L’activation du PDGFR favorise le phénotype agressif des synoviocytes de patients atteints de polyarthrite rhumatoïde via la formation d’invadosomes
Par
Roxane R.Lavoie Programmes d’Immunologie
Mémoire présenté à la Faculté de médecine et des sciences de la santé en vue de l’obtention du diplôme de maitre ès sciences (M.Sc) en Immunologie
Sherbrooke, Québec, Canada 11 Mai 2017
Membres du jury d’évaluation
Pre Claire Dubois, programme d’Immunologie Pre Sophie Roux, programme d’Immunologie
Pre Nathalie Perreault, programme d’Anatomie et biologie cellulaire © [Roxane R.Lavoie, 2017]
RÉSUMÉ
L’activation du PDGFR favorise le phénotype agressif des synoviocytes de patients atteints de polyarthrite rhumatoïde via la formation d’invadosomes
Par
Roxane R.Lavoie Programmes d’Immunologie
Mémoire présenté à la Faculté de médecine et des sciences de la santé en vue de l’obtention du diplôme de maitre ès sciences (M.Sc) en Immunologie, Faculté de médecine et des sciences de la santé, Université de Sherbrooke, Sherbrooke, Québec, Canada, J1H 5N4 La polyarthrite rhumatoïde (PR) est une maladie auto-immune qui mène à une inflammation chronique et à une destruction progressive des articulations. Les effecteurs principaux de cette pathologie sont les synoviocytes de type fibroblastique (FLS). Ces derniers utilisent les invadosomes, des structures riches en actine et en métalloprotéases, afin de dégrader la matrice extracellulaire (ECM). Ce phénotype pro-destructif résulte d’une activation des FLS par différents facteurs de croissance, dont le PDGF et le TGF-β. Les récepteurs à activité tyrosine kinase, dont le PDGFR, sont impliqués dans la pathogenèse de plusieurs maladies, incluant le cancer et la PR. Une activation de ces récepteurs peut mener, entre autres, à la survie, à la différenciation et à la prolifération des cellules. L’étude présentée dans ce mémoire montre que parmi les RTK les plus communs, le PDGFR est spécifiquement phosphorylé chez les cellules synoviales de patients atteints de PR, contrairement aux cellules de patients non arthritiques ou atteints d’arthrose. De plus, l’activation du PDGFR résulte en une augmentation de la formation d’invadosomes par les FLS. Nous avons aussi démontré que la formation d’invadosomes par le PDGFR nécessite l’activation de la voie de signalisation PI3K/Akt faisant intervenir les isoformes α
et δ de la PI3K. De plus, l'inhibition de l’activation du PDGFR ou la neutralisation du
PDGF endogène inhibe la formation des invadosomes et la dégradation de l'ECM par les synoviocytes, ce qui suggère la présence d'une boucle d'activation autocrine impliquant le PDGF. Parmi les isoformes du PDGF, nous avons démontré que le PDGF-B est exprimé de façon significativement plus élevée dans les synoviocytes provenant de patients atteints de PR. Nos données indiquent également une association entre le PDGF et le TGF-β dans la formation des invadosomes. Cette dernière implique la production autocrine de ligands du PDGFR induite par le TGFβ via la signalisation TβR1/Smad et PI3K/Akt. L’inhibition des isoformes de PI3K de classe I indique que le PI3Kα est impliquée de façon sélective dans l'expression de PDGF-B. Ces résultats démontrent que le PDGFR est un RTK nécessaire au phénotype destructeur des cellules synoviales d’arthrite. Ils fournissent aussi des preuves d'une association entre le TGF-β et le PDGFR dans la formation d’invadosomes chez les synoviocytes de patients atteints de la PR.
SUMMARY
Platelet-Derived Growth Factor Receptor Activation Promotes the Prodestructive Invadosome-Forming Phenotype of Synoviocytes from Patients with Rheumatoid
Arthritis
By
Roxane R.Lavoie Immunology Program
Thesis presented at the Faculty of medicine and health sciences for the obtention of Master degree diploma maitre ès sciences (M.Sc.) in Immunology, Faculty of Medicine and Health
Sciences, Université de Sherbrooke, Sherbrooke, Québec, Canada, J1H 5N4
Rheumatoid arthritis (RA) is an autoimmune disease that leads to chronic inflammation and progressive joint destruction. The main effectors of this pathology are fibroblast-like synoviocytes (FLS). They use invadosomes, actin-rich structures that concentrate metalloproteinases to degrade the extracellular matrix (ECM). This pro-destructive phenotype is due to the activation of FLS by various growth factors, including PDGF and TGF-β. Receptor tyrosine kinases, including PDGFR, are involved in the pathogenesis of several diseases, including cancer and RA. Activation of these receptors may lead to cell survival, differentiation and proliferation. The study presented in this thesis shows that among the most common RTKs, PDGFR is specifically phosphorylated in synovial cells of RA patients, unlike cells of non-arthritic or osteoarthritic patients. In addition, activation of PDGFR results in an increase in invadosome formation by FLS. We also shown that formation of invadosome by PDGFR requires the activation of the signaling pathway PI3K/Akt, that specifically involves the α and δ isoforms of PI3K. In addition, inhibition of PDGFR activation or neutralization of endogenous PDGF inhibits the formation of invadosomes and the degradation of the ECM by synoviocytes, suggesting the presence of an autocrine activation loop involving PDGF. Among the PDGF isoforms, we demonstrate that PDGF-B expression is significantly higher in synoviocyte cell lines from RA patients. Our data also indicates an association between PDGF and TGF-β for invadosome formation that involves autocrine production of PDGF-B induced by TGF-β through the Smad/TβR1 and PI3K/Akt pathways. Inhibition of class I PI3K isoforms indicates that PI3Kα is selectively involved in the expression of PDGF-B. These results demonstrate that PDGFR is an RTK necessary for the pro-destructive phenotype of RA FLS. They also provide evidence of an association between TGF-β and PDGFR in invadosome formation by synovial cells from RA patients.
Keywords : PDGFR, rheumatoid arthritis, fibroblast-like synoviocytes, TGF-β, PI3K, invadosome
Résumé ... iv
Summary ... v
Table des matières ... vi
Liste des figures ... viii
Liste des tableaux ... ix
Liste des abréviations ... x
Introduction ... 1 1 Polyarthrite rhumatoïde ... 1 1.1 Généralités ... 1 1.2 Étiologie ... 2 1.3 Diagnostic ... 2 1.4 Biomarqueurs ... 4 1.5 Traitements actuels ... 5 1.5.1 Traitements non-biologiques ... 5 1.5.2 Traitements biologiques ... 6 1.5.3 Traitements alternatifs ... 8 2 Physiologie de l’articulation ... 9
2.1 Caractéristiques des articulations synoviales ... 9
2.1.1 Synoviocytes de type macrophagique ... 11
2.1.2 Synoviocytes de type fibroblastique ... 11
2.2 Les FLS dans la polyarthrite rhumatoïde ... 12
3 Invadosomes ... 15
3.1 Caractéristique générale ... 16
3.2 Biogenèse de la formation des invadopodes ... 19
3.3 Induction de la formation des invadopodes ... 24
3.3.1 Le PDGF (facteur de croissance dérivé de plaquettes) ... 24
3.3.2 Le TGF-β (facteur de croissance transformant) ... 29
Hypothèse/problématique ... 34 Objectifs ... 34 Article ... 35 Avant-propos ... 35 Résumé ... 35 ABSTRACT ... 36 INTRODUCTION ... 37
MATERIAL AND METHODS ... 39
RESULTS ... 44
DISCUSSION ... 58
REFERENCES ... 63
Discussion ... 76
Phosphorylation du PDGFR... 76
Les RTK comme cible thérapeutique ... 78
La PI3K permet la formation des invadosomes ... 79
Conclusion... 90
Remerciements ... 91
LISTE DES FIGURES
MémoireFigure 1 Physiologie de l’articulation. ... 12
Figure 2 Activation des FLS ... 15
Figure 3 Schéma des structures cellulaires d’invasion et de migration ... 16
Figure 4 Initiation de la formation d’invadosome ... 20
Figure 5 Assemblage de l’invadosome ... 21
Figure 6 Maturation de l’invadosome ... 23
Figure 7 Interactions PDGF/PDGFR ... 26
Figure 8 Signalisation non-Smad du TGFβ ... 31
Figure 9 L’expression de PI3KCD, mais pas de PI3KCA, est augmentée dans les échantillons de polyarthrite rheumatoïde ... 84
Article Figure 1. RA-FLS generate ECM-degrading invadosomes. ... 45
Figure 2: RA-FLS display increased invadosome forming ability. ... 47
Figure 3: Phosphorylation of PDGFR is increased in RA synovial cells and tissue sections. ... 49
Figure 4: PDGF activity regulates invadosome formation by RA synoviocytes. ... 51
Figure 5: PI3K/AKT as effectors of invadosome formation in RA synovial cells. ... 53
Figure 6: Involvement of autocrine TGFβ in PDGFR-induced invadosome formation in RA synovial cells. ... 55
Figure 7: Implication of the TβR1/Smad pathway in PDGF-B expression induced by TGFβ. ... 57
LISTE DES TABLEAUX
Tableau 1 Critères d’évaluation de la polyarthrite rhumatoïde par l’EULAR/ACR (2010) .. 3 Tableau 2 Liste des principaux inhibiteurs de la voie PI3K/Akt présentement en essais cliniques de phase II ... 86
LISTE DES ABRÉVIATIONS
AAPC Auto-anticorps dirigés contre les protéines citrullinées ACR American College of Rheumatology
ACREU Service communautaire de recherche et d’évaluation sur l’arthrite
ADAMTS A disintegrin and metalloproteinase with thrombospondin motifs
ADN Acide désoxyribonucléique
AFAP110 Protéine de 110 kDa associée aux filaments d'actines Akt Proteine kinase B (PKB)
ARN Acide ribonucléique
Arp2/3 Protéine reliée à l'actine 2/3
BMP Protéines morphogénétiques osseuses Cdc42 Cell division control protein 42 homolog cellules NK Cellule « Natural Killer »
CIA Arthrite induite au collagène
CMHII Complexe majeur d'histocompatibilité II CPA Cellule présentatrice d'antigène
CTLA-4 Antigène 4 des lymphocytes T cytotoxiques
DAG Diacylglycérol
DMARD Médicaments antirhumatismaux modificateurs de la maladie DPP4 Dipeptidyl peptidase-4
ECM Matrice extracellulaire
EGFR Récepteur du facteur de croissance épidermique EMT Transition épithelio-mesenchymateuse
ERK Kinase régulée par un signal extracellulaire EULAR European League Against Rheumatism FAK Kinase d’adhésion focale
Fc Fragment crystallisable
FGFR Récepteur du facteur de croissance des fibroblastes FLS Synoviocyte de type fibroblastique
Grb2 Growth factor receptor-bound protein 2 Grb7 Growth factor receptor-bound protein 7
HGFR; c-Met Récepteur du facteur de croissance des hépatocytes HLA-DRB1 HLA class II histocompatibility antigen, DRB1 beta chain ICAM-1 Molécule d’adhésion inter-cellulaire
IGF-1 Facteur de croissance 1 analogue à l'insuline IgG Immunoglobuline de type G
IL Interleukine
IP3 Inositol triphosphate
JNK c-Jun N-terminal kinase
LAR Antigène lié aux leucocytes
Lck Protéine tyrosine kinase spécifique des lymphocytes MAPK Protéine kinase activée par des mitogènes
MLS Synoviocyte de type macrophagique
MMP Métalloprotéase matricielle
MT1-MMP Métalloprotéase matricielle membranaire de type 1 N-WASP Protéine du syndrome de Wiskott-Aldrich
Nck1 Région non-catalytique de la protéine adaptatrice de tyrosine kinase 1 NSAID Anti-inflammatoire non stéroïdien
OA Arthrose
p130Cas Substrat associé à Crk
PAK Kinase P21 activée
PCR Protéine C réactive
PDGF Facteur de croissance dérivé des plaquettes
PDGFR Récepteur du facteur de croissance dérivé des plaquettes PDK1 3-phosphoinositide dépendant de la proteine kinase-1
PFA Paraformaldéhyde
Domaine PH Domaine homologue de la pleckstrine PI(3,4,5)P3 Phosphatidylinositol-3,4,5-trisphosphate PI(3,4)P3 Phosphatidylinositol-3,4-trisphosphate PI(4,5)P2 Phosphatidylinositol-4,5-trisphosphate
PI3K Phosphoinositide 3-kinase
PKC Protéine kinase C
PLC Phospholipase C
PR Polyarthrite rhumatoïde
PTEN Phosphatase and tensin homolog PTP Protéine tyrosine phosphatase
PTPN22 Protéine tyrosine phosphatase, non récepteur de type 22
pY Tyrosine phosphorylée
RA Rheumatoid arthritis
ROS Espèce réactive de l’oxygène RTK Récepteur tyrosine kinase
SHIP2 SH2 domain containing inositol 5-phosphatase 2 SHP SH2 domain containing phosphatase
SKF Src family kinases.
STATS Signal transducer and activator of transcription
SYNJ2 Synaptojanin 2
TACE Tumor necrosis factor-α-converting enzyme
TG2 Transglutaminase 2
TGF-β Facteur de croissance transformant-β
TIMP Tissue inhibitor of metalloproteinases
TKS4 Substrats de tyrosine kinases possédant 4 domaines SH3 Tks5 Substrats de tyrosine kinases possédant 5 domaines SH3 TNF Facteur de nécrose tumorale
Tyr Tyrosine
TβR Récepteur du facteur de croissance transformant β UPAR Récepteur de l’urokinase
VCAM-1 Molécule d'adhésion aux cellules vasculaires 1 VEGF Facteur de croissance de l'endothélium vasculaire VS Vitesse de sédimentation
WIP Wiskott-Aldrich Syndrome Interacting Protein
β bêta
γ gamma
INTRODUCTION
1 Polyarthrite rhumatoïdeLa polyarthrite rhumatoïde (PR) est une maladie chronique, autoimmune et inflammatoire qui mène à une destruction progressive des articulations. Elle s’attaque en premier lieu aux articulations des mains et des pieds et peut par la suite affecter presque toutes les articulations synoviales ainsi que plusieurs organes. La PR a une prévalence d’environ 1% en Amérique du nord ainsi qu’en Europe et est trois fois plus commune chez les femmes (Alamanos et Drosos, 2005).
1.1 Généralités
Les manifestations de la polyarthrite rhumatoïde sont très variables, ce qui rend le diagnostic et le traitement difficiles. Au début, les patients présentent des symptômes plutôt généraux, comme de la fatigue, une perte de poids ainsi que des raideurs matinales. Par la suite, des symptômes spécifiques aux arthrites avec une inflammation des articulations et de la douleur, souvent de façon symétrique, se manifestent. Lorsque la PR devient plus sévère, il y a déformation et perte de fonction des articulations. Ceci est associé à une dégradation du cartilage et des os ainsi qu’à une atrophie des tendons et des muscles entourant l’articulation. En conséquence, les personnes atteintes de PR éprouvent des difficultés dans leurs activités quotidiennes, comme marcher, s’habiller et se laver.
La polyarthrite rhumatoïde peut aussi affecter d’autres organes de manière directe (processus rheumatoide, vasculite) ou indirectement (complications de l’inflammation chronique ou des traitements). Des études ont démontré que les patients atteints de polyarthrite rhumatoïde ont un plus grand risque de développer un infarctus du myocarde ainsi qu’une bronchopneumopathie chronique obstructive (Solomon et al. 2003, Shen et al. 2014). Dans environ 20% des cas, les patients vont développer des nodules rhumatoïdes, généralement sur les coudes, les mains et les pieds (« La Société de l’Arthrite », site web). La PR elle-même n’est pas fatale, mais elle diminue de façon significative la qualité de vie des personnes atteintes en plus d’augmenter les risques de troubles systémiques.
La sévérité et la progression de la polyarthrite rhumatoïde peuvent varier entre les patients. L’installation de la PR peut être rapide (quelques jours ou semaines) ou prolongée (quelques semaines ou mois) et ce, à n’importe quel stade de la vie. Habituellement, la PR s’installe chez les gens de 20 à 50 ans.
1.2 Étiologie
Certains facteurs génétiques et environnementaux peuvent influencer le développement de la PR. Des études d'associations pangénomiques ont identifié plus de 100 gènes pouvant être impliqués dans la genèse de la maladie (Kochi et al., 2014; Okada et al., 2014). L'antigène leucocytaire humain (HLA)-DRB1 est le facteur de risque génétique le plus important pour la PR. Il a été reconnu comme un facteur de risque en 1978 après avoir été étudié en raison de son rôle dans la réponse immunitaire (Stastny, 1978). Plusieurs allèles HLA-DRB1 sont reconnue pour être associés à la PR et ont été validés dans de nombreuses populations différentes (Kochi et al., 2014). Le second gène fortement associé à la PR est la protéine tyrosine phosphatase non-récepteur de type 22 (PTPN22). HLA-DRB1 et PTPN22 sont des facteurs de risque génétiques majeurs qui représentent 40 % du risque génétique à développer la PR. En plus des facteurs génétiques, certains facteurs environnementaux peuvent augmenter la susceptibilité à développer la maladie. Le facteur de risque environnemental le plus important est la consommation de tabac. Une étude a démontré une association entre la consommation de tabac et la présence d’auto-anticorps dirigés contre les protéines citrullinées (AAPC) (Pedersen et al., 2006).
1.3 Diagnostic
La PR est diagnostiquée à l’aide de tests sérologiques ainsi que par l’identification des symptômes, qui incluent la présence de synovites avec tuméfaction des articulations et la douleur. Les rayons-x sont utilisés pour orienter le diagnostic et apprécier l'atteinte osseuse et cartilagineuse. L'étude échographique et par IRM des articulations permet d'augmenter la sensibilité pour déceler les synovites. Par contre, il n’existe pas de test permettant de diagnostiquer précisément cette forme de rhumatisme.
Les premiers critères cliniques pour le diagnostic de la PR ont été établis par l’American College of Rheumatology (ACR) en 1987 qui se basait sur les manifestations tardives de la maladie (Arnett et al., 1988). Il est maintenant reconnu que le diagnostic précoce permet de débuter des traitements efficaces précocement, ce qui améliore les résultats cliniques. En 2010, l’ACR et l’European League Against Rheumatism (EULAR) ont mis au point un système d’évaluation des polyarthrites récentes sans érosion osseuse, plus sensible basé sur des marqueurs biologiques et des symptômes qui permettent d'évoquer le diagnostic de PAR à un stade précoce, en l'absence d'élément pour un autre diagnostic (Aletaha et al., 2010) (Tableau 1).
Tableau 1 Critères d’évaluation de la polyarthrite rhumatoïde par l’EULAR/ACR (2010)
Nombre d’articulation atteinte (0-5)
Résultat positif si le pointage est supérieur à 6
・1 grande articulation 0
・2 à 10 grandes articulations 1
・1 à 3 petites articulations (grande non comptée) 2 ・4 à 10 petites articulations (grande non comptée) 3 ・> 10 articulation (au moins une petite) 5
Sérologie (0-3)
・Négatif pour FR et AAPC 0
・Faiblement positif pour FR ou AAPC 2
・Hautement positif pour FR ou AAPC 3
Durée des symptômes (0-1)
・< 6 semaines 0
・≥ 6 semaines 1
Réactifs de la phase aigue (0-1)
・Normal PCR et VS 0
・Anormal PCR et VS 1
Pour un résultat positif de PR, le patient doit atteindre un pointage d’au moins 6 sur une possibilité de 10. FR=facteur rhumatoïde, AAPC=auto-anticorps dirigés contre les protéines citrullinées, PCR=protéine C réactive et VS=vitesse de sédimentation globulaire (Aletaha et al., 2010).
1.4 Biomarqueurs
Les rhumatologues utilisent certains marqueurs biologiques pour orienter le diagnostic de l'arthrite (auto-anticorps) ou apprécier le degré d'inflammation systémique (VS et CRP). Très tôt dans le développement de la maladie, et même parfois avant les premiers symptômes, les auto-anticorps dirigés contre les protéines citrullinées (AAPC) peuvent être détectés. La positivité du facteur rhumatoïde est plus tardive, n'apparaissant souvent qu'après plusieurs mois d'évolution.
Les facteurs rhumatoïdes (FR) sont des auto-anticorps dirigés contre la région Fc des IgG humaines (Artandi et al., 1992). S'ils ne semblent pas conribuer à la pathogénie de la synovite rhumatoïde, leur rôle a été démontré dans la pathogénie de la vasculite rhumatoïde et celle des nodules rhumatoïdes chez les patients atteints de PR en formant des complexes immuns. Les FRs peuvent être détectés dans le sérum de la majorité des patients atteints de PR et la présence de FR chez les patients corrèle avec l’agressivité de la maladie. Cependant, les FRs ne sont pas spécifiques à la PR et se retrouvent dans de nombreuses autres pathologies, comme le lupus érythémateux ou certaines pathologies infectieuses (Popescu et al., 2013). Les AAPCs sont des anticorps produits par les cellules plasmatiques contre des auto-antigènes citrullinés. Ils sont principalement présents chez les patients fumeurs et/ou ayant le HLA-DRB1 (Hill et al., 2003). Par contre, l'expression et la réactivité des AAPC est variable entre les patients atteints de PR mais aussi chez le même patient. Selon les critères d’évaluation de la PR, les patients peuvent être classés positif ou négatif pour les AAPCs, ce qui pourrait influencer leurs réponses aux médicaments (van Dongen et al., 2007).
Deux autres tests sanguins sont effectués chez les patients afin d’évaluer le degré d’inflammation systémique. La protéine C réactive (PCR) est une protéine de phase aiguë produite par le foie et libérée dans la circulation sanguine. Un niveau élevé de PCR est indicatif d'un niveau élevé d’inflammation. Des études ont démontré que le risque accru de maladies cardiaques est corrélé à un niveau élevé de PCR chez les patients atteints de PR (Ridker et Cook, 2004). De plus, il a été démontré que la vitesse de sédimentation
globulaire (VS), une mesure du taux de sédimentation de globules rouges, est proportionnelle au niveau d'inflammation. Les résultats de ce test donnent une autre indication de la sévérité de la PR. Par contre, ce test est non spécifique puisque la VS est influencée par l'inflammation quelqu'en soit son origine, et également par l'anémie ou la présence d'une gammapathie monoclonale.
1.5 Traitements actuels
La polyarthrite rhumatoïde a un impact socio-économique majeur. En effet, nous devons prendre en considération les frais reliés aux traitements ainsi que toutes les dépenses indirectes occasionnées par l’invalidité des patients. En 2013, le service communautaire de recherche et d’évaluation sur l’arthrite (ACREU) a estimé le coût de la polyarthrite rhumatoïde à environ 33 milliards de dollars annuellement. On estime que ce montant pourrait doubler en 2031 (« La Société de l’Arthrite », site web).
Comme avec la plupart des maladies humaines, il est essentiel de traiter la PR le plus tôt possible afin de minimiser les effets dommageables de la maladie (Mc Ardle et al., 2015). Il existe plusieurs façons établies pour traiter les symptômes de la PR. Le premier choix de traitement est l’utilisation de médicaments non-biologiques qui se révèlent efficaces et pour lesquels on connaît les effets secondaires. Plus récemment, les rhumatologues prescrivent également des médicaments biologiques.
1.5.1 Traitements non-biologiques
Les médicaments anti-inflammatoires non stéroïdiens (NSAIDs) sont les premiers traitements offerts aux patients. Ce sont les médicaments les moins puissants et les moins spécifiques mais ils peuvent réduire l’inflammation et la douleur efficacement avec peu d’effets secondaires. Ils vont tout simplement bloquer la production de prostaglandine en inhibant la cyclooxygénase. L’ibuprofène et l’aspirine entrent dans cette catégorie de médicaments. Cependant, les NSAIDs sont connus pour avoir des effets indésirables, y compris un risque accru de thrombose de l'artère coronaire, d'ulcère et d’insuffisance rénale (« American College of Rheumatology », site web). Les NSAIDs sont des traitements
symptomatiques à effet rapide, souvent débutés initialement et en parallèle aux traitements de fond qui ont tous un délai d'action prolongé.
Les médicaments antirhumatismaux modificateurs de la maladie (DMARDs) sont utilisés comme traitement standard suite au diagnostic de PR. Le mécanisme d’action de ces médicaments est encore peu connu, mais ils permettent de ralentir la progression de la maladie ou d’induire une rémission (Aletaha et al., 2007). Par contre, ces médicaments peuvent être toxiques ce qui fait que plusieurs patients vont abandonner le traitement (Aletaha et Smolen, 2002). Les DMARDs sont souvent utilisés en combinaison avec d’autres traitements. Les DMARDs comprennent le léflunomide, l’hydroxychloroquine et le méthotrexate. Ce dernier est le plus souvent prescrit, il agit en inhibant le métabolisme de l’acide folique. Le méthotrexate est le médicament possédant le meilleur ratio efficacité/toxicité. Par contre, il n’est pas efficace chez tous les patients (« American College of Rheumatology », site web).
Les glucocorticoïdes, comme la prednisone, sont aussi utilisés comme traitement symptomatique (à l’instar des NSAIDs) pour leurs potentiels anti-inflammatoires et pour le soulagement de la douleur. Soit, administré oralement ou par injection, ils sont pris en combinaison avec des DMARDs compte tenu du délai d'action de ces derniers, mais seront arrêtés dès que possible. Par contre, ils possèdent des effets secondaires importants et l’arrêt des glucocorticoïdes après un traitement prolongé, peut s’avérer difficile du fait de l’insuffisance de production naturelle de stéroïdes par le corps secondaire à la prise prolongée de corticoïdes exogènes.
1.5.2 Traitements biologiques
Les traitements biologiques ciblent les cytokines pro-inflammatoires, les lymphocytes B, les lymphocytes T ou d’autres cellules immunitaires. Ces médicaments ont révolutionné le devenir des patients atteints de polyarthrite rhumatoïde. Par contre, ces traitements sont dispendieux et sont inefficaces dans une proportion de patients (Weinblatt et al., 2007). Les médicaments biologiques peuvent être utilisés en monothérapie ou en association avec des DMARDs. Ils peuvent avoir des effets secondaires graves, en particulier s’ils sont
utilisés conjointement avec d'autres agents biologiques (Weinblatt et al., 2007). Les principaux effets secondaires sont d'ordre infectieux.
Les thérapies dirigées contre les cytokines ont l’avantage d’avoir un mécanisme d’action rapide et sans déplétion cellulaire. Les premiers traitements à être approuvés et encore les plus couramment prescrits, sont les inhibiteurs du facteur de nécrose tumorale α (TNFα). Cette classe de traitement comprend l’étanercept (forme soluble du récepteur du TNFα) et l’infliximab et l’adalimumab (anticorps monoclonaux). Comme pour tous les traitements de fond, 30% des patients seront résistants à cette approche (Lipsky et al., 2000). Actuellement, il n'y a aucun moyen établi d'identifier les patients qui répondront efficacement à ces traitements. Fait intéressant, certaines études ont suggéré que la détermination du profil d'expression génique des patients avant le traitement pourrait être un moyen de cibler lesquels seront sensibles aux anti-TNFαs (Toonen et al., 2012).
Le tocilizumab, un anticorps monoclonal humanisé dirigé contre le récepteur soluble de l’IL-6, est un agent biologique efficace. Chez les patients ne répondant pas à un ou plusieurs anti-TNFα, il a été rapporté que le tocilizumab produit des taux de réponse efficaces par rapport au placebo (Emery et al., 2008). Dans plusieurs essais cliniques de phase 3, le tocilizumab réduisait le score clinique de la maladie de façon significative (Smolen et al., 2014).
Une autre cible biologique est le récepteur de l'interleukine-1 (IL-1). L’anakinra est un antagoniste du récepteur de l’IL1 (IL1ra) qui entre en compétition avec l’IL-1 endogène pour la liaison au récepteur. L'utilisation de ce médicament en combinaison avec les NSAIDs, le méthotrexate ou les glucocorticoïdes réduit rapidement les symptômes de la maladie chez 40% des patients par rapport aux témoins placebo (Cohen et al., 2002; Cohen, 2004). Cependant, il a été démontré ne pas être efficace chez un certain nombre de patients qui ne répondent pas à la thérapie anti-TNFα (Buch et al., 2004). Globalement peu efficace dans la PR, il demeure toutefois une indiqué pour le traitement des formes systémiques (maladie de Still).
En plus de cibler les cytokines, les thérapies biologiques peuvent cibler les cellules immunitaires. L’abatacept est une protéine de fusion humaine recombinante composée de la partie Fc des IgG et du domaine extracellulaire du CTLA-4 (le CTLA4 est un inhibiteur physiologique de la voie de co-activation CD28/CD80-86). Ce médicament inhibe l'activation des lymphocytes T par le blocage des signaux essentiels de costimulation des cellules présentatrices d'antigène (CPA). Il est le plus souvent utilisé en combinaison avec le methotrexate, à la suite d’une réponse insuffisante aux anti-TNFαs (Genovese et al., 2005).
Le rituximab, quant à lui, est un anticorps monoclonal chimérique qui agit en inhibant le CD20 sur les lymphocytes B, ce qui entraîne une diminution des lymphocytes B en périphérie et au niveau de la membrane synoviale des articulations. Dans une étude de phase 3 avec des patients qui répondent inadéquatement au traitement anti-TNFα, le rituximab a montré une amélioration significative des réponses dans plus de 50% des patients par rapport aux témoins placebo (Cohen et al., 2006).
1.5.3 Traitements alternatifs
Il est fortement suggéré aux patients d’apporter des changements dans leurs styles de vie, comme faire de l’exercice, changer de régime alimentaire ainsi que l’utilisation d’accessoires fonctionnels. La chirurgie peut être nécessaire pour les patients ayant des destructions ostéo-articulaires majeures afin de soulager la douleur, augmenter l’amplitude de mouvement et corriger les déformations articulaires. Les chirurgies peuvent consister en la résection d'une région de la membrane synoviale (synovectomie). Il peut aussi y avoir des chirurgies reconstructices plus invasives, visant à restaurer la structure et la fonction par le nettoyage du cartilage ou un remplacement de l'articulation complète (arthroplastie).
Malgré cette panoplie de médicaments, il n’y a présentement aucune façon de guérir la PR et on en connaît très peu sur ce qui déclenche la maladie. La thérapie de la PR s'est améliorée avec l'introduction d'agents biologiques qui ciblent les cytokines, les lymphocytes B ou les cellules T. Dans certains cas, ces thérapies permettent de réduire en partie les dommages du cartilage et des os. De plus, une proportion significative de patients
continuent d'avoir une inflammation active et une progression de la maladie malgré une thérapie optimisée (Moreland, 2005). Il y a un grand besoin de développer d’autres traitements efficaces. Les thérapies pourraient cibler des molécules ou des voies de signalisation impliquées dans la dégradation du cartilage ainsi que les symptômes associés (Cohen et Fleischmann, 2010; Sweeney et Firestein, 2007). La découverte de voies qui régulent la destruction des tissus dans la PR pourrait conduire à de nouveaux agents thérapeutiques.
2 Physiologie de l’articulation
La classification structurale des articulations se base selon leur morphologie. Les articulations fibreuses permettent très peu de mouvement puisque les os sont joints entre eux par du tissu conjonctif très dense. Dans les articulations cartilagineuses, les os sont unis par du cartilage mais, sont dépourvus de cavité articulaire tout comme les articulations fibreuses. Ce sont les articulations synoviales qui offrent la plus grande liberté de mouvement du corps et ce sont les plus nombreuses (Hui et al., 2012). Elles sont caractérisées par des ligaments, du cartilage hyalin, une cavité articulaire, du liquide synovial et une capsule articulaire.
2.1 Caractéristiques des articulations synoviales
La surface des os des articulations synoviales est recouverte d’un cartilage hyalin. Cette surface lisse et glissante permet d’absorber les chocs et de réduire la friction lors des mouvements. Ces articulations possèdent des ligaments qui permettent de lier les os ensemble mais aussi d’empêcher les mouvements extrêmes qui peuvent endommager l’articulation.
Une des caractéristiques des articulations synoviales est la présence d’une cavité articulaire contenant du liquide présent en quantité infime à l’état normal. Ce liquide synovial a des fonctions biomécaniques, métaboliques et régulatrices. La propriété la plus importante de ce liquide est d’assurer la lubrification de l’articulation (Hui et al., 2012). Il est composé de plasma sanguin provenant des capillaires de la membrane synoviale et de molécules
lubrifiantes sécrétées par les synoviocytes incluant l’acide hyaluronique, la lubricine et les protéoglycans (Barton et al., 2013; Schmidt et al., 2007). Le liquide synovial possède des propriétés et fonctions uniques dans le maintien de l’homéostasie articulaire (Hui et al., 2012). Le liquide synovial contient des molécules solubles comme des morphogènes (dont le TGFβ), cytokines et facteurs de croissance qui assurent la communication entre les différentes populations cellulaires de l’articulation (Hui et al., 2012). Ces molécules régulatrices présentent dans le liquide synovial peuvent être dérivées du plasma ou sécrétées par les chondrocytes, cellules synoviales ainsi que des cellules inflammatoires présentes dans la membrane synoviale. Les cytokines présentent dans le liquide synovial peuvent être catégorisées comme pro-inflammatoires ou anti-inflammatoires selon leurs effets sur le tissu. Les cytokines pro-inflammatoires présentes dans le liquide synovial d’arthropathies variées comprennent l’IL-1α, l’IL-1β, le TNFα, l’IL-6, l’IL-8, l’IL-17 et l’IL-18 et les cytokines anti-inflammatoires comprennent l’IL-4, l’IL-10 et l’IL-13 (Doß et al., 2007; Futani et al., 2002; Goldring, 2000). On retrouve également des facteurs de croissances dans le liquide synovial tels que le PDGF et l’IGF-1 (McInnes et Schett, 2007; Textor et al., 2013). Dans les arthrites, le profil cytokinique est altéré. La plupart des cytokines et facteurs de croissance sont en très faible concentration dans les conditions d’origine mécaniques (OA) mais leurs concentrations sont nettement plus élevées dans le cas d’arthrites, en particulier la PR, avec un profil cytokinique distinct selon le stade évolutif (Raza et al., 2005). Les cytokines vont jouer un important rôle dans la pathogenèse de la synovite et dans la destruction articulaire, c’est pourquoi plusieurs thérapies vont cibler ces cytokines (McInnes et Schett, 2007).
La capsule articulaire entoure la cavité articulaire. La paroi des capsules articulaires se compose de deux couches bien distinctes : un tissu conjonctif résistant et flexible appelé couche fibreuse à l’extérieur, et une interne qui est la membrane synoviale (synovium). Les cellules synoviales, appelées synoviocytes, sont en contact avec la cavité intra-articulaire et sont responsables de la production de composantes du liquide synovial.
2.1.1 Synoviocytes de type macrophagique
Les synoviocytes de type A ou macrophagiques (MLS) sont habituellement ronds et localisés dans la partie supérieure de la membrane synoviale (Iwanaga et al., 2000). Les synoviocytes de type A migrent dans la membrane synoviale où ils deviennent résidants, par contre, on ne sait pas encore si ces cellules se différencient in situ ou avant leurs arrivées. Leur signature moléculaire est similaire aux autres populations macrophagiques résidentes puisqu’ils expriment les marqueurs CD11b, CD68, CD14, CD163 et Fc Rγ. Ils expriment également les molécules du complexe majeur d’histocompatibilité de classe II (CMHII), ce qui leur permet de jouer un rôle dans la présentation d’antigènes dans les premières étapes de la réponse immune (Iwanaga et al., 2000). En conditions inflammatoires, les synoviocytes de type A produisent plusieurs enzymes, dont la cathepsine, qui serait alors relâchée dans la matrice extracellulaire et induirait des dommages tissulaires (Iwanaga et al., 2000). De même que les autres macrophages tissulaires, les synoviocytes de type macrophagique possèdent une activité phagocytaire ainsi qu’une faible capacité proliférative (Bartok et Firestein, 2010).
2.1.2 Synoviocytes de type fibroblastique
Les synoviocytes de type B ou fibroblastique (FLS) sont des cellules mésenchymateuses qui possèdent plusieurs caractéristiques des fibroblastes incluant l’expression du collagène de type IV et V, de la vimentine et du CD90. Les synoviocytes de type B représentent entre 75% et 80% des cellules de la membrane synoviale et ont des propriétés uniques in situ qui les distinguent des autres populations fibroblastiques (Iwanaga et al., 2000). Par exemple, les FLS expriment la cadhérine-11, une molécule d’adhésion, qui permet l’agrégation des FLS in vitro et in vivo. Les souris déficientes en cadhérine-11 se développent normalement mais développent une membrane synoviale non définie dans les articulations diarthrodiales. De plus, les FLS de la couche interne de la membrane expriment l’uridine diphosphoglucose déshydrogénase qui permet de synthétiser l’acide hyaluronique, une composante importante du liquide synovial et de la matrice extracellulaire (Iwanaga et al., 2000). Les molécules d'adhésion de cellule vasculaire (VCAM-1), qui normalement médient l’adhésion des leucocytes à l’endothélium vasculaire, sont également exprimées sur les FLS. Leur rôle dans le tissu demeure incertain mais elles pourraient favoriser
l’ancrage des FLS à la fibronectine de la matrice extracellulaire. Les intégrines, comme α5/β1 qui lie la fibronectine, les molécules d’adhésion intercellulaire (ICAM-1) et le CD55 sont aussi exprimées sur les synoviocytes de type fibroblastique et peuvent servir à les identifier par immunohistochimie (Bartok et Firestein, 2010).
2.2 Les FLS dans la polyarthrite rhumatoïde
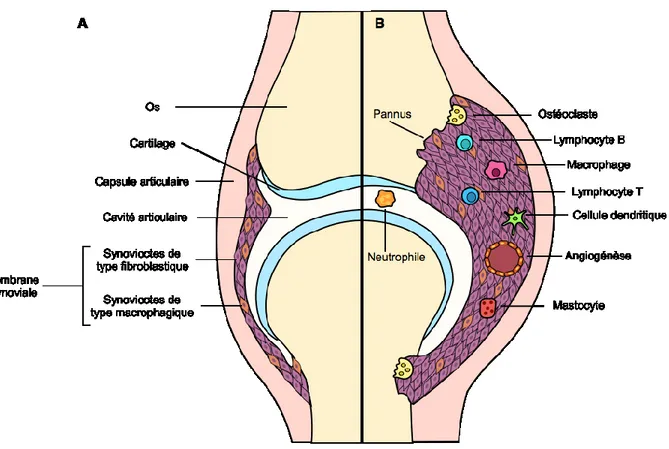
La polyarthrite rhumatoïde est caractérisée par une hyperplasie de la membrane synoviale,
qui est habituellement d’une épaisseur de 2 à 3 couches cellulaires, et une infiltration de nombreuses cellules, dont les macrophages, les lymphocytes T et les lymphocytes B ainsi que les cellules endothéliales et FLS (Figure 1). Cette migration est favorisée par l'expression de cytokines, chimiokines et molécules d'adhésion. Le liquide synovial devient également plus riche en cellules inflammatoires, majoritairement des neutrophiles, ce qui mène à une augmentation du volume (dû au processus inflammatoire) et à une diminution de la viscosité.
Figure 1 Physiologie de l’articulation.
synoviocytes, les FLS et les MLS. L’articulation de patients atteints de PR (B) se caractérise par une inflammation et un épaississement de la membrane synoviale qui envahit le cartilage et l’os, détruisant l’articulation (Strand et al. 2007, numéro de licence 3944821035763)
D’autre part, la membrane synoviale forme un pannus invasif qui a comme fonction de dégrader le cartilage et les os. La formation du pannus se caractérise par une surabondance de FLS causée par un débalancement entre leur prolifération et apoptose. Une augmentation de la différenciation et migration des cellules souches mésenchymateuses peut aussi contribuer à ce débalancement. L’hyperplasie de la membrane synoviale augmente la distance entre les vaisseaux sanguins et les synoviocytes ce qui diminue la diffusion de l’oxygène dans le synovium (Y.-A. Lee et al., 2007). L'hypoxie tissulaire se produit lorsqu’il y a un déséquilibre entre l'offre et la demande d'oxygène. L’hypoxie articulaire induit l’expression du VEGF par différents types cellulaires, ce qui stimule l’angiogenèse ainsi que la perméabilité vasculaire (Brenchley, 2000). Plusieurs études in vitro ont démontrés que l'hypoxie est un facteur important de la régulation de l'angiogenèse, de la migration et de la dégradation de la matrice par les synoviocytes de type fibroblastique (Ahn et al., 2008; Akhavani et al., 2009). Le pannus peut être comparé à un processus tumoral.
Les FLS normaux contribuent à l’homéostasie de l’articulation en produisant des composants importants de la matrice extracellulaire et du liquide synovial. Dans la PR, les FLS sont activés ce qui débalance leurs fonctions homéostatiques. Les synoviocytes arthritiques libèrent une plus grande quantité de métalloprotéinases matricielles (MMPs),
produisent moins de collagénase et synthétisent des cytokines et chimiokines. Ce sont les
FLS qui sont considérés comme les effecteurs principaux de la dégradation puisqu’ils vont promouvoir la croissance du pannus et l’inflammation. Les FLS arthritiques produisent une plus grande quantité de protéases dont les collagénases (MMP1 et MMP13), la stromélysine-1 (MMP3), les aggrécanases (ADAMTS4 et ADAMTS5) et les cathepsines,
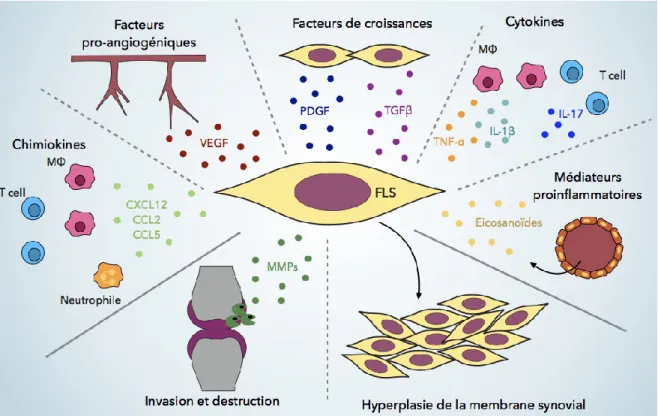
ce qui augmente leurs propriétés destructrices (Bottini et Firestein, 2013). Les synoviocytes
peuvent être activés par plusieurs cytokines, chimiokines, facteurs de croissances et éicosanoïdes qui sont synthétisés par les leucocytes infiltrant ou par les FLS eux-mêmes (Figure 2). Plus précisément, le TNF-α et l’IL-1β produits par les macrophages ainsi que
l’IL-17 produit par les cellules T peuvent activer les cellules synoviales (Noss et Brenner, 2008). En contre partie, les synoviocytes arthritiques synthétisent également des cytokines pro-inflammatoires dont l’IL-6, l’IL-15 et l’interféron de type I, ce qui amplifie l’état inflammatoire de l’articulation. Par ailleurs, les FLS sont les principales sources de RANKL (ligand du récepteur du facteur nucléaire kappa B) dans les conditions pathologiques telles que la PR (Ainola et al., 2008; Hashizume et al., 2008). RANKL se lie à son récepteur, RANK, présent chez les monocytes, ce qui initie la différenciation des ostéoclastes. Les FLS ont donc un rôle essentiel dans l'ostéoclastogénèse, ce qui favorise la destruction osseuse (Jung et al., 2014). Les FLS expriment également des récepteurs aux facteurs de croissance. Le facteur de croissance des fibroblastes (FGF) et le facteur de croissance placentaire (PLGF) sont connus pour être impliqués dans la prolifération et la migration des synoviocytes (Malemud, 2007; Yoo et al., 2015). De plus, le TGFβ et le PDGF sont également connus pour promouvoir l’invasion des synoviocytes et peuvent activer les synoviocytes de façon autocrine puisqu’ils sont produits par ceux-ci (Bartok et al., 2014; Cheon et al., 2002; Lafyatis et al., 1989). L’hypoxie locale va favoriser la production de VEGF et des espèces réactives à l'oxygène (ROS), qui peuvent également
activer les FLS (Kumkumian et al., 1989). Ultimement, l’activation des FLS va mener à
Figure 2 Activation des FLS
Les FLS sont activés par différents facteurs, ce qui favorise la formation d’un pannus et la production de protéases qui dégradent la matrice extracellulaire. (Bottini & Firestein 2013, numéro de licence 3944920589588)
3 Invadosomes
Les invadosomes ont été découverts en 1980 par le Dr. David-Pfeuty et le Dr. Singer. Ces chercheurs ont montré que les fibroblastes transformés à l’aide du virus sarcome de Rous (VSR) (qui contient l’oncogène v-Src) réarrangent leur cytosquelette aux points d’adhésion focaux pour former des grappes circulaires d’α-actinine et de vinculine. Ils nommèrent ces structures rosettes (David-Pfeuty et Singer, 1980). En 1985, Tarone et al. ont montré que ces protrusions contiennent également de l’actine et des protéines phosphorylées sur des résidus tyrosine. Ils renomment alors ces structures des podosomes par homologie à des pieds ancrés dans la matrice extracellulaire (Tarone et al., 1985). Au cours de la même année, les laboratoires du Dr. Cheng et du Dr. Parson ont fait une importante observation. Ils remarquent que dans les fibroblastes VSR, il y a accumulation de la tyrosine kinase v-Src aux sites d’adhésion à l’ECM et dégradation de l’ECM à ces sites. Ils proposèrent le terme invadopode afin de refléter les propriétés dégradatives et adhérentes de ces structures (Chen et al., 1985). Par la suite, plusieurs études ont démontré la présence de ces structures
dans différents types cellulaires dont les ostéoclastes, macrophages, cellules dendritiques, cellules endothéliales, cellules cancéreuses du sein, de la prostate et du colon (Buccione et al., 2004; Gimona et al., 2008; Linder et al., 2011; Schoumacher et al., 2010; Zambonin-Zallone et al., 1988). Afin d’éclaircir la nomenclature, Dr. Courtneidge et Dr. Murphy proposèrent d’attribuer le terme podosome aux cellules normales, le terme invadopode aux cellules cancéreuses et de regrouper ces structures adhésives et dégradatives sous le terme invadosome (Murphy et Courtneidge, 2011). Nous avons récemment démontré au laboratoire que les synoviocytes de type fibroblastique utilisent des invadosomes, afin de dégrader la matrice extracellulaire, dont la matrice cartilagineuse (Lauzier et al., 2011).
3.1 Caractéristique générale
Il est important de faire la distinction entre les podosomes et invadopodes des autres protrusions cellulaires, dont les filopodes et les lamellipodes et des autres structures adhésives, dont les sites d’adhésions focaux (figure 3).
Les lamellipodes et les filopodes sont essentiels pour la motilité cellulaire, l'organisation des domaines membranaires et la phagocytose. Les lamellipodes sont de larges protrusions en forme de feuillet et sont situés au front de migration de la cellule (Small et al., 2002). Les filopodes sont habituellement retrouvés dans la même région que les lamellipodes, ils sont de fines protrusions de la membrane plasmique, riches en actine et agissent comme des antennes pour les cellules afin de sonder leur environnement. Les filopodes ont un rôle important dans la migration cellulaire et dans l'extension des neurites des cellules neuronales (Mattila et Lappalainen, 2008). Les lamellipodes sont composés de filaments d’actine ramifiés tandis que les filopodes sont formés de filaments d’actine parallèles. Ces deux structures ne possèdent aucune activité protéolytique.
Les sites d’adhésion focaux sont des contacts adhésifs entre la cellule et la matrice extracellulaire par l'interaction des intégrines transmembranaires avec leurs ligands extracellulaires. Ces sites ancrent la cellule à l’ECM et peuvent servir de médiateurs dans la signalisation. Fait intéressant, des résultats récents ont indiqué que les sites d’adhésion focaux ont aussi une capacité à dégrader l’ECM, ce qui renforce les similitudes entre les invadopodes/podosomes et ces structures d’adhésion (Wang et McNiven, 2012). Par contre, les études au sujet de l’implication de la kinase d'adhésion focale (FAK) dans la formation des invadosomes sont controversées. D’une part, il a été montré que FAK possède un rôle de régulateur négatif de la dynamique et de la formation d’invadopodes dans les cellules cancéreuses du sein. La phosphorylation de Src par FAK promeut la formation des sites d’adhésions focaux et l’inhibition de la formation des invadopodes (par séquestrations de Src aux sites d’adhésion focaux) (Chan et al., 2009). Inversement, il a été démontré que FAK induit la phosphorylation de Src, ce qui favorise le désassemblage des sites d’adhésions focaux et la formation des invadopodes (Alexander et al., 2008; Hauck et al., 2002).
Lors de l’initiation de l’invasion tissulaire des cellules cancéreuses, les cellules doivent dégrader la membrane basale et la matrice extracellulaire afin d’atteindre les vaisseaux sanguins ou le système lymphatique (Nguyen et al., 2009). Ces invadopodes sont nécessaires à la dégradation de la matrice extracellulaire afin de permettre l’invasion cellulaire mais aussi pendant l’intravasation, une étape importante du processus
métastatique (Stylli et al. 2008, Buccione et al. 2009). Lorsque les cellules sont cultivées sur une matrice, l’invadopode se retrouve au niveau de la surface ventrale de la cellule sous le noyau et à une longueur de 2 à 10 μm. Comparativement, le podosome est plus petit (0,5 à 2 μm) que l’invadopode et on le retrouve plutôt au front de migration de la cellule. Le podosome possède aussi une activité protéolytique via la métalloprotéase matricielle membranaire de type 1 (MT1-MMP) et le récepteur de l’urokinase (UPAR) (Linder 2007, Murphy & Courtneidge 2011). Tandis que l’activité de dégradation des invadopodes est due à la présence de protéases répertoriées en quatres catégories soit : les métalloprotéases régulées par le zinc (la famille des MMPs et la famille des ADAMs), les protéases à sérine et les cathepsines (Stylli et al., 2008). Les membres de la famille des MMPs sont des endopeptidases multifonctionnelles dépendant du zinc qui peuvent dégrader divers composants ECM. Presque toutes les MMPs sont sécrétées dans le milieu extracellulaire, à l'exception de six MMPs ancrées à la membrane plasmique ; MT1-MMP à MT6, appelées respectivement MMP-14, MMP-15, MMP-16, MMP-17, MMP-24 et MMP-25. Les invadopodes sont enrichis en MT1-MMP, MMP-2 et MMP-9 et sont essentiels à la formation et à l’activité de l’invadopode. Parmi les protéases de la famille des ADAMs (A Disintegrin and metalloproteinase), il a été montré que ADAM12 contribue à la fonction de l'invadopode à plusieurs niveaux qui comprennent la dégradation de l'ECM, la modulation de la fonction des intégrines et sa fonction de «sheddase» qui active les facteurs de croissance (Seals & Courtneidge 2003, Moss & Lambert 2002). ADAM12 est localisée aux invadopodes via sont interaction à la protéine d'échafaudage Tks5 et il a été démontré que ADAM12 permet le déclenchement de l'assemblage de l’invadosome (Abram et al., 2003; Albrechtsen et al., 2011). Deux protéases à sérine, la sépharase et DPP4, sont également associées à l’invadopode mais leurs rôles demeurent peu connus. Certaines études suggèrent que la sépharase, via son activité gélatinase, peut contribuer à l’activité globale de l’invadopode (Christiansen et al., 2007; Ghersi et al., 2006). Finalement, il a été démontré que l’expression des cathepsines, des endopeptidases, est augmentée dans plusieurs lignées cellulaires cancéreuses et qu’elle aurait un rôle dans l’invasion tumorale (Brisson et al., 2011; Chen et Platt, 2011; Gillet et al., 2009). Parmi les 11 membres de la famille des cathepsines, la cathepsine K est capable de cliver et de dégrader les collagènes de type I et II (Dejica et al., 2008; Han et al., 2009). Il a été démontré que les cathepsines
sont retrouvées aux podosomes des ostéoclastes et macrophages, par contre, leurs implications dans l’activité des invadopodes reste à déterminer (Jevnikar et al., 2012; Touaitahuata et al., 2014). Jusqu’à présent, il a été démontré que les FLS expriment plusieurs métalloprotéases dont MT1-MMP, MMP-2, MMP-3, MMP-9, ADAM10 ainsi que ADAM17/TACE (Li et al., 2015; Liu et al., 2014; Tolboom et al., 2002; Yuan et al., 2014). De plus, il a été récemment démontré que la cathepsine B régule l’invasion et la migration des FLS dans la PR (Tong et al., 2014).
3.2 Biogenèse de la formation des invadopodes
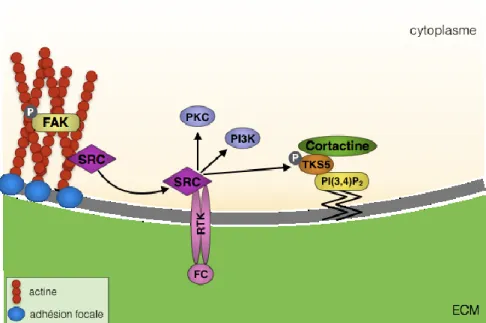
La biogenèse des invadopodes a été bien caractérisée chez les cellules cancéreuses. L’assemblage et le désassemblage des invadopodes est un mécanisme finement régulé qui implique un réarrangement du cytosquelette et l’adhérence cellulaire. Le phénotype invasif des cellules est caractérisé par une perte des sites d’adhésions focaux et par la formation des invadopodes. Une des étapes initiales implique la dissolution des sites d’adhésions focaux via l’implication de la protéine FAK, mais comme mentionné précédemment, d’autres études doivent être effectuées afin d’élucider son rôle exact dans la biogenèse des invadopodes. Par la suite, il est clair que la phosphorylation de la kinase Src est nécessaire pour l’initiation de la formation de l’invadopode (figure 4) (Buccione et al., 2009; Chen, 1989; Tarone et al., 1985). L’activation de Src permet à la kinase de lier ses substrats dont les substrats de tyrosine kinase avec quatre et cinq domaines SH3 (Tks4 et Tks5). Tks4 et Tks5 sont des protéines d’échafaudage spécifiques aux invadopodes. Les études sur ces deux protéines démontrent qu’elles ont des fonctions différentes chez les invadopodes malgré leur forte homologie. L’inhibition de Tks5 empêche la formation de l’invadopode mais n’a aucun effet sur la sécrétion des MMPs. Par contre, l’inhibition de Tks4 mène à la formation de la structure d’actine de l’invadopode mais il n’y a aucune dégradation de l’ECM dû à une perte de la localisation membranaire de la MT1-MMP à l’invadopode (Buschman et al., 2009; Seals et al., 2005). Lorsque phosphorylée, Tks5 est capable de se lier au phosphatidylionostol-3,4-bisphosphate (PI(3,4)P2) via son domaine PX. Le PI(3,4)P2 peut être produit à partir du PI(3)P ou du PI(4)P par la phosphoinositide 3-kinase (PI3K) de classe I ou de classe II. Une fois ancrée au PI(3,4)P2 du site de formation de l’invadopode, Tks5 est responsable du recrutement de la cortactine (Oser et al., 2009). La cortactine est une protéine d’échafaudage qui assure le recrutement de protéines impliquées
dans le remodelage de l’actine et favorise ainsi la réorganisation des filaments d’actine (Yamaguchi et al., 2005). Le domaine terminal NH2 acide de la cortactine favorise sa liaison au complexe Arp2/3 (Weaver et al., 2001). Le complexe Arp2/3 est la machine moléculaire qui génère des réseaux ramifiés de filaments d'actine dans les cellules (Goley et Welch, 2006). Il a été montré que la cortactine forme des agrégats au niveau des sites de formation des invadopodes et serait une des premières étapes de la biogénèse de ces structures (Artym et al., 2006).
Figure 4 Initiation de la formation d’invadosome
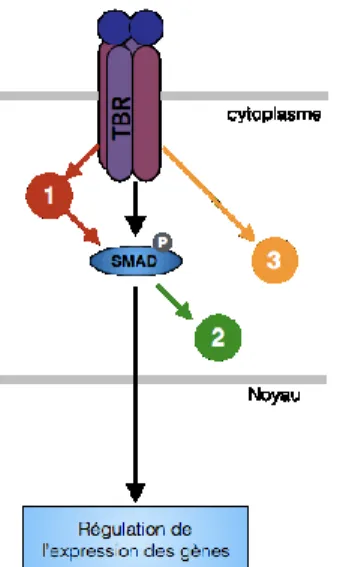
Les cellules établissent des adhérences focales avec la matrice extracellulaire (ECM) grâce à SRC et FAK phosphorylé. Il y a libération de Src, lui permettant de se lier et de phosphoryler TKS5, PI3K et PKC. La phosphorylation de TKS5 lui permet de lier la PI(3,4)P2 et la cortactine. Ce mécanisme
intracellulaire est initié par l’activation des RTKs. FC : facteurs de croissance, P : phosphorylé. (Murphy et Courtneidge, 2011) ; numéro de licence 3998530412186)
Suite à leur recrutement au site de formation de l’invadopode, la Tks5 et la cortactine permettent le recrutement d’autres protéines impliquées dans la réorganisation du cytosquelette d’actine et ainsi permettre l’assemblage de la structure d’actine de l’invadopode (figure 5). Tks5 peut s’associer à plusieurs régulateurs d’actine grâce à son domaine SH3 dont, Nck1, Nck2, N-WASP et Grb2, permettant ainsi leur rapprochement (Oikawa et al., 2008; Stylli et al., 2009). Après la formation des précurseurs d’invadosomes, la cortactine se lie à la cofiline, séquestrant et inhibant son activité d’assemblage de l’actine (Saykali et El-Sibai, 2014). Cependant, lorsque la cortactine
devient phosphorylée sur les résidus tyrosine (Tyr), la cofiline est libérée de sa liaison à la cortactine lui permettant ainsi de générer une amplification des ramifications de l'actine à l'invadosome (Oser et al., 2009). La cortactine peut être phosphorylée par la kinase activée en p21 (PAK), la kinase régulée extracellulairement (ERK) et Src (Weaver, 2008). La cortactine peut aussi se lier à d’autres régulateurs d’actine essentiels à la formation de l’invadosome, tels que, Cdc42, WIP et dynamine (Yamaguchi et al., 2005). On retrouve également plusieurs autres substrats de Src aux invadosomes, dont p130Cas et AFAP110, deux protéines adaptatrices impliquées dans la formation des invadosomes (Lock et al., 1998; Thomas et Brugge, 1997). Cette cascade d’évènements permet l’élongation de la protrusion d’actine dans la matrice extracellulaire (Oser et al., 2009; Yamaguchi et al., 2005). Une fois que le précurseur de l’invadosome est formé, la cortactine est déphosphorylée, ce qui est nécessaire pour la stabilisation de l’invadosome. La déphosphorylation de la cortactine séquestre la cofiline ce qui empêche une polymérisation excessive de l’actine par la cofiline. Quelles phosphatases sont impliquées dans la déphosphorylation de la cortactine dont la phosphatase PTP1B (Stuible et al., 2008).
Figure 5 Assemblage de l’invadosome
L’assemblage de l’invadosome se produit par le recrutement et l'activation des protéines régulatrices de l'actine (Arp2/3, WIP) et la phosphorylation des composantes clés de l'invadopode
(cortactin, Tks5). La phosphorylation de la cortactine (par Src, PAK et ERK) et Tks5 régulent la formation de l'invadosodome. Tks5 s’associe Nck1, Nck2, Grb2 et N-WASP. La cortactin lie WIP et la dynamine. De plus, la phosphorylation de la cortactine provoque la dissociation de la cofiline pour initier la polymérisation de l'actine. P : phosphorylé (Murphy et Courtneidge, 2011) ; numéro de licence 3998530412186)
Une fois l’assemblage de l’invadosome complété, il y aura maturation de l’invadosome et dégradation de la matrice extracellulaire (figure 6). Il a été montré que la sécrétion et la localisation des protéases aux invadopodes sont en grande partie régulées par la cortactine et la Tks4. La cortactine joue également un rôle clé dans la biogénèse de l’invadopode. Clark et al. ont démontré que dans les cellules déficientes en cortactine le nombre d’invadopodes est diminué et ceux-ci ont perdu leur capacité à dégrader l’ECM. Inversement, la surexpression de la cortactine augmente considérablement la dégradation de l’ECM (Clark et al., 2007). En plus d’avoir un impact sur la formation d’invadopodes, la sécrétion de MMP-2 et MMP-9, ainsi que la relocalisation membranaire de la MT1-MMP sont étroitement corrélées au taux d'expression de cortactine (Clark et al., 2007). C’est la protéine adaptatrice Tks4 qui permet le recrutement de la MT1-MMP aux invadopodes (Buschman et al., 2009). En effet, il a été démontré que Tks4 n’intervient ni dans la formation de l’invadopode, ni dans la sécrétion de MMPs. Par contre, Tks4 permettrait la localisation et la stabilisation de la MT1-MMP aux invadopodes, qui pourrait alors activer les MMPs (Buschman et al., 2009). La MT1-MMP est une métalloprotéinase essentielle à la dégradation de la matrice extracellulaire. Elle est composée d’un peptide signal, d’un propeptide, d’un domaine catalytique, de deux domaines charnières, d’un domaine hémopexine, d’un domaine transmembranaire et d’une queue cytoplasmique courte de 20 acides aminés. Au cours du processus de sécrétion, son peptide signal est clivé par une peptidase et le propeptide par la furine de sorte que l'enzyme exprimée à la surface cellulaire est sous forme active (Roghi et al., 2010). Une fois à la membrane plasmique, la MT1-MMP peut être internalisée par la voie d’endocytose dépendante de la clathrine ou par une voie dépendante des cavéoles (Poincloux et al., 2009; Steffen et al., 2008). Par la suite, la MT1-MMP s’accumule dans des vésicules intracellulaires où la protéase sera soit dégradée par les lysosomes, soit recyclée à la surface de la cellule ou aux invadosomes (Paz et al., 2014; Takino et al., 2003; Wang et al., 2004). En plus de dégrader des composantes de l’ECM, y compris la fibronectine, la gélatine, la vitronectine, ainsi que le collagène de
types I, II et III, la MT1-MMP permet l’activation d’autres métalloprotéinases (Overall et Dean, 2006). La MT1-MMP active est inhibée par les inhibiteurs tissulaires de métalloprotéinases (TIMP) TIMP-2, TIMP-3 et TIMP-4, mais pas par TIMP-1. Parmi ceux-ci, TIMP-2 a un double rôle puisqu’il inhibe MT1-MMP et favorise l'activation de la proMMP-2. Afin d’activer la MMP2, la MT1-MMP forme un homodimère via son domaine hémopexine et l’une des deux enzymes se lie à TIMP-2 via son domaine catalytique. Le domaine C-terminal de TIMP-2 se lie alors au domaine hémopexine de la proMMP-2, en positionnant la proMMP-2 de façon optimale pour son activation par l’autre MT1-MMP active dans le complexe (Itoh et Seiki, 2004). La MMP-2 ainsi que la MMP-9 peuvent dégrader le collagène de type IV qui est l'une des composantes de la membrane basale la plus abondante (Poincloux et al., 2009).
Figure 6 Maturation de l’invadosome
L'invadosome favorise la dégradation de l'ECM en sécrétant la MMP-2 et la MMP-9 et en recrutant la MT1-MMP à l'extrémité de la structure. Pendant la maturation, les invadopodes favorisent la dégradation de l'ECM en coordonnant la sécrétion de la matrice. La déphosphorylation de la cortactine permet la stabilisation de l’invadosome. P : phosphorylé (Murphy et Courtneidge, 2011) ; numéro de licence 3998530412186)
Les invadopodes sont des structures dynamiques qui sont renouvelées mais les mécanismes précis de leur désassemblage ne sont pas bien connus. La formation de l’invadopode fait
intervenir plusieurs kinases pour permettre le recrutement et l’activation d’effecteurs impliqués dans le remodelage de l’actine. La phosphorylation de Tks5 par Src favorise la formation de l’invadopode alors que la phosphorylation de AFAP110 et de la cortactine permet le désassemblage des invadopodes (Dorfleutner et al., 2008). AFAP110 est une protéine adaptatrice de Src qui se lie à l'actine filamenteuse et donc, qui localise les kinases impliquées dans les changements de l'organisation du cytosquelette d'actine. La phosphorylation étant une modification post-traductionnelle réversible, le recyclage fait donc intervenir plusieurs phosphatases. Par exemple, la protéine phosphatase PTP-PEST se retrouve aux sites potentiels de formation d’invadopode et bloque leur formation (Diaz et al., 2009). De plus, la protéine phosphatase PTEN, connue pour être un suppresseur de tumeur, bloque l’invasion cellulaire dépendante des invadopodes en inactivant la protéine Src via le blocage de l’activité de la protéine Stat3 (Mukhopadhyay et al., 2010). Par contre, certaines phosphatases peuvent avoir un effet positif sur la formation des invadopodes dont la protéine phosphatase PTP-ε qui déphosphoryle la tyrosine 527 de Src afin de l’activer (Granot-Attas et al., 2009).
3.3 Induction de la formation des invadopodes
La formation des invadopodes implique le recrutement des multiples protéines, suivie de la polymérisation de l'actine et de l'allongement de la membrane plasmique. Les intégrines (β1, β2 et αV) et les facteurs de croissances (dont le PDGF, l’EGF et le TGFβ), peuvent initier la cascade de signalisation impliquée dans la formation des invadopodes (Beaty et al., 2013; Desmarais et al., 2009).
3.3.1 Le PDGF (facteur de croissance dérivé de plaquettes)
Le PDGF est un facteur de croissance impliqué dans la survie, la prolifération et la motilité de plusieurs types cellulaires. Le PDGF a été isolé pour la première fois des plaquettes humaines. Le PDGF et son récepteur correspondant (PDGFR) ont d’importants rôles dans la régulation des fonctions physiologiques humaines. Il a été montré que le PDGF joue un rôle dans la formation des vaisseaux sanguins, la régulation de la cicatrisation et le maintien de la pression des fluides interstitiels (Berk et al., 1986; Robson et al., 1992; Rodt et al.,
1996). Une augmentation de l’expression du PDGFR a été observée dans le cancer du sein et l’inhibition du PDGFR et du c-Kit avec l’imatinib (inhibiteur de plusieurs tyrosine-kinases dont c-Kit, Abl, SCF et PDGFR), supprime la croissance et l’invasion des cellules cancéreuses (Roussidis et al., 2007). De plus, il a été montré que l’expression de l’ARN messager du PDGF est significativement plus élevée dans le liquide synovial des patients atteints de polyarthrite rhumatoïde comparativement aux patients atteints d’arthrose. Il a été démontré que le PDGF augmente l’infiltration des monocytes/macrophages dans la membrane synoviale des patients atteints de polyarthrite rhumatoïde, ce qui contribue à l’hyperplasie de celles-ci (Lafyatis et al., 1989; Ross et al., 1990).
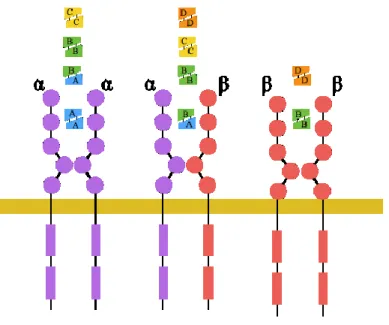
Le PDGF est une molécule dimérique composée des chaines polypeptidiques A, B, C et D. L’association des chaines polypeptides homologues forment des homodimères (PDGF-AA, PDGF-BB, PDGF-CC et PDGF-DD) et les chaines polypeptidiques A et B peuvent s’associer pour former l’hétérodimère (PDGF-AB) (Heldin et al., 1998). Tous les isoformes de PDGF sont donc sous forme dimérique et possèdent par conséquence deux épitopes de liaison aux récepteurs ; ainsi chaque dimère de PDGF se lie à deux récepteurs simultanément (Fretto et al., 1993; Herren et al., 1993). Ces isoformes du PDGF ont des effets sur les cellules cibles en liant, avec différentes spécificités, les deux types de récepteurs de la tyrosine kinase structurellement apparentés appelés récepteur au PDGF α (PDGFRα) et récepteur au PDGF β (PDGFRβ). La plupart des récepteurs à activité tyrosine kinase sont activés par la liaison de leurs ligands, ce qui mène à la dimérisation ou à l’oligomérisation des récepteurs. Les différents ligands vont induire la dimérisation de différents types de PDGFR (Figure 7). Le PDGFRα lie la chaîne du ligand A, B et C, tandis que PDGFRβ lie uniquement à la chaîne du B et D (Andrae et al., 2008; Heldin et al., 1998). Les récepteurs α et β contiennent chacun cinq domaines extracellulaires et un domaine intracellulaire à activité tyrosine kinase. Les trois domaines extracellulaires les plus distaux de la membrane plasmique sont impliqués dans la liaison du ligand. La liaison du PDGF permet de former un pont entre les deux récepteurs. Le complexe récepteur-ligand est stabilisé par l’interaction directe entre les quatrièmes domaines extracellulaires des deux récepteurs (Heldin et al., 1998).