DÉVELOPPEMENT DU MOTIF SULFAHYDANTOÏNE
COMME SOURCE DE COMPOSÉS BIOACTIFS
Mémoire
Camille Lapointe Verreault
Maîtrise en chimie
Maître ès Sciences (M. Sc.)Québec, Canada
Résumé
Le présent mémoire se divise en deux parties. La première décrit le développement d’une nouvelle méthodologie de synthèse de peptides analogues contraints par une sulfahydantoïne. La deuxième partie décrit la synthèse de nouveaux composés dérivés du motif sulfahydantoïne au potentiel antibactérien.
Le premier chapitre est consacré à la description des agents thérapeutiques peptidiques et de leurs applications. Il est sujet également de l’intérêt de développer de nouveaux peptides contraints. Le deuxième chapitre décrit la synthèse du motif sulfahydantoïne ainsi que la synthèse de peptides analogues contraints sur support solide et en solution. Le troisième chapitre traite des résultats conformationnelles des peptides contraints obtenus à l’aide de la spectropolarimétrie de dichroïsme circulaire et par la spectroscopie de résonance magnétique nucléaire.
Le quatrième et dernier chapitre porte sur la synthèse de nouveaux composés dérivés de la sulfahydantoïne et leur caractérisation. Finalement, les résultats des tests biologiques réalisés avec les composés sont présentés.
Table des matières
Résumé ... iii
Liste des tableaux ... vii
Liste des figures ... ix
Liste des abréviations ... xiii
Remerciements ... xvii
Chapitre 1 : Introduction... 1
1.1 Introduction générale ... 1
1.2 Les protéases ... 2
1.3 Agents thérapeutiques peptidiques ... 4
1.4 Les hétérocycles ... 7 1.5 Le motif sulfahydantoïne ... 8 1.6 Contraintes peptidiques ... 10 1.6.1 Tournant-β ... 11 1.6.1.1 β-Hairpin ... 13 1.6.2 Brins-β et feuillets-β ... 14 1.6.3 Hélices-α ... 15 1.6.4 Peptides cycliques ... 16
1.7 Stratégie de contrainte peptidique ... 18
Chapitre 2 : Synthèse d’un peptide modèle contraint ... 23
2.1 Préambule ... 23
2.2 Rappel bibliographique sur la synthèse de sulfahydantoïnes ... 23
2.3 Stratégie de synthèse de tripeptides analogues contraints par la sulfahydantoïne sans carbonyle en N5 ... 30
2.3.1 Synthèse de la sulfahydantoïne ... 30
2.3.2 Stratégie de substitution en N5 ... 33
2.3.2.1 Préparation d’analogues alkylants d’acides aminés. ... 33
2.3.2 Ozonolyse ... 36
2.4 Synthèse sur support solide ... 37
2.4.1 Support solide ... 38
2.5.1 Travaux antérieurs ... 39
2.5.2 Synthèse d’un pentapeptide contraint (79) ... 39
2.5.3 Synthèse d’un décapeptide contraint ... 43
2.7 Conclusions ... 45
Chapitre 3 : Études conformationelles des peptides contraints ... 47
3.1 Préambule ... 47
3.2 La spectropolarimétrie de dichroïsme circulaire (DC) ... 47
3.3 Résultats des études conformationnelles par DC ... 49
3.3.1 Pentapeptide contraint et modèle... 50
3.3.2 Décapeptide contraint et modèle... 54
3.4 La spectroscopie de résonance magnétique nucléaire (RMN) ... 56
3.4.1 Études conformationnelles par RMN ... 57
3.4.1.1 COSY ... 58
3.4.1.2 NOESY ... 60
3.4 Conclusion ... 63
Chapitre 4 : Synthèse et caractérisation d’inhibiteurs de β-lactamases ... 65
4.1 Préambule ... 65
4.2 Les lactivicines ... 66
4.3 Les sulfahydantoïnes comme potentiel antibactérien ... 68
4.4 Synthèses des sulfahydantoïnes ... 70
4.5 Tests d’inhibition de β-lactamases ... 74
4.5.1 Tests enzymatiques ... 75 4.5.2 Résultats ... 76 4.6 Conclusion ... 81 Conclusions générales ... 83 Partie expérimentale ... 85 Références ... 99
Liste des tableaux
Tableau 1. Essais de substitution en position N5 sur la sulfahydantoïne 68
Tableau 2. Essais de substitution sur la sulfahydantoïne par
N-Boc-2-bromo-1-aminoéthyle pour obtenir 72
Tableau 3. Déplacements des CHα et NH du pentapeptide contraint 79
Tableau 4. Corrélation entre les groupements NH et les groupements de la chaîne
latérale
Liste des figures
Figure 1. Exemples d’inhibiteurs de la protéase du VIH-1 d’importance clinique Figure 2. Représentation conformationnelle du site actif des protéases
Figure 3. Limitations dues à la flexibilité des molécules peptidiques Figure 4. Exemples d’hétérocycles biologiquement actifs
Figure 5. Le motif sulfahydantoïne
Figure 6. Restriction des angles ψ et Φ d’un acide aminé
Figure 7. Structure générale du motif sulfahydantoïne comme élément de
reconnaissance développé par Groutas
Figure 8. a) Composés mimétiques des tournants-β du groupe de Kahn b)
conformation des tournants-β de type I et II
Figure 9. Composé mimétique des tournants-β du groupe de Burgess
Figure 10. Dipeptide D-Pro-L-Pro et incorporation dans une chaîne peptidique Figure 11. Peptidomimétique de brin-β de Smith
Figure 12. a) Terphényle b) Composés mimétiques de Hamilton Figure 13. Macrocycles de Deslongchamps
Figure 14. Acide α-kaïnique
Figure 15. Étude par dichroïsme circulaire des peptides contraints et modèles
correspondant développé dans notre laboratoire
Figure 16. Stratégie de contrainte peptidique par l’incorporation d’une
sulfahydantoïne proposée par notre laboratoire
Figure 17. Tripeptide contraint sulfonyle N-Boc-L-Val-Phe-[SO2]-D-Ala-OMe et
son diagramme de Ramachandran correspondant
Figure 18. Lieu d’épimérisation lors de l’introduction du motif sulfahydantoïne dans
une chaîne peptidique
Figure 19. Schéma de synthèse de Hanewacker et al. Figure 20. Schéma de synthèse de Timberlake et al. Figure 21. Schéma de synthèse de Muller et al. Figure 22. Schéma de synthèse de Kohn et al. Figure 23. Schéma de synthèse de Dewynter et al.
Figure 24. Schéma de synthèse de Groutas et al. Figure 25. Schéma de synthèse de Diederich et al. Figure 26. Schéma de synthèse de Albericio et al. Figure 27. Schéma de synthèse de Voyer et al.
Figure 28. Stratégie de synthèse en solution pour la préparation d’un pseudo
tripeptide contraint sans carbonyle sur N5
Figure 29. Estérification de l’acide aminé de départ et réaction avec CSI/t-BuOH Figure 30. Aménagement fonctionnel et réactivité du CSI
Figure 31. Reaction de Mitsunobu sur la sulfahydantoïne démontrant la O- et la
N-alkylation
Figure 32. Mécanisme de la réaction de Mitsunobu et préparation de 64 Figure 33. Cyclisation du sulfamide menant à la sulfahydantoïne 60
Figure 34. Synthèse des électrophiles pour l’étape d’alkylation au départ de l’acide
aminé L-phénylalanine
Figure 35. Alkylation en position N5 de la sulfahydantoïne
Figure 36. Alkylation en position N5 de la sulfahydantoïne
Figure 37. Réaction d’ozonolyse pour obtenir le peptide contraint 73
Figure 38. Synthèse d’un dipeptide 77 sur résine oxime nécessaire pour la
synthèse de 79
Figure 39. Synthèse d’un pentapeptide contraint 79
Figure 40. Synthèse sur support solide du pentapeptide analogue modèle 81 sans
sulfahydantoïne
Figure 41. Synthèse d’un tétrapeptide contraint 84 Figure 32. Synthèse d’un décapeptide contraint Figure 43. Analogue non contraint 87 de 86
Figure 44. Courbes de DC associées aux principales structures secondaires de
peptides : conformation aléatoire, feuillet-β et hélice-α
Figure 45. Spectre DC de la sulfahydantoïne 68 dans le TFE et l’acétonitrile Figure 46. Spectre DC du pentapeptide contraint 79 et modèle 81 dans le TFE Figure 47. Spectre DC du pentapetides contraint et de son analogue modèle à
Figure 48. Spectre DC du pentapeptide contraint et de l’analogue modèle dans
l’acétontrile
Figure 49. Spectre DC du pentapeptide contraint dans le TFE vs l’ACN
Figure 50. Spectre DC du décapeptide contraint 86 et modèle 87 dans le TFE Figure 51. Spectre DC du décapeptide contraint 86 et modèle 87 à différents
pourcentages d’eau dans le TFE
Figure 52. Spectre COSY du pentapeptide contraint 79 dans le DMSO deutéré à
10 mg/mL
Figure 53. Agrandissement du spectre COSY du pentapeptide contraint 79 a)
zone des NH b) zone des CHα
Figure 54. Spectre NOESY du pentapeptide contraint 79
Figure 55. Spectre NOESY du pentapeptide contraint, zone des pics croisés entre
les NH et les autres protons
Figure 56. Antibiotiques dans la classe des β-lactames
Figure 57. Lactivicine (LTV) et Phénoxyacétyl lactivicine (PLTV) Figure 58. Mécanisme d’action de la LTV et de la PLTV
Figure 59. Structure de base des sulfahydantoïnes antibactériens de Groutas où L
= groupe partant
Figure 60. L’acide carboxylique et point de substitution latérale des
sulfahydantoïnes nécessaires à l’activité anti β-lactamase
Figure 61. Mécanisme d’action hypothétique des sulfahydantoïnes comme
inhibiteur de protéases à sérine
Figure 62. Synthèses des sulfahydanoïnes au potentiel antibactérien ciblées Figure 63. Préparation de sulfahydantoïnes substituées par divers réactifs Figure 64. Ozonolyse des sulfahydantoïnes pour conduire aux composés cibles Figure 65. Schéma de type Michaelis-Menten de l’inhibition compétitive
Figure 66. Test d’inhibition de la β-lactamase TEM-1 avec les sulfahydantoïnes 98
à 103 à 1mM
Figure 67. IC50 de 102b
Figure 68. Activité enzymatique de la TEM-1 en présence de différents
Figure 69. IC50 des composés a) 102b et b) 101b
Liste des abréviations
Abréviation Nom
ACN Acétonitrile
ADN Acide désoxyribonucléique
Aib Acide α-aminoisobutyrique
Ala Alanine
ARN Acide ribonucléique
BOC tert-Butyloxycarbonyle
CCM Chromatographie sur couche mince
COSY Correlation spectroscopy
mCPBA Acide m-(chloro)perbenzoïque
CSI Isocyanate de chlorosulfonyle
DBU 1,8-Diazabicyclo[5,4]undéc-7-ène DC Dichroïsme circulaire DCC Dicyclohexylcarbodiimide DCE 1,2-dichlorométhane DIAD Diisopropylazodicarboxylate DIEA Diisopropyléthylamine DMAP (4(N,N-Diméthylamino)pyridine) DMF (N,N-diméthylformamide) DMS Diméthylsulfure DMSO Diméthylsulfoxyde E Enzyme EDC 1-(3-Diméthylaminopropyl)-3-éthylcarbodiimide hydrochlorure FMOC 9-Fluorénylméthyloxycarbonyle HBTU O-(Benzotriazol-1-yl)-N,N,N′,N′ tétraméthyluronium hexafluorophosphate
HOBT N-Hydroxybenzotriazole
I Inhibiteur
IC50 Concentration maximale pour 50% d’inhibition
Leu Leucine
LTV Lactivicine
NOESY Nuclear Overhauser Effect spectroscopy
P Produit
PBP Penicillin Binding Protein
pH Potentiel hydrogène
Phe Phénylalanine
PLTV Phénoxyacétyl lactivicine
Pro Proline
RMN Résonance magnétique nucléaire
S Substrat
Ser Sérine
SM Spectrométrie de masse
SOSS Synthèse organique sur support solide
TFA Acide trifluoroacétique
TFE Trifluoroéthanol
THF Tétrahydrofurane
UV Ultraviolet
Val Valine
« The future belongs to those who believe in the beauty of their dreams »
Remerciements
En tout premier lieu, je tiens à remercier très sincèrement mon directeur de recherche, le professeur Normand Voyer, pour m’avoir accueilli dans son groupe de recherche et pour avoir eu confiance en moi. Merci pour ce projet passionnant et rempli de défi, pour ta bonne humeur et ton optimisme inépuisable.
Dans un autre temps, je veux remercier Mélanie Tremblay, qui a été ma mentore dans ce projet. Merci également à Sophie Gobeil et Joëlle Pelletier (tests biologiques sur les dérivés de la sulfahydantoïne) qui m’ont si gentiment accueilli dans leur laboratoire. Ce fut une collaboration très enrichissante. Merci à Pierre Audet, qui m’a grandement aidé lors de la caractérisation par RMN.
Je voudrais également remercier mes collègues de laboratoire, pour leur agréable compagnie et leur soutien lors de ces deux dernières années : Sébastien Cardinal, François Otis, Charles Racine-Bethiaume, Pierre-Alexandre Paquet-Côté. Je ne peux également passer sous silence mes quatre bonnes amies qui ont été présentes tout au long de ma maîtrise, qui m’ont soutenu, encouragé et surtout, mis du bonheur dans chaque journée : Anne-Catherine Breton, Ariane Beaupré, Amélie Champagne et Katéri Ouellet.
Un merci spécial à mes parents, qui ont toujours été derrière moi, qui ont toujours eu une grande confiance en moi et qui m’ont donné le soutien moral et financier pour le faire.
Chapitre 1 : Introduction
1.1 Introduction générale
La chimie médicinale a pour but le design, la synthèse et la production de molécules ayant un potentiel d’agent thérapeutique1. La découverte d’un nouveau médicament implique la synthèse de dizaines de milliers d’analogues d’un composé faiblement actif visant l’augmentation de l’activité originale, de la biodisponibilité et de la sélectivité ainsi que la diminution de la toxicité. On estime que la découverte d’un médicament nécessiterait l’équivalent de 1 000 années pour un seul chercheur et plus de 10 000 composés synthétisés par des chimistes organiciens hautement qualifiés. Ce qui représente une somme importante d’argent et de temps pour l’industrie pharmaceutique1. La venue de la chimie combinatoire et de la synthèse en parallèle permettant la synthèse de librairies de composés plus rapidement est le résultat d’un changement fondamental dans la façon d’entreprendre la découverte de nouveaux médicaments. En effet, ces techniques permettent la synthèse et l’analyse de cent à mille fois plus de composés que l’approche traditionnelle un à un. Par exemple, un seul chimiste peut synthétiser plus de 1 000 analogues d’un produit cible en seulement quelques semaines alors que le travail de dix chimistes pendant un an aurait été nécessaire par la méthode habituelle2. Jusqu’à maintenant, plusieurs ingrédients actifs ont déjà été découverts à l’aide de différentes librairies.
Le design de composés peptidomimétiques avec de nouveaux motifs contraints est un domaine de recherche très actif avec un potentiel important pour le développement d’agents thérapeutiques peptidiques et non peptidiques. Ils représentent les composés de départ pour la conception d’agent thérapeutique. En effet, une multitude de peptides ont été découverts dans les 50 dernières années. Ils contrôlent une série de fonctions vitales tels les métabolismes, la réponse
immunitaire, la digestion, la douleur et la respiration. Ils sont également impliqués dans de nombreuses pathologies3.
1.2 Les protéases
Les quatre principales classes de protéases enzymatiques : métallo, sérine, cystéine et aspartique, catalysent sélectivement l’hydrolyse des liens peptidiques qui lient entre eux les acides aminés d’une chaîne peptidique. Le contrôle qu’elles effectuent sur la synthèse, le repliement et la fonction des protéines leur permet de réguler certains processus physiologiques tels que : digestion, fertilisation, croissance, différenciation, migration, défense immunitaire, apoptose, etc4.
Les protéases appartenant à ces classes sont également essentielles à la propagation de nombreuses maladies. Il existe plus de 50 protéases humaines pour lesquelles une ou plusieurs mutations génétiques ont été trouvées. Ces mutations peuvent conduire à des maladies héréditaires. Aussi, les inhibiteurs de ces protéases représentent une utilité thérapeutique prometteuse dans le traitement de maladies telles que le cancer, les maladies inflammatoires, respiratoires, cardiovasculaires, neurodégénératives ainsi que les infections virales. C’est pourquoi le développement d’agents thérapeutiques peut être une approche efficace contre ces enzymes. D’ailleurs, les organismes infectieux utilisent des types de protéases similaires qui, une fois installés dans la cellule hôte, exploitent les nutriments de l’hôte et agissent avec l’aide de la cellule pour propager l’infection5.
Déjà, il existe plusieurs inhibiteurs de protéases utilisés en clinique. À titre d’exemple (figure 1), le Saquinavir (1) produit par Hoffmann-La Roche, le Ritonavir (2) produit par les Laboratoires Abbott, l’Indinavir (3) produit par Merck, le Tripinavir (4) produit par Boehringer-Ingelheim et l’Amprenavir (5) produit par GlaxoSmithKline6,7 sont des médicaments abondamment prescrits pour le traitement des infections au VIH.
Figure 1. Exemples d’inhibiteurs de protéases de VIH-1 d’importance clinique
La plupart des protéases ont une séquence de clivage spécifique, déterminée par les caractéristiques d’hydrophobicité/hydrophilicité et la dimension du site actif, ce qui définit la possibilité de liaisons avec les acides aminés de la chaine latérale du substrat. La nomenclature standard utilisée pour désigner les résidus substrat/inhibiteur qui se lient aux sites enzymatiques correspondants est illustrée à la figure 2.
Figure 2. Représentation conformationnelle du site actif des protéases8
Il a été démontré que pour la plupart des protéases, le complexe inhibiteur/substrat se forme universellement en une conformation étendue ou brin-β. Le squelette peptidique se retrouve donc dans un arrangement linéaire. Cette conformation étant nécessaire pour obtenir de la reconnaissance par les protéases, il est intéressant de développer de nouveaux inhibiteurs conformationellement restreints qui adopteraient la conformation du groupement récepteur. La plupart des inhibiteurs peptidiques de protéases qui ont déjà été développés sont des molécules relativement flexibles ou extrêmement rigides. Le développement de nouvelles molécules peptidiques avec des restrictions conformationnelles pourrait mener à des inhibiteurs de protéases plus sélectifs et puissants.
1.3 Agents thérapeutiques peptidiques
De plus en plus de chercheurs et d’entreprises s’intéressent aux médicaments peptidiques. En effet, les difficultés traditionnellement associées aux peptides sont davantage surmontées, c’est pourquoi on retrouve de plus en plus de peptides en études cliniques. Les peptides jouent un rôle déterminant dans les processus biologiques. Plusieurs petits peptides sont un facteur clé dans la régulation d’une
grande variété de fonctions biologiques, d’action hormonale, de neurotransmission et d’inhibition enzymatique. Il existe une multitude de possibilités de séquences primaires ainsi que de structures tridimensionnelles, ce qui contribue à la diversité des rôles biologiques joués par les peptides. Les peptides et les protéines retrouvés dans la nature afin d’assurer plusieurs fonctions biologiques vitales constituent des candidats idéaux comme agents thérapeutiques. L’avantage qu’ils peuvent apporter en tant que médicament est qu’ils peuvent constituer un médicament puissant, spécifique et non toxique.
Par contre, certaines contraintes comme une faible stabilité, un court temps de demi-vie, une mauvaise biodisponibilité et une digestion rapide par les enzymes du corps restent encore à être surmontées. Davantage de compagnies pensent à intégrer les peptides dans leur stratégie de développement de médicaments. En effet, une fois les limitations des peptides résolus, on pourrait assister à la naissance d’une superclasse de médicaments qui pourraient enrichir significativement le pipeline de l’industrie pharmaceutique.
La recherche pharmaceutique des peptides a connu une hausse depuis les années 80. À cette époque, il y avait seulement environ 5 peptides qui entraient en étude clinique, alors que dès les années 90 il y a eu augmentation à 10 par année. Aujourd’hui, on peut compter près d’une vingtaine d’ajouts à chaque année. Le développement de médicaments peptidiques a des applications dans plusieurs domaines thérapeutiques tels l’oncologie, les problèmes métaboliques et particulièrement les maladies cardiovasculaires. Par contre, seulement 10% des candidats peptidiques au stade clinique sont capables d‘atteindre spécifiquement les cibles intracellulaires9-11.
Une bonne connaissance de la conformation active d’un peptide bioactif est un grand pas dans la compréhension de ses fonctions biologiques. Le design d’analogues contraints de peptides bioactifs constitue une des stratégies les plus utilisées par les chimistes médicinaux, soit la découverte de structure
tridimensionnelle active et le développement de nouveaux agents pharmaceutiques aux actions prolongées avec de meilleures propriétés thérapeutiques12,13.
Par contre, tel que mentionné, les peptides ont des limitations quant à leur utilisation in vivo. Ils ont une faible stabilité métabolique, une mauvaise absorption après ingestion orale, une pharmacocinétique inadéquate et des effets secondaires indésirables importants.
Figure 3. Limitations dues à la flexibilité des molécules peptidiques14
La flexibilité conformationnelle des peptides est un facteur important dans la cause des effets secondaires. En effet, lorsqu’en milieu aqueux, ces molécules sont très flexibles, ce qui a pour effet de diminuer leur activité inhibitrice et favoriser leur interaction avec d’autres récepteurs biologiques menant à des effets secondaires. De même, cette flexibilité empêche la bonne définition de la conformation active (figure 3). Afin de contrer cette flexibilité, il est possible d’imposer une restriction conformationnelle sur le peptide. D’ailleurs, on retrouve dans la nature plusieurs contraintes utilisées afin de réduire la flexibilité, incluant l’incorporation d’un cycle dans la structure.
1.4 Les hétérocycles
Les hétérocycles sont par définition une classe de composés où au moins un atome d’un cycle carboné est remplacé par un hétéroatome. Les hétérocycles les plus courants contiennent un atome d’azote ou d’oxygène. Ils représentent des structures de bases intéressantes dans la modification de pharmacophores pour l’obtention de composés bioactifs plus efficaces et sélectifs. La restriction de la mobilité conformationnelle ainsi que leurs propriétés comme donneur et/ou de sites accepteurs de lien hydrogène rend les hétérocycles très intéressants pour le domaine de la chimie médicinale15. Ils sont d’ailleurs utilisés dans plusieurs librairies combinatoires. Connu comme étant des agents thérapeutiques utiles, on retrouve une grande diversité structurale d’hétérocycles.
Plusieurs librairies d’hétérocycles ont conduit à de nouveaux inhibiteurs d’enzymes. Les hydantoïnes(6), les dicétopipérazines(7), les pyrrolidines(8), les imidazoles(9), les β-lactames(10) et les isoxazoles(11) sont quelques exemples d’hétérocycles faisant parti de librairies (figure 4)1.
1.5 Le motif sulfahydantoïne
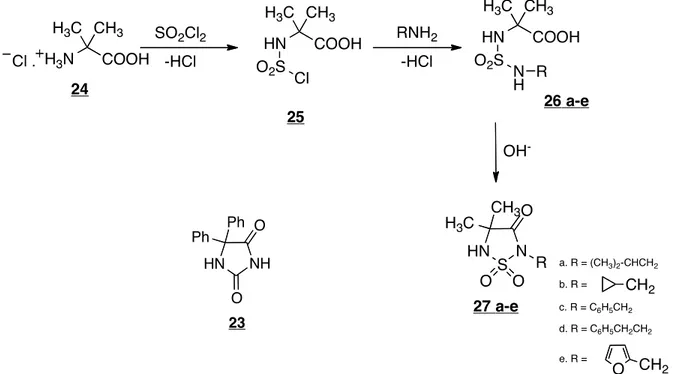
Les sulfahydantoïnes (figure 5) s’avèrent être un hétérocycle très intéressant.
Figure 5. Le motif sulfahydantoïne
En effet, la sulfahydantoïne représente un motif original. Incorporée dans une chaîne peptidique, elle amène une restriction conformationnelle importante du squelette peptidique. En effet, son intégration dans un acide aminé ou un peptide induit une restriction importante des angles Ψ et Φ (figure 6). De plus, le groupement sulfonyle, en plus d’être un site électrophile réactif, rapproche les deux atomes d’azote et contraint ces deux angles afin de conduire à des peptides plus rigides16.
Les sulfahydantoïnes ont d’abord été ciblées comme des hétérocycles intéressants en raison de leurs propriétés potentielles d’antiépileptiques, d’antihypertenseurs, de succédanés du sucre et pour leur affinité pour les protéines MHC de classe
II18,19. Groutas les a également reconnues comme étant des inhibiteurs de
protéases à la sérine20.
Groutas a également rapporté la synthèse en solution d’une librairie de 1,1-dioxyde-3-oxo-1,2,5-thidiazolidine ayant une très bonne activité inhibitrice pour les protéases à la sérine mais plus particulièrement de l’élastase humaine, de la cathepsine G et de la protéase 320. D’ailleurs, ces protéases sont reconnues pour jouer un rôle important dans les maladies inflammatoires comme la bronchite, l’emphysème, le psoriasis, etc. Elles représentent donc d’importantes cibles thérapeutiques. C’est pourquoi des inhibiteurs sélectifs de ces enzymes pourraient conduire à des thérapies potentiellement efficaces contre ces maladies et autres problèmes associés.
Groutas et al. ont utilisé une analyse cristalline du complexe d’un inhibiteur d’ovomucoïde de dinde et de l’élastase des leucocytes humains pour le design des inhibiteurs, ce qui a permis de démontrer que le motif sulfahydantoïne est un élément de reconnaissance hautement efficace20. Son arrangement spatial bien défini permet une optimisation des interactions favorables avec les sites Sn et Sn1
du site actif de l’enzyme. De plus, une affinité spécifique pour les différentes protéases à la sérine a aussi été démontrée à l’aide d’études de sélectivité. Les groupements alkyles et aromatiques sont reconnus pour favoriser l’inhibition des protéases à la sérine neutre. Ce qui correspond bien à la spécificité connue des protéases à la sérine pour leurs substrats20,22.
Figure 7. Structure générale du motif sulfahydantoïne comme élément de
reconnaissance développé par Groutas
1.6 Contraintes peptidiques
Les chimistes médicinaux s’intéressent au peptidomimétisme depuis plus d’un quart de siècle maintenant. Le peptidomimétisme a été conçu afin de contourner les problèmes associés aux peptides naturels, soit leurs pharmacocinétiques inadéquates, leur faible biodisponibilité et leurs nombreux effets secondaires. Cette stratégie sert également à améliorer la sélectivité et l’efficacité des peptides, d’où l’imitation de peptides a un grand potentiel dans la découverte de nouveaux médicaments13. Elle se décrit par un composé, une petite structure non-peptidique aux propriétés du peptide d’origine pouvant imiter ou bloquer les effets biologiques d’un peptide. Agissant comme ligand d’un enzyme, il peut servir de substrat ou d’inhibiteur. Le design de composés peptidomimétiques commence par le développement d’une relation structure-activité qui pourrait définir une séquence active ou un pharmacophore majeur conduisant à l’identification du ou des résidus responsables des effets biologiques. Alors, une contrainte structurale est insérée afin de rechercher les mêmes caractéristiques d’arrangements 3D. Depuis les 50 dernières années, une multitude de peptides biologiquement actifs ont été découverts11.
Les hélices-α, les feuillets-β et les tournants sont les trois classes de structures secondaires principales des peptides et des protéines. Il existe de nombreuses stratégies de contraintes peptidiques. Plusieurs techniques consistent à obtenir des conformations d’hélices-α et de feuillets-β, mais les techniques de restriction les plus souvent rencontrées sont l’induction ou la stabilisation des conformations
en tournants. Ceci est dû, entre autre, au fait que les tournants peuvent être induits dans un petit peptide. Par ailleurs, une grande partie des recherches sur les peptides contraints a démontré que les conformations étendues sont des sites idéaux pour une bonne reconnaissance puisqu’ils représentent un arrangement hautement accessible autour d’une protéine compacte. Dans le domaine de la peptidomimétique, le concept de restriction conformationnelle a été exploré dans une multitude de directions. La section suivante en présente quelques exemples12.
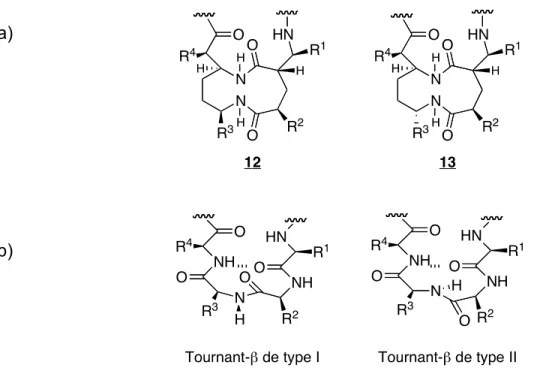
1.6.1 Tournant-β
Les tournants, qui constituent une structure secondaire souvent retrouvée à la surface des protéines, présentent plusieurs groupements fonctionnels polaires. Ils représentent donc des sites de reconnaissance potentiels pour régir plusieurs processus biologiques. Ils sont aussi reconnus pour être impliqués dans plusieurs interactions substrat/récepteur. La conformation tournant-β est impliquée entres autres dans les interactions protéine-protéine. Elle sert à lier deux structures secondaires ensemble, par exemple une portion hélice-α à un feuillet-β. On retrouve deux types de tournant-β correspondant, les types I et II ainsi que leur conformation inverse soient le type I’ et le type II’. Ces deux types se distinguent par une différence d’angle de 180°. C’est seulement une petite partie des protéines qui sera impliquée lors des interactions et constituera la majorité de l’affinité. La mimétique des tournants-β permet la synthèse de librairies de petites molécules représentant la majeure partie du motif de reconnaissance23,24.
Plusieurs chercheurs se sont penchés sur cette problématique au cours des dernières années. Les récentes études basées sur une peptidomimétique minimaliste, soit l’imitation d’une chaîne d’acide aminé par des composés présentant les mêmes propriétés sans contenir la chaîne peptidique, ont été inspirées par les travaux de Smith, Hirchmann et Nicolaou entre autres25.
En effet, le groupe de Michael Kahn s’est intéressé à cette stratégie et a ainsi synthétisé deux motifs (12 et 13), qui miment respectivement les types I et II des tournants- β (figure 8).
a)
b)
Figure 8. a) Composés mimétiques des tournants-β du groupe de Kahn b)
Conformation des tournants-β de type I et II26,27
Le chercheur Kevin Burgess s’est lui aussi penché sur la question. Son groupe de recherche s’intéresse particulièrement à la mimétique simple de structure secondaire. Ce qui a conduit au design d’un composé analogue des tournants-β de type I (14).
1.6.1.1 β-Hairpin
Une dernière approche de peptidomimétisme souvent rencontrés dans la littérature consiste en la mimétique des structures en tête d’épingle (β-hairpins). Cette structure secondaire est formée par deux brins-β antiparallèles liés par des liens hydrogène et rejoints par un tournant ou une boucle serrée. La peptidomimétique de cette conformation pourrait mener à une nouvelle thérapeutique d’épitopes. Cette approche touche à plusieurs cibles thérapeutiques, dont les anticorps et les récepteurs des cytokines, les peptides antimicrobiens, les inhibiteurs de protéases, les interactions protéine-protéine et les interactions protéine-ARN.
Une des approches utilisées est le design de macrocycle adoptant la conformation β-hairpin par le moyen d’un pont. Plusieurs modèles de dipeptides ont été étudiés afin d’induire cette conformation à la chaîne peptidique. Le dipeptide D-Pro-L-Pro (figure 10) s’avère être un pont très efficace. En effet, ce dipeptide adopte une conformation de tournant-β, ce qui est idéal pour permettre l’induction de la conformation. De plus, le groupe de recherche de Marshall a bien démontré que le dipeptide D-Pro-L-Pro adopte bien une conformation de tournant-β de type II. Quant au groupe de recherche de Kopple, ils ont démontré que ce même dipeptide permet à un hexapeptide d’adopter une conformation β-hairpin39-41.
1.6.2 Brins-β et feuillets-β
Le brin-β était souvent reconnu comme étant une conformation peptidique mal définie. Récemment, il a été identifié comme un élément fondamental de reconnaissance des processus physiologiques reconnus par plusieurs récepteurs biomoléculaires30. D’ailleurs, il a été démontré que toutes les familles de protéases lient leurs inhibiteurs/substrats dans la conformation de brin-β étendu où la structure peptidique (ou la molécule équivalente non-peptidique) se retrouve dans un arrangement conformationnel linéaire30. De plus, les brins-β sous forme de feuillets-β sont non seulement des éléments de stabilisation de la structure des protéines mais aussi des motifs clés de reconnaissance qui se lient à d’autres protéines ou à l’ADN. Les feuillets-β sont utilisés par les protéines entres autres pour se lier à l’ADN. Le peptidomimétisme des brins-β pourrait donc permettre la découverte de ligands compétitifs pour les récepteurs se liant normalement à des feuillets-β. Cependant, la formation de feuillets-β peut parfois être indésirable. Par exemple, l’agrégation de ces derniers joue un certain rôle dans les maladies d’Alzheimer et Creutzfeldt-Jakob c’est pourquoi une intervention sur la formation des feuillets-β représente une nouvelle stratégie thérapeutique potentielle en cours de validation30, 31.
Il existe plusieurs méthodes de peptidomimétique des brins-β. D’ailleurs la nature utilise également ces méthodes comme les ponts disulfures, les liens doubles, les acides aminés N-méthyle, les aromatiques et les hétérocycles ainsi que la cyclisation. Plusieurs chercheurs se sont penchés sur la question dont Amos B. Smith III. Ce dernier a travaillé, en collaboration avec d’autres chercheurs sur le développement d’une nouvelle structure peptidomimétique non-peptidique ayant des propriétés pharmacocinétiques améliorées. Deux objectifs différents ont fait l’objet de recherche, soit le design d’agoniste/antagoniste non-peptidique et le développement de nouveaux inhibiteurs de protéases. Dans le deuxième cas, les recherches visaient le développement de nouveaux composés qui mimeraient la conformation brin-β d’un peptide d’origine. Leur design se base sur la structure non-peptidique pyrrolinone afin d’imposer une conformation brin-β aux inhibiteurs.
Figure 11. Peptidomimétique de brin-β de Smith25,30
1.6.3 Hélices-α
Le domaine de recherche sur l’imitation de structures synthétiques et plus spécifiquement d’une région de la surface des protéines comme les hélices-α connait un moins grand succès que pour les conformations précédentes. Cette conformation peut également permettre un contrôle des interactions protéine-protéine. Plusieurs stratégies de contraintes existent, dont la stabilisation d’un peptide de longueur moyenne en hélice-α. Le chercheur Andrew D. Hamilton s’est penché sur cette problématique32. Son groupe de recherche a développé un composé complètement non-peptidique où l’on retrouve les mêmes fonctionnalités des chaînes latérales ainsi que les mêmes distances et angles que dans les hélices-α. Leurs travaux ont mené à une nouvelle famille de protéomimétiques basés sur un modèle de terphényle fonctionnalisé (figure 12). Ce dernier représente une structure qui imite deux tournants de la myosine et démontre certaines analogies fonctionnelles du lien avec une grande affinité à la calmoduline33.
a)
b)
Figure 12. a) Terphényle b) Composés mimétiques de Hamilton33
Les complexes protéine-hélices-α présentent plusieurs interactions critiques impliquant les chaines latérales des résidus i, i + 3 et i + 7 qui sont souvent retrouvés le long d’une face de l’hélice. Les Tertphényles représentent un modèle très intéressant pour cette peptidomimétique puisqu’ils représentent une structure simple et facile à synthétiser32-34.
1.6.4 Peptides cycliques
Restreindre le degré de liberté d’une molécule peut mener à l’amélioration de son interaction avec le composé cible ainsi qu’à améliorer leur affinité. Le peptidomimétisme par macrocyclisation est une autre technique qui a eu du succès dans ce domaine. Les recherches du groupe de Pierre Deslongchamps portent sur le développement d’une nouvelle classe de macrocycles.
Figure 13. Macrocycles de Deslongchamps35,36
Ce macrocycle est un tripeptide cyclisé, lié par une attache (Téther). Cette stratégie présente plusieurs avantages dont la possibilité d’utilisation d’une multitude d’acides aminés pour former le tripeptide ainsi que plusieurs possibilités de composés pour former l’attache de ce macrocycle, pouvant ainsi mener à une large variété de composés37.
Le groupe de recherche du professeur Stephen Hannessian a orienté ses recherches sur l’incorporation d’un peptide cyclisé dans un composé inhibiteur potentiel. Leurs travaux sont inspirés de l’acide α-kaïnique qui représente aussi un acide L-glutamique contraint. Ils ont ainsi fait le design de plusieurs nouveaux motifs d’inhibiteurs mono- et bicyclique38.
20
Figure 14. Acide α-kaïnique38
Le potentiel important du développement de nouvelles stratégies de contraintes peptidiques est bien démontré par ces derniers exemples. Plusieurs méthodes de contrainte du squelette peptidique existent dans la littérature. Les types de contraintes sont classés en deux grandes catégories, soit les contraintes globales et les contraintes locales. Les contraintes globales consistent en une formation de
liaisons covalentes entre deux terminaisons du squelette peptidique, entre deux chaînes latérales ou entre une terminaison et une chaîne latérale. Les contraintes locales constituent quant à elles une introduction d’une contrainte sur le squelette peptidique ou sur les chaines latérales afin de modifier les propriétés du peptide d’origine42,43.
1.7 Stratégie de contrainte peptidique
Toutes ces techniques de restriction ont certaines limitations. Elles sont difficilement adaptables à la synthèse supportée et sont généralement limitées à la formation de chaînes recourbées.
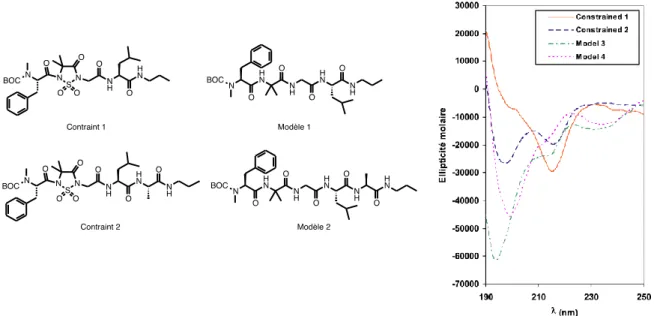
Les études récentes de notre groupe de recherche ont démontré que les peptides contraints par le motif sulfahydantoïne adoptent une conformation ouverte70. Quatre peptides ont été synthétisés dans nos laboratoires pour fin d’analyses, soit deux peptides contraints par le motif sulfahydantoïne et leur peptide modèle correspondant (figure 15). Des analyses par dichroïsme circulaire ont révélé un changement de structure entre les peptides contraints et modèles (voir figure 15). Puisque ces peptides ont démontré que le motif sulfahydantoïne induisait bien une contrainte dans une chaîne peptidique, nous avons décidé de poursuivre les études sur de nouveaux modèles de peptides.
Figure 15. Étude par dichroïsme circulaire des peptides contraints et modèles
correspondant développé dans notre laboratoire
De plus, comme il a été mentionné plus haut, l’introduction du motif sulfahydantoïne dans une chaîne peptidique induit une restriction importante des angles ψ et ϕ. Aussi, la présence du groupement sulfonyle a pour effet de rapprocher les deux atomes d’azote pour ainsi donner un peptide beaucoup plus rigide (figures 16 et 17). Peu de peptides rapportés dans la littérature sont contraints dans leur forme ouverte et pourtant beaucoup de peptides d’un grand intérêt adoptent cette conformation dans leur site actif.
21 Figure 16. Stratégie de contrainte peptidique par l’incorporation d’une
Des études antérieures sur un tripeptide analogue contraint de Val-Phe-Ala réalisées à l’aide de la modélisation moléculaire (figure 17), ont permis d’obtenir les valeurs des angles ψ et ϕ imposés par la présence du motif sulfahydantoïne et d’ainsi situer ce dernier sur le diagramme de Ramachandran (figure 17)47.
22
Figure 17. Tripeptide contraint sulfonyle N-Boc-L-Val-L-Phe-[SO2]-D-Ala-OMe et
son diagramme de Ramachandran correspondant47
L’introduction du motif sulfahydantoïne dans une chaîne peptidique permet donc l’induction d’une conformation ouverte unique sur cette même chaîne peptidique tout en rigidifiant latéralement la chaîne. Aussi, cette stratégie est adaptable à la synthèse supportée en plus d’être générale pour toutes séquences16.
Notre groupe de recherche s’est penché sur le développement d’une stratégie pour incorporer le motif sulfahydantoïne dans une chaîne peptidique. Plusieurs obstacles ont été rencontrés lors de ce développement. Finalement, une stratégie de synthèse en solution a été développée dans nos laboratoires afin d’obtenir des molécules peptidomimétiques originales.
La deuxième partie de ce mémoire est dédiée à l’élaboration d’une de ces synthèses et son adaptation à la synthèse sur support solide (chapitre 2). Des études conformationnelles par dichroïsme circulaire et RMN sur des peptides contraints seront également présentées (chapitre 3).
Chapitre 2 : Synthèse d’un peptide modèle
contraint
2.1 Préambule
Nous avons démontré que l’introduction du motif sulfahydantoïne dans une chaîne peptidique mène à l’épimérisation du stéréocentre de la sulfahydantoïne (figure 18)5. Cette épimérisation serait entres autres causée par la présence du groupement carbonyle de l’acide aminé voisin en N5. Il était d’abord essentiel de développer un tripeptide analogue contraint où il y aurait absence du carbonyle en N5 et donc d’épimérisation pour pouvoir ensuite l’introduire dans une chaîne peptidique. De plus, l’absence du groupement carbonyle devrait également aider à éliminer l’ouverture du cycle de la sulfahydantoïne lors de la synthèse sur support solide, une réaction indésirable que nous avons également observée. Nous avons donc développé une méthode afin d’éliminer le groupement carbonyle problématique.
Figure 18. Lieu d’épimérisation lors de l’introduction du motif sulfahydantoïne dans
une chaîne peptidique
2.2 Rappel bibliographique sur la synthèse de sulfahydantoïnes
Peu de synthèses de sulfahydantoïnes chirales ont été rapportées. La première synthèse du motif sulfahydantoïne chiral en solution fut décrite par Dewynter et
al.46. Par ailleurs, d’autres groupes ont aussi rapporté des synthèses de sulfahydantoïnes, mais avec des motifs achiraux ou racémiques.
2.2.1 Hanewacker et al.19
Hanewacker et al. se sont intéressés au motif sulfahydantoïne pour son analogie à la phénytoïne (23), un composé aux propriétés antiépileptiques. Ils ont travaillé sur la synthèse d’une série de sulfahydantoïne (1,1-dioxyde-1,2,5-thiadiazolidin-3-ones) en positions 1, 4 et 4 au départ de l’acide α-aminoisobutyrique (Aib) (24). Les sulfahydantoïnes (27 a-e) ont été synthétisées par cyclisation en milieu basique des sulfamides (figure 19).
Figure 19. Schéma de synthèse de Hanewacker et al.19
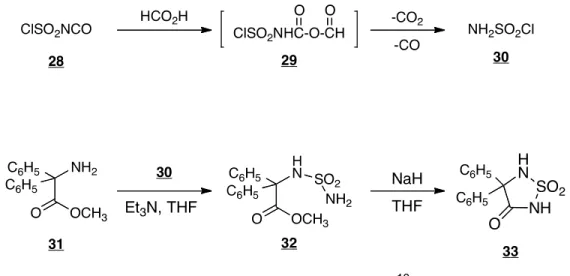
2.2.2 Timberlake et al.18
Timberlake et al. se sont également intéressés aux sulfahydantoïnes pour les mêmes raisons que Hanewacker, soit leur analogie à la phénytoïne. Ils ont quant à eux utilisé l’isocyanate de chlorosulfyle afin d’introduire le groupement sulfamide et finalement les sulfahydantoines sont obtenus à la suite d’une cyclisation en milieu basique (NaH). De plus, à la suite de leurs études cristallographiques, ils ont découvert que le motif sulfahydantoïne n’était pas plan.
Figure 20. Schéma de synthèse de Timberlake et al.18 2.2.3 Muller et al.44
Muller et al. ont travaillé sur le développement de nouveaux édulcorants. Ce qui a mené leur recherche sur la synthèse de 1,1-dioxyde-1,2,5-thiadiazolidin-3-ones à partir d’acides aminés racémiques et de l’isocyanate de chlorosulfonyle afin d’évaluer leur goût sucré. Ils ont ainsi découvert que les sulfahydantoïnes sont dix fois moins sucrés que le sucrose.
2.2.4 Kohn et al.45
Les 1,1-dioxyde-3-imino-1,2,5-thiadiazolidines 38 ont servis de composés de départ à la synthèse de Kohn et al. des 1,1-dioxyde-1,2,5-thiadiazolidin-3-ones (figure 22). Ces derniers sont préparés à l’aide d’un traitement au HCl éthanolique de 38 permettant d’obtenir l’ester éthylique intermédiaire 39 qui, après un traitement basique, résulte en sulfahydantoïnes 37.
Figure 22. Schéma de synthèse de Kohn et al.45
2.2.5 Dewynter et al.46-48
Dewynter et al. ont effectué la synthèse des sulfahydantoïnes chirales par cyclisation des sulfamides 41 en milieu basique à reflux (figure 23). Cette synthèse de sulfahydantoines fut la première rapportée par nos groupes.
2.2.6 Groutas et al.49-51
Comme il a été mentionné précédemment, Groutas et al. ont également synthétisé des sulfahydantoïnes fonctionnalisées à partir d’acides aminés chiraux. Cette synthèse a permis l’obtention de nouveaux inhibiteurs sélectifs de protéases à la sérine. La première synthèse publiée par Groutas emploie la même méthode que Dewynter et al. décrite précédemment, soit la synthèse du sulfamide à partir du CSI suivi d’une cyclisation en milieu basique dans le THF (figure 24). La réalisation d’une amination réductive pour introduire un alkyle en N5 avant la cyclisation de la sulfahydantoïne est ingénieux.
Figure 24. Schéma de synthèse de Groutas et al.49-51
2.2.7 Diederich et al.52
En 1999, Diederich et al. publiait une synthèse de sulfahydantoïnes à partir d’acides aminés racémiques inspirée de la synthèse de sulfamide de Dewynter et
al.. À la suite de cette synthèse, ils ont mesuré, par titrage, le pKa du proton en
position 5 de la sulfahydanoïne 51, soit 11,9, ce qui a permis d’identifier ce composé comme un fort donneur de pont hydrogène.
Figure 25. Schéma de synthèse de Diederich et al.52 2.2.8 Albericio et al.53,54
Albericio et al. ont travaillé sur le développement de nouveaux herbicides. Intéressés par le motif sulfahydantoïne comme composé modèle, ils ont rapporté la première synthèse de sulfahydantoïnes sur support solide (résine de Wang) (figure 26). Groutas avait déjà présenté cette même stratégie, dans le cas d’une synthèse en solution.
Les composés ayant un potentiel en agrochimie ont un pKa similaire à celui des
acides carboxyliques. Albericio a donc, dans le cadre de ses recherches, mesuré les pKa des analogues synthétisés, qui sont d’environ 3,5.
2.2.9 Voyer et al.16,55
Peu de synthèses de sulfahydantoïnes sur support solide ont été décrites dans la littérature. Albericio et al. fut le premier groupe à rapporter ce type de synthèse. Notre laboratoire s’est également intéressé à la synthèse de sulfahydantoïnes chirales sur support solide ayant comme objectif l’obtention de librairies de composés biologiquement actifs. Cette méthode vise la synthèse de sulfahydantoïnes hautement et sélectivement substituées. Cette stratégie de synthèse utilise la résine oxime comme support solide (figure 27). L’acide aminé est d’abord couplé à la résine. Le schéma de synthèse suit le même protocole que Groutas et al. ont utilisé19. La cyclisation s’effectue lors du clivage dans des
conditions douces et anhydres.
Figure 27. Schéma de synthèse de Voyer et al.16,55
Notre laboratoire a aussi travaillé sur une approche de synthèse de la sulfahydantoïne en solution qui sera présentée au prochain point.
2.3 Stratégie de synthèse de tripeptides analogues contraints par la
sulfahydantoïne sans carbonyle en N5
Notre stratégie de synthèse de tripeptides analogues contraints par la sulfahydantoïne s’inspire de la méthode de synthèse de Dewynter et al. (figure 28)56.
Figure 28. Stratégie de synthèse en solution pour la préparation d’un pseudo
tripeptide contraint sans carbonyle sur N5.
Par cette méthode, l’hétérocycle 60 est premièrement synthétisé56. Ensuite, une substitution sélective de l’azote N5 suivie d’une ozonolyse sont effectuées afin d’obtenir le tripeptide analogue contraint 59.
2.3.1 Synthèse de la sulfahydantoïne
Cette synthèse part d’un ester d’acide aminé réalisé par une simple réaction d’estérification à l’aide du chlorure de thionyle. Cette réaction permet d’utiliser une grande variété d’acide aminé de départ, menant ainsi à plusieurs analogues sulfahydantoïnes (figure 29).
L’analogue sulfonylé d’une urée est obtenu à l’aide du réactif clé utilisé dans cette synthèse, l’isocyanate de chlorosulfonyle, CSI (figure 30). On retrouve deux sites électrophiles sur ce composé, ce qui le rend très utile dans la synthèse de ce genre de fonction.
Figure 30. Aménagement fonctionnel et réactivité du CSI.
Un dérivé chlorosulfonamide, instable, protégé par un groupement BOC est obtenu en plaçant le CSI en présence de t-BuOH. Ce composé peut être additionné in situ à un ester d’acide aminé afin d’obtenir l’analogue sulfonylé d’une urée (63), un composé stable14.
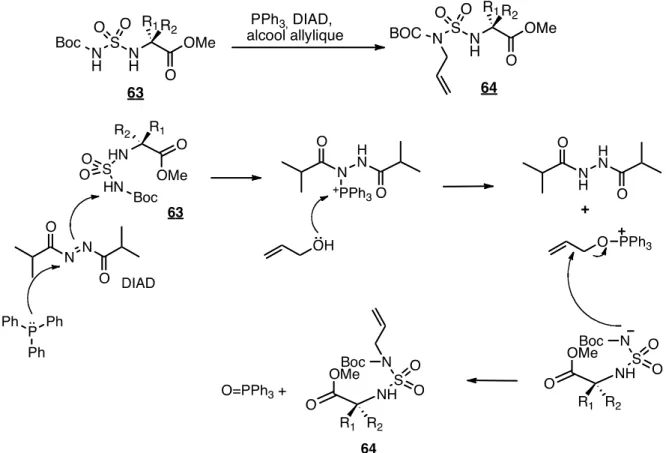
Nous avons observé la présence de produit O-alkylé lors de la réaction de Mitsunobu lorsque celle-ci était réalisée après la réaction de cyclisation avec différents alcools utilisés (figure 31). Afin d’éviter la présence de produit O-alkylé lors de la réaction de Mitsunobu, nous voulons réaliser cette réaction avant la cyclisation57. Cette réaction est utilisée dans cette synthèse dans le but d’effectuer une alkylation sur un azote à l’aide d’un alcool. La disponibilité d’une grande variété d’alcool commercial permet la synthèse de plusieurs analogues.
42
Figure 31. Réaction de Mitsunobu sur la sulfahydantoine démontrant la O- et la
Lors de la réaction de Mitsunobu (figure 32)58, la triphénylphosphine attaque en premier lieu le diisopropyl azodicarboxylate, DIAD. Ensuite, l’azote peut aller chercher un proton acide, généralement d’un acide carboxylique. Selon Albericio et
al. (53), le pKa du proton sur l’azote en N2 du dérivé sulfamide est légèrement plus
bas que celui d’un acide carboxylique. Diederich et al.52 a quant à lui déterminé que le pKa du proton sur l’azote en N5 est d’environ 11. Ce qui permet d’observer
une réaction sélective sur l’azote N2 afin d’obtenir le sulfamide alkylé sélectivement en position 2 (64). Une double Mitsunobu peut être observée si la réaction n’est pas arrêtée à temps. Les rendements obtenus sont de 60-79%.
Figure 32. Mécanisme de la réaction de Mitsunobu et préparation de 64.
Les sulfahydantoïnes sont finalement obtenues suite à la déprotection du groupement BOC suivie d’une cyclisation réalisée en milieu basique à reflux (figure 33). Les rendements obtenus pour cette réaction sont de 73-96 %.
Figure 33. Cyclisation du sulfamide menant aux sulfahydantoïnes 60.
Nous avons noté qu’aucune racémisation n’est observée lors de cette cyclisation malgré les conditions basiques utilisées.
2.3.2 Stratégie de substitution en N5
2.3.2.1 Préparation d’analogues alkylants d’acides aminés.
Pour préparer nos peptides contraints avec la sulfahydantoïne sans racémisation nous avons besoin d’alkiler en position N5 avec des analogues d’acides aminés sans carbonyles. Des composés électrophiles ont d’abord été synthétisés avec différents groupements partants à l’aide de méthodes connues dans la littérature (figure 34). En premier lieu, une réduction de l’acide aminé de départ a été réalisée à l’aide du BH3/THF afin de réduire le carbonyle. L’alcool a été substitué par un
bromure par une réaction de bromation à l’aide du N-bromosuccinimide59. L’iodure a été introduit par l’iode60. Finalement le groupement O-mésylate a été introduit à l’aide du chlorure de l’acide méthylsulfonique61. Les rendements obtenus pour ces réactions sont d’environ 30-50 %.
Figure 34. Synthèse des électrophiles pour l’étape d’alkylation au départ de l’acide
aminé L-phénylalanine.
La substitution en position N5 a été tentée en utilisant la nucléophilicité de cet azote sur la sulfahydantoïne. Plusieurs essais de substitution ont été réalisés dans différentes conditions et à l’aide de composés aux groupements partants variés (67-69) (figure 35)62. Les résultats sont rapportés au tableau 1.
Figure 35. Alkylation en position N5 de la sulfahydantoïne
Tableau 1. Essais de substitution en position N5 sur la sulfahydantoïne (68).
Aucune réaction tentée à l’aide des électrophiles (67-69) n’a fonctionné. Dans tous les cas, les produits de départ étaient récupérés à la suite d’une purification sur colonne de chromatographie sur silice. Le cycle aromatique présent sur la chaîne latérale pourrait expliquer l’absence de réaction par un effet d’encombrement stérique empêchant ainsi la substitution. L’azote N5 étant aussi encombré, un nouvel essai de substitution a été réalisé à l’aide du N-Boc-2-bromo-1-aminoéthyle (figure 36). Ce composé commercial a été sélectionné dans le but de réduire, voir éliminer tout encombrement stérique de l’électrophile. Les résultats sont rapportés au tableau 2.
Figure 36. Alkylation en position N5 de la sulfahydantoïne
Tableau 2. Essais de substitution sur la sulfahydantoïne par
N-Boc-2-bromo-1-aminoéthyle pour obtenir 72.
La réaction a été réalisée avec succès. De plus, celle-ci a été faite plusieurs fois et les rendements sont similaires, ce qui a démontré sa répétabilité. Par contre, l’optimisation de la réaction n’a pas été effectuée, ce qui explique les faibles rendements. Le but du projet étant en premier lieu la synthèse de nouveaux peptides contraints par le motif sulfahydantoïne ainsi que l’effet de cette contrainte sur la structure du peptide, l’optimisation de cette réaction a été jugée non nécessaire au moment de la recherche. Éventuellement, une optimisation serait envisagée.
2.3.2 Ozonolyse
L’alcool allylique utilisé lors de la réaction de Mitsunobu dans la synthèse de la sulfahydantoïne a été choisi dans le but d’obtenir une glycine au C-terminal par une réaction d’ozonolyse.
L’ozone a été produit à l’aide d’un ozonisateur à partir d’oxygène. Un papier KI a été utilisé comme indicateur de présence d’ozone. L’ozonolyse a été suivie d’une réaction à l’oxone afin de former l’acide et ainsi obtenir la glycine au C-terminal du tripeptide analogue contraint 73.
Le composé 72 a été dissout dans un minimum de CH2Cl2 puis la solution est
amenée à -78°C. Ensuite, un courant d’ozone est bullé dans la solution jusqu’à une coloration bleue ou jusqu’à une réaction complète démontrée par CCM. Finalement, un traitement au DMS est effectué pendant la nuit pour obtenir l’aldéhyde correspondant (figure 37). L’acide (73) est obtenu à la suite d’une réaction avec l’Oxone dans le DMF qui est suivie par SM63-65.
Figure 37. Réaction d’ozonolyse pour obtenir le peptide contraint 73
2.4 Synthèse sur support solide
La synthèse organique sur support solide (SOSS) est une méthode très utile dans la synthèse d’oligomère peptidique. Cette méthode apporte plusieurs avantages tel la réduction du nombre d’étapes à seulement trois : addition des réactifs, filtration et lavage de la résine. Ce qui facilite de beaucoup les synthèses et permet l’automatisation. De plus, les synthèses sont réalisées plus rapidement grâce à l’élimination des étapes de purification. Seul le composé final, après clivage, nécessite d’être purifié. Aussi, il est possible par cette méthode d’atteindre des rendements très élevés puisqu’une plus grande concentration (excès) des réactifs est utilisée. Finalement, plusieurs composés peuvent être synthétisés en parallèle simultanément en SOSS66.
2.4.1 Support solide
Le choix du bon support solide est important afin d’obtenir des synthèses efficaces. Il existe plusieurs supports pouvant être utilisés. Merrifield67 fut le premier à utiliser un copolymère de styrène. Le polystyrène est le polymère le plus souvent utilisé.
Polystyrène (PS) :
Les résines sont des supports solides, qui, avec de bons solvants, sont en structure de gel permettant la pénétration des réactifs et du solvant dans les billes, dans lesquelles les réactions chimiques ont lieu.
Dans le cadre de nos synthèses, le support solide utilisé est la résine oxime. C’est Kaiser et DeGrado qui ont développé cette résine dans les années 8068. Ce support solide permet de travailler en stratégie BOC, une stratégie efficace au clivage facile qui ne génère aucun sous-produit nuisible aux réactions subséquentes. De plus, l’utilisation de ce groupement protecteur permet de conserver le motif sulfahydantoïne intact lors de l’élongation de la chaîne, puisque sa déprotection se fait dans des conditions acides. Le premier acide aminé est lié à la résine par un lien ester d’oxime qui se forme entre le support solide et le carbonyle de l’acide aminé. Ainsi, une élongation de la chaîne peptidique est possible puisque ce lien est assez stable, en plus d’être suffisamment labile pour permettre un clivage par un nucléophile dans des conditions relativement douces69.
2.5 Intégration du motif sulfahydantoïne dans un peptide plus complexe
2.5.1 Travaux antérieurs
La synthèse de peptides contraints par le motif sulfahydantoïne a déjà été réalisée dans notre laboratoire. Dans ce cas, la chaîne peptidique de ces peptides n’a pas été modifiée. Lors du clivage par un nucléophile, une ouverture et/ou un déplacement du cycle étaient observés. De plus, l’acide aminé Aib a été utilisé lors de la synthèse de la sulfahydantoïne afin d’éliminer le problème de racémisation. En effet, la présence du groupement carbonyle de l’acide aminé voisin en N5 rend le proton alpha chiral de la sulfahydantoïne plus acide provoquant ainsi son épimérisation. Puisque lors de ces travaux il a été démontré que le motif sulfahydantoïne induisait bien une conformation étendue sur la chaîne peptidique, il a été envisagé de refaire la synthèse de ces deux peptides, cette fois en éliminant le groupement carbonyle voisin du N5, afin d’éliminer le réarrangement ou l’ouverture du cycle ainsi que toute racémisation. Les résultats pourront ainsi être comparés avec ceux pour les peptides contraints 74 et 7569.
2.5.2 Synthèse d’un pentapeptide contraint (79)
La première étape de la synthèse est le couplage d’un acide aminé sur la résine oxime. Ce premier couplage s’effectue dans des conditions classiques de couplage peptidique en utilisant le N,N'-Diisopropylcarbodiimide (DIC). La résine oxime est placée dans une ampoule à synthèse peptidique. La fonction acide de
l’acide aminé est activée pour ensuite être fixée à la résine. La réaction de couplage s’effectue pendant 3 heures (figure 38).
Afin de déterminer le taux de substitution de la résine, un test de Kaiser quantitatif à la ninhydrine est réalisé71. Ce test permet d’ajuster les quantités de chaque réactif utilisé au cours de la synthèse. L’estimation du taux de substitution peut aussi se faire à la suite d’un clivage d’une quantité précise de résine à l’aide de la propylamine69. De façon générale, un taux de substitution d’environ 0.6 mmol/g est obtenu. Finalement, afin de s’assurer qu’aucun site oxime non substitué ne réagisse lors de l’élongation de la chaîne peptidique, une réaction d’acétylation avec l’anhydride acétique est réalisée afin de les bloquer.
Le couplage du deuxième acide aminé a été effectué avec 3 équivalents d’acide aminé protégé, 3 équivalents de HBTU, 3 équivalents de HOBt et 2 équivalents de DIEA pendant 2 heures. Le groupement protecteur BOC a été enlevé avec une solution d’acide trifluoroacétique (TFA) à 50% dans le dichlorométhane. Ces réactifs sont utilisés afin de maximiser les conditions de couplage et de réduire les risques de racémisation.
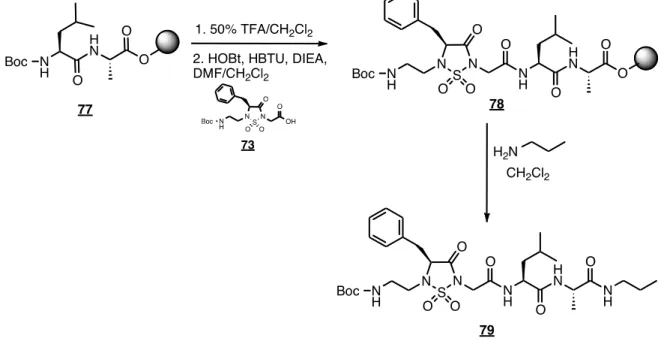
Figure 38. Synthèse d’un dipeptide 77 sur résine oxime nécessaire pour la
synthèse de 79.
Ensuite, le couplage du tripeptide analogue contraint 73 sur un acide aminé s’effectue dans les conditions standards en utilisant HOBt et HBTU. Ce dernier a été couplé sur le synthon Leu-Ala préalablement attaché à la résine oxime dans les conditions décrites plus haut (figure 39).
Lors de l’étape de clivage du peptide de la résine, il y a une possibilité d’ouverture du cycle de la sulfahydantoïne, ce qui peut nuire à la formation de peptides contraints désirés. En effet, le cycle pourrait subir une ouverture lors du clivage car le carbonyle voisin en N5 contribue à augmenter le caractère électrophile du sulfonyle et ainsi favorise l’ouverture du cycle15. L’absence de ce groupement carbonyle sur le pentapeptide contraint 79 devrait éliminer ce phénomène. Ceci est confirmé par le RMN 1H du composé final, où l’on observe bien les signaux du pentapeptide contraint 79 désiré. De plus, aucune racémisation n’est observée dans les spectres RMN.
Figure 39. Synthèse d’un pentapeptide contraint 79.
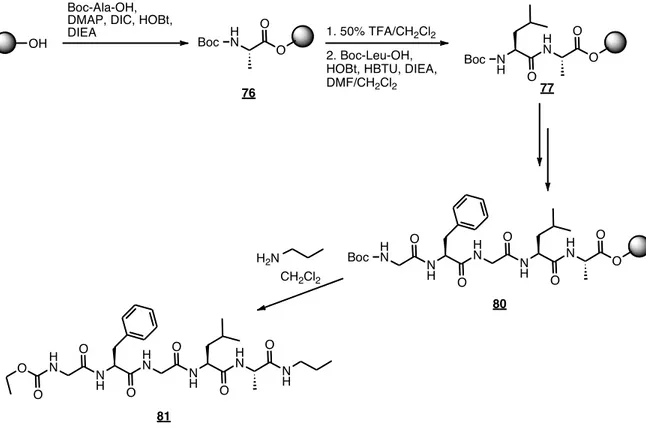
À des fins de comparaisons lors d’analyses conformationnelles, un peptide analogue non-contraint 81 a également été synthétisé (figure 40). Il est composé de la même séquence d’acide aminé, sans toutefois contenir le motif sulfahydantoïne ni la portion modifiée du N-terminal. Aussi synthétisé sur support solide, chaque acide aminé a été couplé dans des conditions standards décrites précédemment.
Figure 40. Synthèse sur support solide du pentapeptide modèle 81 sans
sulfahydantoïne
La synthèse d’un tétrapeptide contraint 84 a également été tentée (figure 41). Par contre, le couplage du tripeptide contraint sur un acide aminé n’a pas été réussi. On observe sur le spectre SM l’absence de masses caractéristiques pour ce peptide. De plus, lors du couplage, un test de Kaiser qualitatif a permis d’affirmer que le couplage n’a pas fonctionné. En effet, la coloration bleue lors de ce test indique que le N-terminal est toujours libre. Puisque nous avions le peptide contraint 79, nous n’avons pas poursuivi la synthèse de 84.
Figure 41. Synthèse d’un tétrapeptide contraint 84
2.5.3 Synthèse d’un décapeptide contraint
Afin de pousser davantage les études d’impact conformationnelle du motif sulfahydantoïne sur un peptide, nous avons également synthétisé un décapeptide contraint. Ceci a été réalisé en couplant un motif sulfahydantoïne sur l’octapeptide
85 préparé dans notre laboratoire pour des visées antimicrobiennes. Ces types de
peptides sont composés de leucines ainsi qu’un acide aminé modifié avec un éther-couronne. Ce motif a été repris afin de synthétiser un décapeptide contraint au N-terminal pour une sulfahydantoïne.
Le peptide 86 a préalablement été synthétisé sur support solide à l’aide de la résine de Wang72. Chaque couplage d’acide aminé sur cette résine est effectué dans les mêmes conditions standards décrites pour la résine oxime. Dans ce cas les couplages s’effectuent sur une heure72. Une fois le peptide clivé de la résine pour obtenir 85, il était possible de faire un couplage en solution de ce dernier avec
le dipeptide sulfahydantoïne 73 pour conduire à 86 (figure 42). Le spectre de masse a bien démontré la présence du pentapeptide analogue contraint 86.
Figure 42. Synthèse d’un décapeptide contraint
Le peptide non contraint correspondant à 86 (87) a également été synthétisé par les mêmes méthodes de synthèse (figure 43).
![Figure 17. Tripeptide contraint sulfonyle N-Boc-L-Val-L-Phe-[SO 2 ]-D-Ala-OMe et son diagramme de Ramachandran correspondant 47](https://thumb-eu.123doks.com/thumbv2/123doknet/6895683.193782/38.918.121.751.264.589/figure-tripeptide-contraint-sulfonyle-boc-diagramme-ramachandran-correspondant.webp)