
[1.1] AGjc = RT ( ln<f>m + 1 + ( Xms ~* Xsp ^ ^s
+ %mp(*p " V -
+ 1)/*)
ou AGjc est le changement en énergie libre pour la conversion d'une mole de monomère liquide en une mole1 de polymère amorphe liquide, R est la constante des gaz, T est la température en degrés Kelvin, n est le nombre moyen de segments dans la chaîne et Vm et Vs sont les volumes molaires du monomère et du solvant respectivement. <j> est la fraction volumique sous les conditions d'équilibre, et % est le paramètre d'énergie libre entre n'im porte lesquels de deux composants, et les indices m, s, p se rapportent au monomère, solvant et polymère respectivement. Si n est grand et <t>p pas trop petite, le dernier terme de 1 ' équa tion [1.1] est négligeable et on obtient:
[1.2]
AGic = RT(ln*m + ^ + (%ms " Xsp(Vm/Vs))4>«s
+ Xmp ( ^p “ ^m) )
AG^c représente le changement en énergie libre lors de la po lymérisation d'une mole de monomère liquide en une mole1 de polymère amorphe liquide d'une longueur de chaîne infinie. La variation observée de <j>m, montre l'effet de l'environnement
sur le comportement de la polymérisation è 1'équilibre. Afin
de mettre en évidence cet effet de 11 environnement, les équa tions [1.1] et [1.2] peuvent être transformées pour obtenir la relation de <f>m en termes de propriétés thermodynamiques du milieu.
Dans leurs travaux sur la polymérisation de l'a- méthylstyrène, Ivin et Léonard (2) ont prouvé expérimenta
lement que <J)m peut être exprimée par la relation:
C1-3] V = + %
oti 4>^ est la valeur de <|>m quand <j>p tend vers zéro et B est la pente obtenue en portant en graphique <f>m en fonction de <j>p. Dans tous les cas "n" est passablement grand. Une valeur de "n" supérieure à 150 est acceptable et 1'hypothèse d'une lon gueur de chaîne infinie est une bonne approximation.
L'équation [1.2] peut être transformée pour obtenir une expression (3); laquelle peut être comparée avec 1'équation
[1.3] . Tout d'abord on développe le ln<j)m en série :
[1.4] ln<|>m = lna + (<j)m - a) /a + (4>m - a) 3/2a3 + (<j>m - a) 3/3
oti 0<4>m<2a. Si la valeur de la constante- est telle que
(<j>m - a)2/2a2 est négligeable en regard des deux premiers ter mes de l'équation [1.4], on peut alors écrire :
[1.51 ln<j>m = lna + <J>m/a - 1
Se rappelant que <j)g = 1 - <j>m - <j>p et en utilisant 1 ' équation [1.5] pour ln<}>m, après réarrangement de l'équation [1.2], l'é quation suivante, pour la variation de (|>m avec <j)p, est obtenue :
10
C1.6]
- (AGlc/RT) + lna - 3 XmP+ 6 ' m 8 + X mp 1/a6 + xmp - 1/3
-( Ooù 3 = X - Xcr, (V /V ) . Mises à part les fractions volu- ms sp m o
miques <J>m et
4>^f
tous les termes apparaissant dans 11 équation [1.6] sont constants a une température donnée. Comparant les équations [1.3] et [1.6] il s'ensuit alors que:[1.7] B = (Xmp - 8)/(8 + Xmp - 1/&)
A partir de cette relation, la variation de la concentration du monomère à 11 équilibre avec la concentration du polymère peut être exprimée en termes de la constante "a" et des inter actions entre les composants du système, mesurées par les paramètres thermodynamiques g et Xmp* est une constante pour une polymérisation donnée et pour une température donnée, "a" peut alors être remplacée par <j)^ dans l'équation [1.5]. Idéalement les valeurs de "a" et <t>0 coincident. De nos
con-m
sidérations précédentes et en comparant les équations [1.3] et [1.6] on obtient finalement:
[
1
.
8
]
(AGlc/RT) + lna + g rm 8 + X_ mp l/a aUtilisant cette définition de "a", 11 équation [1.6] peut être réécrite pour donner:
m mp
- 1/ a) ] 4>
mp p
11
Si AGlc/RT, g et xmp sont connus, 1'équation [1.8] peut être calculée pour "a". Connaissant maintenant "a", il est alors possible de calculer la variation de <|>m avec <f>^ utilisant 1'équation [1.6]' et de comparer avec les résultats expéri mentaux.
Puisque les résultats expérimentaux sont inexis tants pour le calcul de Xms en termes de 11 énergie libre mo laire d'excès de mélange; 1'entropie d'excès de mélange étant considérée comme nulle, on calcule x suivant la relation:
ms Cl-» Xms = âHM/t<RT*P
où AHm est la chaleur intégrale du mélange monomère-solvant, et x^ et <f>^ sont la fraction molaire du monomère et la fraction volumique du solvant respectivement, dans ce mélange binaire.
1.2.2) Effet de la température
Les effets de température (3) se font surtout sentir sur les fractions volumiques 4>m et <j)p. On peut estimer ces effets par la variation de "a" et de "B" avec la température
(T). La variation de "a" avec T est obtenue en dérivant "a", donnée par 1'équation [1.8], par rapport a T. Si la tempé rature varie de è T2, "a" variera alors de a^ è a^. Utili sant la relation de Gibbs-Helmholtz et intégrant de è ?2 on obtient finalement:
12
On doit noter ici, que les paramètres % varient très peu avec la température (3)(%<1/T). Nous pouvons donc poser comme hy pothèse que les paramètres % sont constants.
Possédant maintenant la valeur de "a", on peut alors calculer la valeur de "B" en utilisant l'équation [1.7]. Si Xmp'XSp'AHiç et AHm sont connus, une seule détermination de <j>m avec <J> correspondant à une température donnée est alors re quise. Puisque maintenant <j>m et <|> sont connues,
<p^
peut être calculée suivant la relation:El.ll]
= (1 - <f> - <p ) / (1 - 6)
63 al p p
Les valeurs de AH^ et de <|V sont alors substituées dans 1 '.équa tion [1.9] pour obtenir les valeurs de Xms* L'équation [1.2] nous donne la valeur de AG1 /RT et substituant cette valeur dans 1'équation [1.83 on obtient la valeur de "a". Après avoir calculé "B" è partir de l'équation [1.7], il est alors possible d'écrire: <J>^ = a + B^ pour une température donnée. La varia tion de <J>m avec ^ è toute autre température peut être obtenue en utilisant les équations [1.10] et [1.7]. Si la valeur de AGjc/RT est connue è une température donnée, il est alors pos
sible, en utilisant le processus décrit précédemment, d'obtenir le diagramme entier de (J) en fonction de <j> è différentes tem
ni P
pératures sans avoir è exécuter è chaque fois la détermination de 4>m et *p.
CHAPITRE II
PARTIE EXPERIMENTALE
Pour mener è bien une polymérisation par voie ioni que, on doit opérer sous vide ou sous atmosphère inerte. Dans le cas présent nous avons utilisé un système è vide et nous
-4
avons opéré è une pression inférieure è 10 mm de Hg; pression au-dessus de laquelle il est pratiquement impossible d'amorcer une polymérisation.
2.1) LE SYSTEME A VIDE
Le système è vide est constitué d'un système conven tionnel muni d'une série de robinets et d'une pompe mécanique è succion couplée è une pompe à diffusion pour atteindre une pression inférieure à 10 4 mm de Hg. La région d'opération proprement dite est enroulée d'un fil de chrome1 qui sert
DISTILLATION
PURIFICATION
FIL DE
CHROMEL
ROBINETS
BENZENE
POMPES
15
d'élément chauffant dans le but d'aider au dégazage du verre et d'empêcher la condensation des vapeurs lors des distillations
(Voir Fig. 1)
Le système se divise en trois parties distinctes: --- la région de pompage reliée à la région d'opération par le
robinet "A"
--- la région de purification et d'emmagasinage dans laquelle on conserve le solvant (benzène) et le monomère (THF) avant
les distillations finales
--- la région de distillation et de préparation des ampoules è polymérisation "AP"
Ces deux dernières régions constituent ce que l'on a dénommé plus haut la région d'opération proprement dite.
2.2) LES REACTIFS
2.2.1) L'amorceur
On a utilisé comme amorceur, le triéthyloxonium hexa- fluorophosphate (EtgO+PFg) qui nous a été gracieusement fourni par M. P. Dreyfuss de la compagnie B.F. Goodrich Brecksville Ohio. Ce produit a été préalablement libéré de son eau d'hydra tation qui peut détruire les centres actifs lors de 1'amorçage de la polymérisation. Cette purification s'effectue par disso lution et précipitation répétées trois ou quatre fois dans le chlorure de méthylène et l'éther anhydre respectivement; cette opération est suivie d'un séchage final dans une étuve è vide è la température de la pièce pendant au moins une demi-heure.
16
Après chacune des précipitations, on vidange le solvant (chlorure de méthylène) et le précipitant (éther anhydre) par simple décantation. On conserve ensuite 11amorceur dans un dessicateur sous vide en présence de P^O^ dans une chambre froide è -10 °C. Dans ces conditions le produit peut être uti lisé pendant une période d1environ 6 mois; après quoi il faut le repurifier.
2.2.2) Le solvant et le monomère
Le benzène (solvant) que nous avons utilisé nous provient de la compagnie Macco sous le nom de Benzène (Thio phene Free) numéro 6441. Bien qu'étant passablement pur, il a fallu quand même le distiller sous vide, tout d'abord en pré sence d'hydrure de calcium, puis ensuite dans un ballon conte nant un miroir de sodium. De cette façon il est immédiatement utilisable comme solvant dans les polymérisations.
Pour ce qui est du THF (monomère), nous l'avons obte nu de la compagnie Anachemia Ac-8914 et nous l'avons purifié de
la même façon que précédemment. Nous avons en plus ajouté du naphtalène au distillât. Le naphtalène forme un complexe de couleur verte avec le sodium et nous indique si le THF est suf fisamment pur pour être utilisé. Le miroir de sodium agit comme agent purifiant. Lorsque le sodium est épuisé par les impuretés contenues dans le THF, la formation du complexe sodium-naphtalène n'est plus possible et une coloration jaune apparaît. Il faut alors redistiller sur un nouveau miroir de sodium. De plus il
17
est préférable de recouvrir les ballons avec une feuille d'alu minium.
2.3) LA POLYMERISATION
La polymérisation s1 effectue dans des ampoules appe lées "ampoule à polymérisation" (AP sur la Fig. 1) préalable ment calibrées et lavées à l'acide sulfo-chromique. Les pro blèmes de viscosité nous ont obligés à utiliser des ampoules ayant une section passablement grande (environ 2.35cm2) qui a eu pour effet d'abaisser la précision sur la détermination du volume de la solution à 1'équilibre. Mais étant donné que la variation de volume durant la polymérisation n'est pas telle ment grande, nous avons remédié % cette imprécision en utilisant une ampoule à distillation (AD). Cette ampoule, beaucoup plus petite que la première, est graduée et calibrée (section 0.2005 cm2) avec du mercure dans un bain thermostaté. Les mesures de volumes se font "à l'aide d'un cathétomètre qui nous donne les distances verticales en centimètres (±0.005 cm) entre le ménis que du liquide et la graduation sous le ménisque. Il est alors
facile de déduire le volume exact en appliquant la relation: section x hauteur = volume si on prend bien soin de placer 1'am poule è distillation dans un bain à température connue. (Pré cision sur le volume de ±2%).
On prépare les solutions en déposant tout d'abord une quantité déterminée de 1'amorceur dans 1'ampoule à polymé risation et on fait le vide pendant environ une demi-heure.
18
On baigne ensuite l'ampoule 'à distillation dans l'azote liquide, on ferme les robinets "P" et "A" et on ouvre les robinets "D" et "B" pour distiller le benzène. On ferme ensuite les robinets "D" et "B", on retire l'azote liquide et lorsque le benzène a atteint son point de fusion, on plonge 1'ampoule è distillation dans un bain è température connue. On ajuste alors le volume du liquide en distillant 1'excédant dans les pièges des pompes en ouvrant les robinets "D" et "A". On ferme ensuite le robinet "D" et on évacue le système en gardant le robinet "A" ouvert pendant environ cinq minutes. En maintenant "A" fermé, on bai gne 1'ampoule à polymérisation dans l'azote liquide, on ouvre les robinets "P" et "D" et on distille tout le contenu de 1'am poule è distillation en chauffant si nécessaire. On suggère ici de laisser les robinets "P" et "D" ouverts au moins cinq minutes après le tarissement de 1'ampoule è distillation.
Ayant répété la même technique avec le THF, on gèle la solution dans l'azote liquide et on scelle 1'ampoule dans sa partie amin cie sous l'action des pompes en ouvrant les robinets "P" et "A". Aprè-s avoir détaché 1 ' ampoule è polymérisation du système è vide, on la plonge, après fusion de la solution, dans un bain d'eau è 50°C pendant 30 minutes pour dissoudre complètement 11amorceur et rendre la solution homogène. On place ensuite cette ampoule dans un bain thermostaté è la température désirée et après dix minutes, on mesure une hauteur avec le cathétomètre qui corres pond ê la distance entre le ménisque de la solution et la marque de référence "R" sur 1'ampoule à polymérisation. Cette locali sation du ménisque de la solution par rapport è la marque de
19
référence nous apporte le repère du volume initial de la so lution à la température du bain thermostaté.
2.4) DETERMINATION DES CONCENTRATIONS DE MONOMERE ET DE POLYMERE
Avec les coefficients d'expansion de chacune des substances (monomère et solvant) on calcule leur volume à la température du bain thermostaté. En négligeant le volume oc cupé par 1'amorceur on obtient alors le volume de la solution initiale et on détermine par la suite la concentration initiale du monomère.
Lorsque 1'équilibre monomère-polymère est atteint après environ 4 jours nous reprenons la mesure de la distance verticale entre le ménisque de la solution et la marque de ré férence. On observe généralement une légère diminution de vo lume due a la formation du polymère qui a une densité plus gran de que celle du monomère. On ouvre alors 1'ampoule è l'air
libre tout en la gardant dans le bain et on ajoute ensuite du méthanol pour "tuer" les centres actifs afin d'empêcher toutes variations des concentrations de polymère ê 1'équilibre. Il ne reste alors qu'è évaporer la solution à sec dans un ballon
préalablement pesé pour recueillir le polymère. En déterminant la quantité de polymère, on peut par différence obtenir le poids du monomère è 1'équilibre, puisque l'on connaît le poids du
monomère initial, et effectuer le calcul de la concentration, monomère et du polymère è 1'équilibre étant donné qu'il
Bien entendu il faut connaître le volume de la solution à 1'équilibre. Ce volume (vg0i.) est calculé en soustrayant du volume initial déjà calculé (Vg -^ ) variation de vo lume AV due à la polymérisation suivant la relation: Vg -j. Vg0i - AV. On peut déterminer AV puisque l’on connaît la
section de 1'ampoule dans la région du ménisque de la solu tion.
2.5) DETERMINATION DES MASSES MOLAIRES
Nous avons déterminé les masses molaires de nos polymères par viscosimétrie à l'aide d'un viscosimètre d'Ostwald, utilisant le benzène comme solvant et opérant à 30 °C. Les valeurs des constantes de viscosité "K" et "a", suivant ces conditions, nous ont été fournies par Brandrup et Immergut (10).
CHAPITRE III
LES RESULTATS
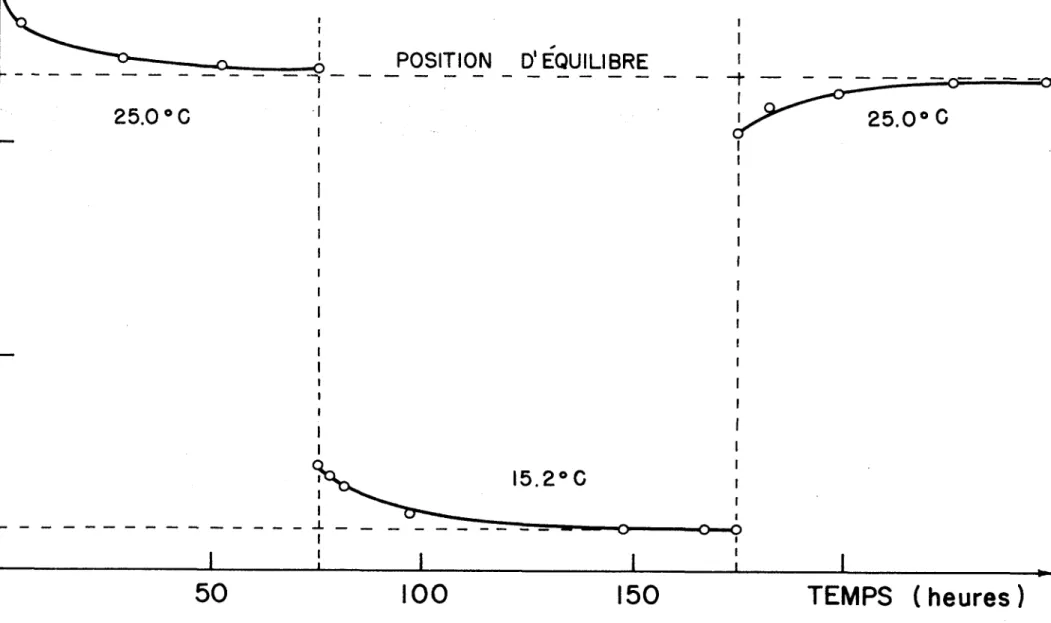
3.1) POSITION D'EQUILIBRE ET REVERSIBILITE DU SYSTEME
Lorsqu'une polymérisation est en marche, on ob serve une diminution du volume de la solution. En effet, il se produit un polymère d'une densité de 1.036 g/cm^ (12) à
3
partir d'un monomère d'une densité de 0.888 g/cm (13). Nous pouvons alors suivre la réaction et connaître è quel moment le système a atteint 1'équilibre# c'est-è-dire après combien de temps il n'y a plus de variations de volume.
La méthode la plus simple pour suivre ces varia tions de volume consiste è utiliser un "dilatomètre" (Fig. 2). Le dilatomètre comporte en sa partie inférieure un bulbe con tenant la majeure partie de la solution (environ 6 ml). Ce
2
22
hauteur
CAPILLAIRE
H
A
U
T
E
U
R
(c
m
)
3
POSITION D'EQUILIBRE
25.0° G
TEMPS ( heures )
FIGURE 3: LA REVERSIBILITE DU SYSTEME ET LA CINETIQUE DE POLYMERISATION
K)
24
lequel se déplace le ménisque de la solution. Une marque de référence au bas du capillaire nous permet de suivre le dé placement du ménisque tout au long de la polymérisation.
Comme toujours, ce déplacement s1 observe è l'aide d'un cathé- tomètre. Nous avons noté qu'il nous faut un minimum de 4 jours pour que l'équilibre monomère-polymère soit atteint è 15.2°C et un minimum de 3 jours pour que 1'équilibre soit atteint à 25°C (Fig. 3). Etant donné qu'il est beaucoup plus difficile de suivre une variation de volume dans une ampoule large comme les ampoules è polymérisation que nous avons utilisées par rap port a un système comme le dilatomètre, toutes les polymérisa tions qui ont suivi se sont effectuées dans un bain thermostaté durant une période de 5 jours pour s'assurer que 1'équilibre monomère-polymère était atteint. De plus, on peut voir sur la Fig. 3 que cet équilibre est réversible puisque l'on retrouve la même position d'équilibre à 25°C. Cette réversibilité du système s'observe par une diminution du volume de la solution
(polymérisation) lorsque l'on passe de 25°C è 15.2°C, et une augmentation du volume de la solution (dépolymérisation) lors que l'on passe de 15.2°C è 25°C.
3.2) LES MESURES DE VOLUMES ET LE CALCUL DES CONCENTRATIONS
Nous avons mentionné à la partie expérimentale que les volumes initiaux du THF et du benzène sont déterminés a une température connue qui est celle du bain d'eau dans lequel nous avons plongé l'ampoule a distillation, (température de la pièce). Il faut maintenant connaître ces volumes a la
tempé-rature de polymérisation. Utilisant les coefficients de dila tation thermique de chacune des substances, on obtient alors :
[3.1] VTHF ” VTHF(1 + aTHFAt)
[3.2] VB = V»(l + aDAt) B
oh At est la différence entre la température de polymérisation et la température de la pièce exprimée en degrés Celcius, V° est le volume de la substance à la température de la pièce, V est le volume de la substance è la température de polymérisa tion, a est le coefficient de dilatation thermique, et les in dices THF et B représentent le tëtrahydrofuranne et le benzène respectivement. Combinant les équations [3.1] et [3.2] on ob tient alors le volume initial de la solution (VgQ^ ):
[3.3] VSol. ” VB + VTHF
négligeant le volume occupé par 11amorceur qui entre en solu tion. On calcule maintenant la concentration initiale du mono mère par la relation:
C3'4] CTHF = (12'316 * VTHF>/VSol.
oh la constante 12.316 représente la concentration molaire maxi mum du THF.
Lorsque 1'équilibre monomère-polymère est atteint, on évapore la solution a sec et on pèse le polymère. On peut dé duire alors le volume du monomère è 11 équilibre suivant la rela tion:
26
[3*5] VTHF " ( (VTHF X dTHF) " WPTHF)//dTHF
oh. WpTHF représente le poids en grammes du polytétrahydrofu- ranne sec et dTHF est la densité du THF (0.888 g/cm^).
Connaissant la section des ampoules h polymérisa tion on calcule le volume de la solution h l'équilibre (par la méthode décrite h la partie expérimentale). Il est alors facile de calculer la concentration du monomère à l'équilibre CM]e!
C3.6] EM]e = (12.316 x VÿHF)/V*cl_
l'étoile (*) indiquant que ces volumes sont "à la température de la polymérisation à 1'équilibre.
La concentration du polymère se calcule en termes de motif de base du polymère. On sait que le motif de base est re présenté par un cycle ouvert du THF de masse molaire 72.1 g. On écrit alors:
C3.7] [P] = (1000/72.1)(WpTHF/V*olJ
3 -1
Le facteur 1000/72.1 cm g mole rapporte le tout en concen tration molaire. [P] est la concentration du polymère è 1'équi libre.
Nous avons compilé dans les tableaux I è. VII qui sui vent, les poids, les volumes et les concentrations des diffé rentes substances dans leurs conditions initiales et a 1'équi libre pour les différentes températures d'opération.
27
Il est à noter que les chiffres apparaissant au haut des colonnes dans les tableaux qui suivent ê 1'indication "Am poules" correspondent au numéro des ampoules à polymérisation utilisées au laboratoire et les indices "prime" (') indiquent que les ampoules ont été réutilisées & la même température.
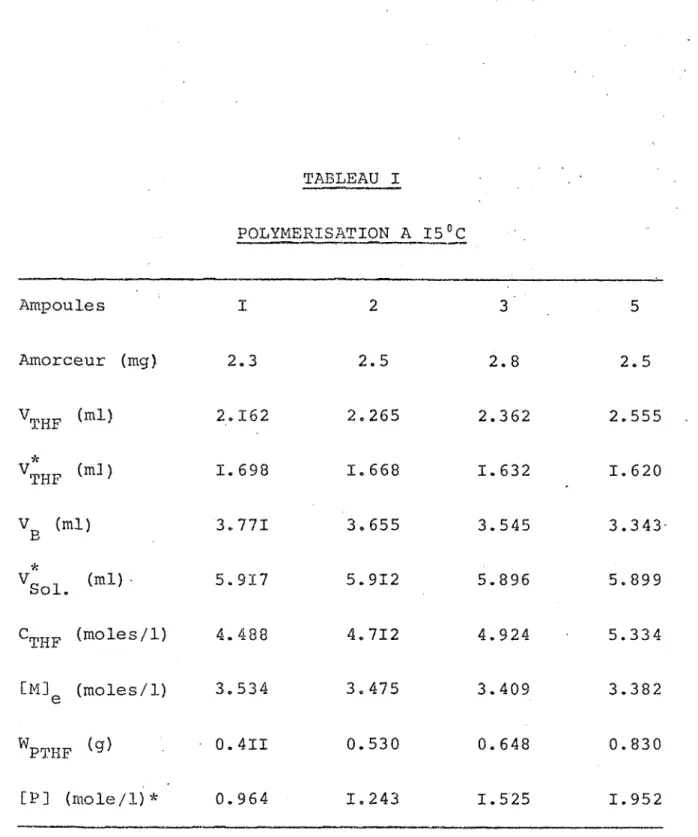
TABLEAU I POLYMERISATION A 15°C Ampoules I 2 3 5 Amorceur (mg) 2.3 2.5 <N CO
2.5
VTHF (ml)2.162
2.265
2.362
2.555 V* THF (m3 )1.698
1.668
1.6321.620
Vg (ml)
3.771
3.655
3.545
3.343
4i (ml) -5.917
5.912
5.896
5.899
CTHF (moles/1)4.488
4.712
4.924
5.334 [M] e (moles/1)3.534
3.475
3.409
3.382
WPTHF • 0.4II0.530
0.648
0.830
[P] (mole/1) *0.964
1.243
1.5251.952
(mole/1)* signifie mole de motif de base par litre
Me a été calculée en considérant le volume molaire du THF comme constant (densité a été prise à 20°C).
TABLEAU II POLYMERISATION A 20°C Ampoules I 2 3 Amorceur (mg) 2.0 2.3 2.2 V THF (ml) 2.178 2.264 2.357 * VTHF (ml) I. 888 1.875 1.815 VB (ml) 3.813 3.737 3.593 * (ml) VSol 5. 968 5.955 5.915 CTHF (moles/1) 4.477 4.645 4.878 1
—
1S
( D (moles/1) 3.897 3.877 3.780 WPTHF ^ 0.257 0.345 0.481 [P] (mole/1)* 0.596 0.803 1.126 4 5 2.0 1.9 2.477 2.593 1.795 1.807 3.482 3.380 5.924 5.926 5.II8 5.346 3.732 3.756 0.605 0.693 I.4I6 1.631 to<o
TABLEAU III POLYMERISATION A 25°C Ampoules I 2 3 4 5 I' 2' Amorceur (mg ) 1.9 1.9 H 00 2.0 2.1 2.0 ro to VTHF ^ml * 2.174 2.264 2.371 2.471 2.541 2.733 2.869 * VTHF (mlj 2.105 2.II3 2.086 2.096 2.045 2.080 1.994 VB (ml) 3.780 3.651 3.575 3.491 3.464 3.267 3.127 * V_ _ (ml ) 5.931 5.903 5.899 5.868 5.850 5.930 5.905 Sol. CTHF (moles/1) 4. 497 4.714 4.9II 5.104 5.263 5.601 5.894 [M] (moles/1 ) 4.371 4. 408 4.355 4.399 4.305 4.320 4.160 e WPTHF ^ 0.061 0.133 0.253 0.332 0.439 0.580 0.777 [P] (mole/1) * 0.143 0.313 0.594 0.786 1.040 1.356 1.825 U) o
TABLEAU IV POLYMERISATION A 30°C Ampoules I 2 3 4 5 9 10 Amorceur (mg )
3.0
2.0
3.0
1.7
2.0
2.1
2.0
V THF (ml )2.399
2.577
2.645
2.745
2.836
2.991
3.138
*
V THF (ml )2.289
2.429
2.378
2.268
2.299
2.2122.140
Vg (ml)3.528
3.525
3.392
3.289
3.189
3.0512.904
vsol (ml.) 5.9276.099
6.034
6.032
6.032 5.9425.882
c
THF
(moles/1 ).4.985
5.201
5.395
5.602
5.787
6.096
6.396
[M]
e (moles/1 ). 4.7564.905
4.854
. 4.631
4.6944.584
4.481 WPTHF (9 ^0.098
0.132
0.237
0.424
0.477
0.692
0.886
[P] (mole/1 ) *0.228
0.299
0.5440.974
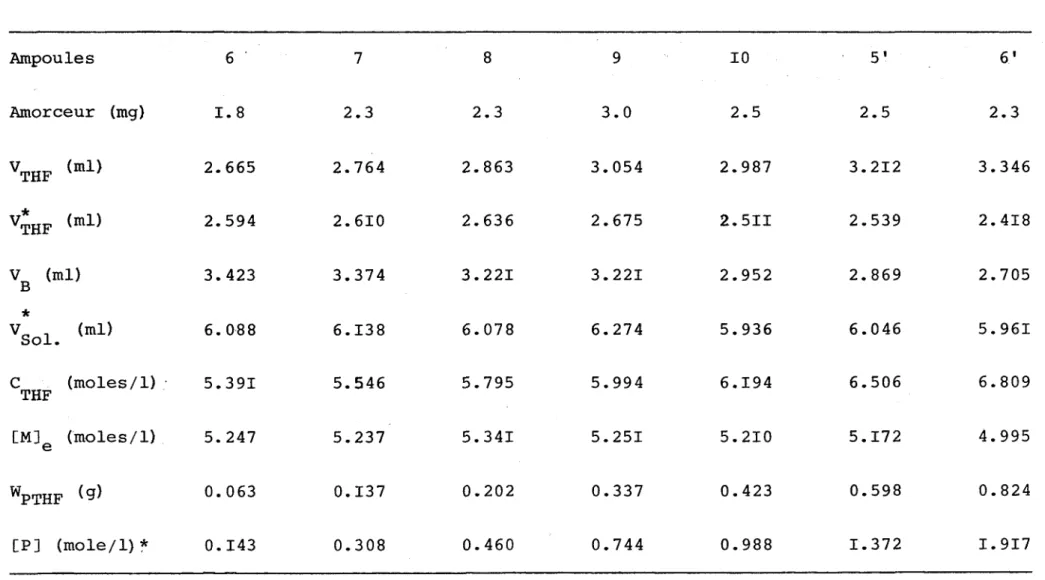
1.095 I. 615 2.089TABLEAU V POLYMERISATION A 35 °C Ampoules 6 7 8 9 10 5' 6' Amorceur (mg) H 00 2.3 2.3 U)
0
2.5 2.5 2.3 V THF (ml) 2.665 2.764 2.863 3.054 2.987 3.212 3.346 V* THF (ml) 2.594 2.610 2.636 2.675 2.5II 2.539 2.418 V (ml) B 3. 423 3.374 3.221 3.221 2.952 2.869 2.705 *v
Sol (ml) 6. 088 6.138 6.078 6.274 5.936 6.046 5.961c
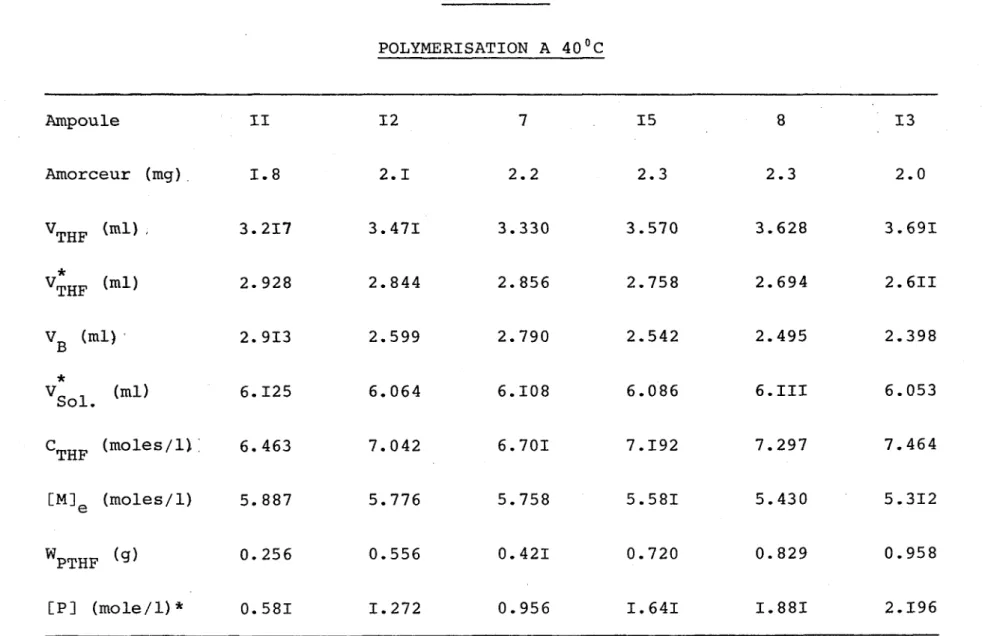
THF (moles/1) 5.391 5.546 5.795 5.994 6.194 6.506 6.809 CM]e (moles/1) 5. 247 5.237 5.341 5.251 5.210 5.172 4.995 WPTHF (9) 0.063 0.137 0.202 0.337 0.423 0.598 0.824 [P] (mole/1) * 0.143 0.308 0.460 0.744 0.988 1.372 1.917 U) K)TABLEAU VI POLYMERISATION A 40°C Ampoule II 12 7 15 8 13 Amorceur (mg) . H 00 2.1 2.2 2.3 2.3 2.0 VTHF (ml) 3.217 3.471 3.330 3.570 3.628 3.691 * VTHF (ml) 2. 928 2.844 2.856 2.758 2.694 2.6II VB (ml) 2. 913 2.599 2.790 2.542 2.495 2.398 * V Sol (ml) 6.125 6.064 6.108 6.086 6.III 6.053 CTHF (moles/1) ; 6. 463 7.042 6.701 7.192 7.297 7.464 [M]e (moles/1) 5. 887 5.776 5.758 5.581 5.430 5.312 WPTHF (9) 0.256 0.556 0.421 0.720 0.829 0.958 [P] (mole/1) * 0. 581 1.272 0.956 1.641 I. 881 2.196
TABLEAU VII POLYMERISATION A 50 °C Ampoules I 2 3 4 3' 7 8 4' Amorceur (mg ) 2. 4 2.3 2.5 2.3 2.0 2.4 2.2 2.5 V THF (ml ) 3.5II 3.709 3.809 3.909 4.066 4.132 4.193 4.307 * V THF (ml) 3.386 3.390 3.444 3.400 3.437 3.392 3.303 3.298 VB (ml) 2. 657 2.457 2.364 2.264 2.120 2.055 1.970 1.869 * V
Sol (ml ) 6.167 6.163 6.I7I 6.170 6.II6 6.175 6.054 6.105
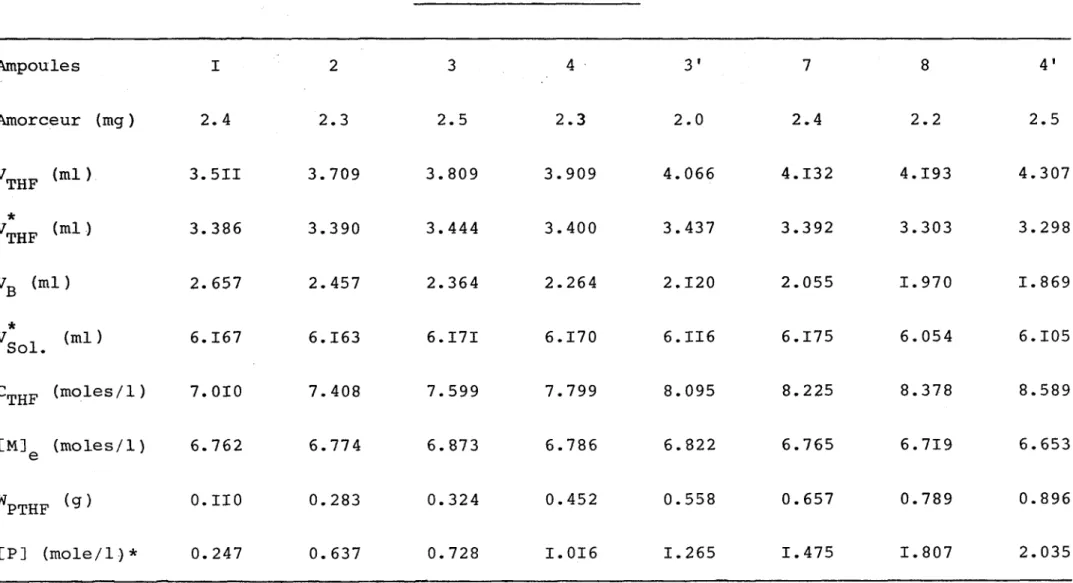
CTHF (moles/1) 7.010 7.408 7.599 7.799 8.095 8.225 8.378 8.589 [Ml e (moles/1) 6.762 6.774 6.873 6.786 6.822 6.765 6.719 6.653 WPTHF ^ 0.110 0.283 0.324 0.452 0.558 0.657 0.789 0.896 CP] (mole/1)* 0. 247 0.637 0.728 1.016 1.265 1.475 1.807 2.035
35
3.3) DETERMINATION DES MASSES MOLAIRES
La détermination des masses molaires s1 est effec tuée A l'aide d'un viscosimètre d'Ostwald dans lequel on a mesuré des temps d'écoulement pour le benzène et les solu tions benzéniques.
Si on opère en solution diluée, on peut poser comme hypothèse que la densité de la solution est pratiquement égale è la densité du solvant. Alors la relation:
[3.8] nr = (teg0i. x dSol. )/ (teB x devient;
[3.9]
nr
= (tegol-)/(teB)où "te" est le temps d1écoulement (en sec.) pour un volume dé-3
terminé, "d" est la densité (en g/cm ), nr est la viscosité relative et les indices Sol. et B se rapportent a la solution benzénique et au benzène respectivement.
Nous savons par ailleurs (9) que la viscosité spé cifique
"ns"
, est en relation avec la viscosité relative sui vant 1'expression:[3.10] 0S = 0r - 1
On défini ensuite la viscosité intrinsèque "[n]" en termes de viscosité spécifique et de viscosité relative suivant les re
lations:
[3.11]
[n]
= lim (n /C) OO s36
C3.12] En3 = lint (lnn /C)
OO
r
Si on porte en graphique la variation de ns/C ou de lnnr/C en fonction de la concentration "C", l'ordonnée à l'origine nous donnera directement la valeur de En!• Flory (9) défini la vis cosité intrinsèque en termes de la masse molaire "M" du poly mère:
[3.13] En! ■= KM01
oh K et a sont les constantes de viscosité ayant les valeurs de 1.31 x 10-3 dl/g et 0.6 respectivement pour le polytétra- hydrofuranne dans le benzène è 30°C (10). Dans le tableau suivant nous avons compilé les différentes valeurs obtenues par cette méthode avec l'ampoule "5" à 15°C.
TABLEAU VIII
DETERMINATION DE LA MASSE MOLAIRE
CPTHF te ns Or Ve (lnnr)/C (g/di) (sec) 99. 8 mm mm _ (dl/g) (dl/g) 0.729 387.5 2. 881 3.881 3.952 1.861 0.486 263. 0 1. 634 2. 634 3.362 2.018 0.364 213.0 1.133 2.133 3.108 2.077 0. 265 176.6 0. 769 1.769 2.902 2.151 0. 194 153.3 0.536 1.536 2.757 2.211
Nous obtenons: En3 = 2.3 2 dl/g et M = 260,500
i,
4.2
MASSE MOLAIRE
260,500
POUR
POUR
C (g/IOOml)
FIGURE 4: LA VARIATION DE n?/C ET DE lnnr/C EN FONCTION DE C
U)
00
o
k
39
Une méthode plus pratique est celle proposée par A.A. Berlin (11) qu'il a dénommée "la méthode par un point". Cette méthode nous donne la viscosité intrinsèque par extra polation è. partir d'une mesure de la viscosité relative en utilisant qu'une seule concentration d'un échantillon donné. Ceci réduit au minimum les mesures de viscosité et l'auteur prévoit une erreur maximale de ±2%. Bien que ne mettant pas en cause la véracité de cette précision, nous avons cru bon de vérifier l'efficacité de cette méthode. Nous avons obtenu com me viscosité intrinsèque les valeurs de 2.32 dl/g et 2.34 dl/g avec les viscosités relatives de 2.133 et 1.769 tirées du ta bleau VIII comparativement à la viscosité intrinsèque de 2.32 dl/g obtenue avec la première méthode. Estimant ces valeurs satisfaisantes nous avons donc adopté la "méthode par un point" pour calculer la masse molaire de tous les autres échantillons de polymère. Les résultats obtenus sont compilés dans les ta bleaux IX et X.
TABLEAU IX
DETERMINATION DES MASSES MOLAIRES
Température Echantillon C te nr [R] Masse molaire n
(°C) (g/dl) (sec) (dl/g) (x 10"3) 15 1 0.440 191.6 1.920 1.627 143 1990 15 2 0.408 197.1 1.975 1.839 176 2441 15 3 0.412 189.4 1.898 1.704 155 2150 15 5 0.364 213.0 2.133 2.320 260 3620 20 1 0. 368 122.4 1.226 0.569 24.9 345 20 2 . 0.464 119.3 1.195 0.393 13.4 186 20 3 0.584 192.5 1.925 1.237 90.9 1260 20 4 0.392 117.5 1.177 0.425 15.3 212 20 5 0. 488 171.9 1.722 1.202 86.6 1202 o
TABLEAU IX (suite) 25 3 0.448 113.5 1.137 0.291 8.2 113 25 4 0.484 181. 8 1.822 1.350 105 1458 25 5 0.372 150.8 1.511 1.177 83.6 1160 30 3 0. 428 136.5 1.368 0.764 40.7 564 30 4 0.356 157.7 1.580 1.370 108 1494 30 5 0. 404 186.4 1.868 1.691 153 2122 30 10 0.348 211.9 2.123 2.412 27:6 3836
TABLEAU X
DETERMINATION DES MASSES MOLAIRES
Température (°C) Echantillon C (g/dl) te (sec) nr Enl (dl/g) Masse molaire (x 10-3) n 35 8 0.524 131.6 1.318 0.547 23.3 323 35 9 0. 648 150.8 1.511 0.676 33.2 460 35 10 0. 628 169.7 1.700 0.911 54.6 757 35 5 0. 432 175.9 1.762 1.421 114 1588 35 6 0. 440 192.2 1.926 1.636 145 2008 40 11 0.542 151.5 1.518 0.816 45.5 630 40 7 0.460 135.9 1.362 0.701 35.3 489 40 12 0. 400 148.4 1.487 1.029 66.9 927 .T* M
TABLEAU X 40 8 40 13 50 2 50 3 50 4 50 3' 50 7 50 8 4' (suite) .256 149.5 , 196 160.1 . 236 108.2 .360 121.6 . 288 138.7 .444 142. T . 404 151.6 .416 164.8 356 137. 4
0
0
0 0 0 0 0 00
50 1.498 1.670 150 2079 1.604 2.576 308 4281 1.084 0.345 10.8 150 1.218 0.563 24.5 339 1.390 1.196 85.9 1191 1.430 0.846 48.3 669 1.519 1.097 74.4 1032 1.651 1.293 97.8 1357 1.377 0.939 57.4 796 4*. u>CHAPITRE IV
LES CALCULS ET LA DISCUSSION DES RESULTATS
Dainton et Ivin postulaient en 1958 (7) que la con centration du monomère a 11 équilibre était une constante pour un système è une température donnée. Plus tard, Ivin et Léonard
(8), dans leurs travaux sur 1'a-méthylstyrène, ont noté qu'il y avait une variation décroissante de la concentration du mono mère a 1'équilibre avec 1'augmentation de la concentration du polymère mais que cette variation est faible.
Si on porte en graphique la concentration du THF S. 1'équilibre en fonction de la concentration du polymère (PTHF)
on obtient une droite de pente négative très près de zéro (Fig. 5). On s'aperçoit qu'avec 1'augmentation de la température, la con centration du monomère è 1'équilibre croît si bien que l'on ob tient la valeur de 12.316 M à 85°C qui constitue ce que l'on appelle la "température plafond" de la polymérisation du THF
[
M
]e
(m
o
le
s
/t
)30° c
1.5 (moles01/! ) 2.0
FIGURE-5: LA VARIATION DE CM]A EN FONCTION DE [P]
4^ Un
46
sans solvant (4). Par définition, il est impossible d'obtenir du polymère au-dessus de cette température. A une température donnée, on devrait s'attendre normalement que la masse molaire du polymère croisse a mesure que la concentration du polymère augmente è la condition d'avoir la même concentration effective d'amorceur. C'est donc dire, qu'en se référant a la Fig. 5 que le nombre "n" (nombre de segments dans la chaîne polymérique) devrait décroître de droite à gauche pour enfin devenir 0 à
[P] = 0. Nous avons mentionné à la partie théorique que nous devions avoir une droite si nous portons en graphique la con centration du monomère a 1'équilibre en fonction de la concen tration du polymère si "n" est supérieur è 150; sinon, on a ce que l'on appelle un "effet de courte chaîne" (3). On remarque que les points ê faible concentration de polymère s'écartent des droites a 25, 30, 35 et 50°C; et que cette déviation est vraisemblablement due è un effet de courte chaîne. Pour ce qui est du troisième point qui s'écarte de la droite a 40°C, nous pensons qu'une mauvaise détermination du volume initial de la
solution ou une erreur de lecture du poids du polymère pourrait être la cause de cette déviation.
De nombreux facteurs peuvent faire varier la lon gueur des chaînes polymériques lors d'une polymérisation amor cée par une substance instable telle que le triéthyloxonium hexafluorophosphate (EtgO^PFg). Tout d'abord, la concentration de 1'amorceur joue un très grand rêle sur la longueur des chaî nes polymériques puisque le nombre de molécules d'amorceur nous donne directement le nombre de chaînes polymériques et plus les
47
chaînes sont nombreuses, plus elles sont courtes. On comprend bien que si l'ampoule à polymérisation n'est pas propre, il y aura une certaine quantité d'amorceur qui sera perdue par ré action avec les impuretés, ce qui aura pour effet de diminuer la concentration de l'amorceur et par ricochet augmenter la longueur des chaînes. On sait que:
[4.1] n = [P]/[L.E.]
et qu'une diminution de la concentration des centres actifs [L.E.] entraînera une augmentation du nombre de segments dans la chaîne et par conséquent une augmentation de la longueur de la chaîne polymérique. De plus, l'amorceur étant instable (il n'est utilisable que pendant une période de 6 mois) on comprend bien que son utilisation après 4 mois ne donne pas la même ef ficacité que son utilisation après une semaine. On produit aussi des courtes chaînes lorsque l'on opère a faibles concen trations initiales de monomère. Puisqu'il y a très peu de mo nomère qui se transforme en polymère; la concentration initiale du monomère se trouvant très près de la concentration du mono mère è l'équilibre, et qu'il y a plusieurs chaînes d'amorcées, on obtiendra forcément des chaînes polymériques assez courtes.
Tous ces facteurs contribuent è produire une irrégu larité dans la détermination des masses molaires compilées dans les tableaux IX et X. On a pu calculer par exemple que l'amor ceur, après environ 4 mois sans repurification, a une efficacité de 35%; c'est-ê-dire que 35% de sa quantité iniliale a été uti
48
que 1'ampoule a polymérisation est parfaitement propre).
4.1) LE CALCUL DE AGlc/RT EN UTILISANT LES FRACTIONS VOLUMIQUES
Comme nous l'avons mentionné au chapitre I, la va leur de AG^C/RT et de g à une température donnée est obtenue par 1'ordonnée à l'origine et par la pente respectivement si on porte en graphique la variation de <j>m en fonction de <j>
(équation [1.6]). On peut aussi obtenir la valeur des pentes et des ordonnées h 1'origine avec les données recueillies è la Fig. 5 en utilisant les relations:
[4.2]
^ = [M]°Vm
[4.3] B = (Vm/Vp)b
où [M]g est la concentration du monomère à 1'équilibre lorsque [P] tend vers.zéro; V et V sont les volumes molaires du
mono-m p
mère et du polymère respectivement et b est la pente obtenue à partir de la variation de [M]e en fonction de [P], On se rap pellera que <j>^ = lim cf> et que
<j>^
est représentée par "a" dans 1'équation [1.6].Nous avons donc obtenu, è partir de la Fig. 5 et des équations [4.2] et [4.3], les valeurs de et de B qui appa raissent au tableau XI. Utilisant maintenant les valeurs de et de B, et posant comme hypothèse que le paramètre d'interac tion entre le monomère et le polymère (xmp) est égal è 0.4, nous obtenons les valeurs de AG]_C/RT et de 6 (tableau XII) .
TABLEAU XI
PENTES ET ORDONNEES A L'ORIGINE
Température (°C) CM]* (moles/1)
<
b B 15 3.672 0.298 -0.156 -0.181 20 3.990 0.324 -0.163 -0.189 25 4.473 0.363 -0.150 -0.175 30 4.893 0.397 -0.198 -0.231 35 5.436 0.441 -0.221 -0.257 40 6.133 0.4 89 -0.358 -0.417 50 6.997 0.566 -0.149 -0.17450
TABLEAU XII
AG-^/RT ET 6 EN FONCTION DE T
Température AGlc/RT
e
(°C)
Nos résultats Dreyfuss Moyen15
-0.
447-0.353
-0.400-0.167
20 -0.347 -0.275 -0.311 -0.134 25-0.167
-0.193
; -0.180-0.014
30 -0. 153-0.114
-0.133 -0.116 35-0.055
-0.026-0.040
-0.10840
-0.159 0.0660.046
-0.488
50
0.298
0.215
0.252
0.198
AG Je / RT
FIGURE 6: LA VARIATION DE
LG-,
../RT EN FONCTION PE 1/TA:
Les résultats de Dreyfuss
B: Nos résultats avec <j>m et <f>
52
Les valeurs de AG^/RT obtenues à 25 et 50 °C s'écartent de la droite expérimentale (Fig. 6, droite "B"). Pour ce qui est de la valeur à 25°C, cette déviation est probablement due à une trop grande marge d'erreur sur la pente de la droite ob tenue à partir des concentrations de monomère ([M]e) et de po lymère (CPI) (Fig. 5); ët étant donné que cette pente est déjà assez faible, 1'erreur en est d'autant plus marquée. Une va riation de 25% dans la pente n'affecte pratiquement pas la va leur de 1'ordonnée à 1'origine mais influence considérablement le calcul de AG^c/RT qui est fonction de 3 (obtenu par la pente).
4.2) LA VARIATION DE [M]° AVEC LA TEMPERATURE
On peut noter que les valeurs de [M]^ obtenues à par tir de la Fig. 5 sont en accord avec la relation de Dainton et Ivin (7) (équation [4,4]) puisque l'on obtient une droite si on porte en. graphique la variation du ln[M]^ en fonction de 1/T,
(Fig. 7). La relation de Dainton et Ivin est la suivante :
[4.4] ln[M]° = (AH /R) (1/T) - (AS°/R)
ou AHp est la chaleur de polymérisation sous des conditions gé nérales d'expérimentâtion et ASp est le changement d'entropie pour [M]^ = 1 mole/1. La Fig. 7 montre que le point à 25°C est bien sur la droite, ce qui signifie que la déviation de AG-^/RT à cette température à la Fig. 6 est due essentiellement à 1'er
reur sur la pente (Fig., 5).. Il n.'en est pas de même pour le point à 50°C. Nous retrouvons la même anomalie è la Fig. 6 et
In [Mlle
+ DREYFUSS
o NOS RÉSULTATS
FIGURE 7: LA VARIATION DE [M]^ EN FONCTION DE 1/T
in
54
nous n'avons pas pu expliquer pourquoi il y a une déviation marquée sur tous les graphiques & cette température puisque la droite obtenue è partir de [M]e et [P] (Fig. 5) passe par tous les points (en rejetant les deux premiers qui illustrent l'ef fet de courte chaîne).
Il fallait aussi s'attendre que nos valeurs expéri mentales de [M3g soient passablement différentes de celles de Dreyfuss (Fig. 7). En effet, ayant cgaéré un même système (po lymérisation du THF) dans des conditions différentes (avec ou sans solvant) nous ne pouvions pas obtenir les mêmes valeurs de [M]° (5).
Par ailleurs, se rapportant a la Fig. 6, étant donné que A G^c/RT est indépendant du solvant et est une constante pour un système (polymérisation du THF) a une température donnée, nous sommes heureux de constater le léger écart entre les va leurs obtenues et celles publiées par Dreyfuss (4). Nous avons compilé les moyennes de ces valeurs dans le tableau XII.
4.3) L'INFLUENCE DU CHOIX DE LA VALEUR DE SUR LA DETERMI NATION DE AH-],. ET DE AS-,,.
La thermodynamique classique nous permet d'écrire :
[4.5] AGlc/RT = (AH1c/R) (1/T) - ASlc/R
où AH^c et AS^C sont les changements d'enthalpie et d'entropie respectivement lors de la conversion d'une mole de monomère
li-55
guide en une mole de motif de base de polymère amorphe liquide de masse molaire infinie. Nous avons porté en graphique nos valeurs de AGjc/RTf ainsi que celles de Dreyfuss (tirées du
tableau XII) en fonction de 1/T et nous avons obtenu deux droites rapprochées comme le démontre la Fig. 6 (les droites "A" et "B").
Il est difficile de choisir une valeur pour Xmp' puis que rien n'a été déterminé définitivement bien que l'on sache qu'elle doit se situer entre 0.3 et 0.5. Cette dernière valeur est attribuée dans un cas idéal, c'est-à-dire en opérant dans un solvant thêta (*). Nous avons voulu montrer l'importance du choix de la valeur du paramètre xmp sur la détermination de AHlc et de ASlc en présentant le tableau XIII.
TABLEAU XIII
L'INFLUENCE DU CHOIX DE LA VALEUR DE x__ SUR LA DETERMINATION DE AH1<? ET DE AS^
. AHfc (cal/mole)
ASic (cal/mole0K)
Xmp = °'3
Xmp = °'4
xmp = 0,3
Xmp = °'4
Nos résultats -3734 -3448 -12.4 -11.5
Dreyfuss -3300 -3036 -10.7 -9.8
(*) Un solvant thêta est celui qui ne fera pas varier les di mensions de la pelote polymérique lorsque le polymère passe en solution. Un mauvais solvant fait contracter la pelote, et un bon solvant la fait dilater.
56
4.4) LE CALCUL DE AG1 /RT EN UTILISANT LES CHALEURS INTEGRALES DE MELANGE
Comme nous l'avons mentionné au chapitre I (Effet de la température), les valeurs de
xmg
sont calculées & partir de cjig et des chaleurs intégrales de mélange (AHM) & l'aide de l'é quation [1.9]. Utilisant les valeurs de AHM (en fonction de la fraction molaire du THF: x^) pour le mélange THF-benzène à 25°C, publiées par Erva en 1955 (6) (Fig. 8) et la définition de en termes de x^:14.61
0' = U - + x^(Vm/Vs))nous pouvons tracer la courbe qui nous donne la variation de
xms
en fonction de <j>g (Fig. 9). Puisque (j)g = 1 - <j)^ (3), utilisantles valeurs de <J>^ présentées dans le tableau XI et se servant de la courbe de la Fig. 9 nous obtenons les valeurs de Xms aux tem pératures de polymérisation. Nous avons défini g (page 10) en fonction des paramètres d1interaction et des volumes molaires; et posant comme hypothèse que xSp = 0.3 et utilisant 11 équation
[1.81 on obtient finalement:
(AG, _/RT - ln(l - (i>‘) - Xms " 0.724) [4.7] *1 --- —--- --- —--- * 1
Xms t 0.124
Il faut noter ici que = 0.919 et que v = 0.4.
Nous pouvons calculer maintenant les valeurs de AGlc/RT puisque nous connaissons les valeurs de
xms
et de aux tempé ratures de polymérisation. (Ces valeurs sont compilées dans leCHALEUR DE MELANGE
-o. 57
-0.59
-0.61
-0.63
-0.65
FIGURE 9: LA VARIATION DE XmS EN FONCTION DE
Un
59
tableau XIV). On peut constater a la Fig. 6 que la droite "C" obtenue à partir des données du tableau XIV s'écarte passable ment des droites "A" et "B". Nous croyons donc que la méthode de calcul de AG^C/RT "à partir des chaleurs intégrales de mélan ge n'est pas tout & fait au point si on en juge par la déviation de la droite "C" par rapport aux droites "A" et "B". De plus, on peut remarquer que les chaleurs de mélange nous sont fournies à 25 °C, ce qui entraîne un choix erronné des valeurs aux autres températures.
Nous avons calculé les valeurs de AH^C et de AS^C à partir des données de AG^/RT recueillies au tableau XIV. Nous avons obtenu les valeurs de -4078 cal mole-"*" et -12.3 cal mole-^
n — 1
K pour AH^C et AS^ respectivement. Nous observons quand même un assez bon accord entre ces résultats et ceux compilés au ta bleau XIII à xmp = 0.4 malgré 1'inexactitude de cette méthode comparée "à celle décrite à la partie 4.1 de ce chapitre.