ETUDE PHARMACOGENETIQUE D'UN
HYPOLIPÉMIANT
Mémoire présenté
à la Faculté des études supérieures de l'Université Laval dans le cadre du programme de maîtrise en pharmacie (avec mémoire)
pour l'obtention du grade de maître es sciences (M.Se.)
FACULTE DE PHARMACIE UNIVERSITÉ LAVAL
QUÉBEC
2009
L'acide fénofibrique (AF), la forme circulante active du fénofibrate, module le profil lipidique. En effet, à dose thérapeutique, il augmente le HDL-cholestérol de 15 à 25% et réduit les triglycérides sanguins de 35 à 50%. La pharmacocinétique de ce médicament est sujette à une variablité interindividuelle d'origine inconnue. Mon projet de maîtrise avait pour but d'approfondir les connaissances relatives au métabolisme de l'AF, en regard de sa voie principale de biotransformation, soit la glucuronidation par les enzymes UDP-glucuronosyltransférases (UGT). Mes travaux ont contribué à l'identification des trois isoformes UGT principalement impliquées dans la formation de l'AF-glucuronide (AF-G), soit les UGT 1 A3, UGT1A9 et UGT2B7, où ce dernier semble avoir le rôle majeur. De plus, cette étude a permis l'identification de variations génétiques (polymorphismes) de ces gènes associées à une altération de l'activité de conjugaison de l'AF. À ce jour, nos résultats suggèrent que certains polymorphismes des UGT pourraient être responsables d'une partie de la variabilité interindividuelle observée dans la pharmacocinétique de l'AF.
Abstract
Fenofibric acid (FA), the active moiety of fenofibrate, modulates the lipid profile. Indeed, the major effects of FA are to lower serum triglycerides by 35 to 50 % and rise HDL-cholesterol by 15 to 25%. This drug is subject to large interindividual variability in its pharmacokinetics and the origin of this variation is unknown. My master's degree project was to study the metabolism of the FA, especially its main way of biotransformation, glucuronidation by UDP-glucuronosyltransferase (UGT) enzymes. My work contributed to the identification of the three UGT isoforms mainly involved in the formation of FA-glucuronide (FA-G), namely UGT 1 A3, UGT1A9 and UGT2B7, amongst which UGT2B7 appears to play a major role. Furthermore, this study allowed the identification of genetic variations (polymorphisms) in these genes that are associated with changes in FA conjugation activity. So far, our results suggest that certain polymorphisms in UGTs could explain part of the interindividual variability observed in FA pharmacokinetics.
Avant-Propos
Ce mémoire de maîtrise, intitulé « Étude pharmacogénétique d'un hypolipémiant », présenté à la faculté des études supérieures de l'Université Laval pour l'obtention du grade de maître es sciences, est rédigé sous la forme d'insertion d'articles.
L'article principal, présenté au chapitre III, intitulé « Identification des UGT humaines impliquées dans le métabolisme du fénofibrate et l'influence des variants génétiques », dont je suis la première auteure, sera soumis pour publication dans le périodique Drug Metabolism and Disposition. J'ai effectué toutes les expériences reliées à ce manuscrit. Tout d'abord, avec l'aide d'un chimiste de l'équipe du Dr. Guillemette, Patrick Caron, j'ai contribué à l'élaboration de la méthode d'analyse par chromatographic liquide de haute performance (HPLC) de l'acide fénofibrique glucuronide. J'ai ensuite effectué tous les essais enzymatiques pour identifier les enzymes UGT impliquées dans le métabolisme de l'acide fénofibrique et les comparer aux isoformes variantes, suite à leur production par transfection stable. Ensuite, j'ai fait les analyses informatiques des cinétiques obtenues pour les enzymes impliquées et leurs isoformes variantes. J'ai également effectué les essais enzymatiques avec les échantillons de foie humain pour vérifier la variabilité interindividuelle et j'ai aussi réalisé les analyses statistiques s'y rattachant. De plus, j'ai généré les résultats d'immunobuvardage pour déterminer l'expression des protéines UGT. Finalement, j'ai participé à la rédaction du manuscrit et préparé toutes les figures du papier. Chantai Guillemette a conceptualisé l'étude, analysé les résultats, rédigé et corrigé le manuscrit avec les auteurs principaux. Marie-Odile Benoit-Biancamano, étudiante au doctorat, a participé à la rédaction et revu les analyses statistiques. Robert J. Straka, de l'Université de Minnesota à Minneapolis, est le collaborateur dans la suite du projet qui porte sur une étude pharmacocinétique du fénofibrate chez des sujets avec les profils génétiques déterminés. Finalement, Michael H. Court, Lisa L. Von Molthe et David J. Greenblatt, de l'Université Tufts à Boston, nous ont donné accès aux échantillons de foie humain (n = 48). Le manuscrit inséré dans ce mémoire est une version préliminaire de l'article qui sera soumis pour publication.
Pendant ma maîtrise, j'ai également participé à d'autres projets en faisant les génotypages. Ainsi, j'ai joint en annexe trois autres articles.
Le second article, présenté en annexe 1, intitulé « Étude pharmacogénomique de la glucuronosyltransférase UGT 1 A3 humaine conjugant les estrogènes », a été publié dans la revue Pharmacogenetics and genomics en juillet 2007. Ce manuscrit, dont Bertrand Caillier et Johanie Lépine sont les premiers co-auteurs, a été réalisé grâce à la participation de plusieurs personnes. Bertrand Caillier, étudiant à la maîtrise, a généré les résultats des études fonctionnelles des variants du promoteur de UGT 1 A3, incluant la détermination des fréquences allèliques, la prédiction des haplotypes et leur fréquence, les études transcriptionnelles et les analyses de retard sur gel. Johanie Lépine, étudiante au doctorat, a généré les résultats des études fonctionnelles des variants de la région codante de UGT 1 A3, incluant le génotypage, la détermination des fréquences allèliques, la prédiction des haplotypes et leur fréquence, les essais enzymatiques suite à l'amplification de chacune des isoformes par transfection stable, de même que le calcul des affinités, vélocités et clairances de chacune de ces isoformes. Bertrand et Johanie, ont écrit la version initiale de ce manuscrit, préparé les figures ainsi que la liste des références. Quant à moi, j'ai réalisé le génotypage du gène UGT1A3 dans une cohorte de 249 Caucasiens Français avec Vincent Ménard, également étudiant à la maîtrise. Louis Perusse est l'un des investigateurs impliqués dans la Quebec Family Study qui nous a donné accès aux échantillons d'ADN des Caucasiens Français. Alain Bélanger et Olivier Barbier, chercheurs au Centre de recherche du CHUQ, ont collaboré à cette étude avec ma directrice de recherche Chantai Guillemette. Elle a également fait le design et la conceptualisation de l'étude, l'analyse des données de même que la rédaction et la révision de l'article. Le manuscrit inséré dans ce mémoire est identique à celui publié.
Le troisième article, présenté en annexe 2, intitulé « Association de polymorphismes des gènes UGT1A8 et UGT2B7 à une formation variable des glucuronides phénolique et acyl de l'acide mycophénolique », a été publié dans le périodique Drug Metabolism and Disposition en août 2006. Olivier Bernard, étudiant à la maîtrise, est le premier auteur de l'article et il a effectué la majorité des expériences reliées à ce manuscrit, soit tous les
contribué à sa soumission au périodique. Kim Journault, une assistante de recherche au laboratoire du Dr. Guillemette, a préparé des lignées stables surexprimant les variants génétiques UGT1A8. Quant à moi, j'ai effectué le génotypage du gène UGT1A8 dans une cohorte de 254 Caucasiens Français. Enfin, Louis Pérusse est l'un des investigateurs impliqués dans la Quebec Family Study qui nous a donné accès aux échantillons d'ADN des Caucasiens Français. Chantai Guillemtte a fait le design et la conceptualisation de l'étude, l'analyse des données de même que la rédaction et la révision de l'article. Le manuscrit inséré dans ce mémoire est identique à celui publié.
Le quatrième article, présenté en annexe 3, intitulé « Caractérisation des variants communs des UGT1A8, UGT1A9 et UGT2B7 présentant des capacités altérées pour l'inactivation des metabolites 4-hydroxylés mutagéniques de l'estradiol et de l'estrone », a été publié dans la revue Cancer Research en janvier 2006. Ce manuscrit, dont Jean Thibaudeau et Johanie Lépine sont les premiers co-auteurs, a été réalisé grâce à la participation de nombreuses personnes. Jean Thibaudeau, étudiant à la maîtrise, a fait les premières séries d'essais enzymatiques, leurs compilations et les analyses statistiques s'y rattachant. Johanie Lépine, étudiante au doctorat, a poursuivi les essais enzymatiques produisant les résultats finaux inclus dans la version publiée. Elle a de plus généré les résultats d'immunobuvardage et d'immunohistochimie pour l'étude de l'expression des protéines UGT dans l'endomètre et la glande mammaire de femmes ménopausées. Jean et Johanie, ont écrit la version initiale du manuscrit, préparé les figures ainsi que la liste des références. Yannick Duguay, étudiant au doctorat, a caractérisé l'activité transcriptionnelle d'un variant du promoteur dans une lignée cancéreuse d'endomètre humain. Georges Pelletier, chercheur au Centre de recherche du CHUQ, a collaboré à l'interprétation des résultats obtenus suite aux analyses d'immunohistochimie. Marie Plante et Bernard Têtu, de l'Hôtel-Dieu de Québec, sont respectivement le médecin clinique et le pathologiste avec qui nous travaillons dans le cadre de notre étude sur le cancer de l'endomètre, ce qui a permis l'étude d'échantillons utérins. Jacques Brisson et Simon Jacob, de l'Hôpital Saint-Sacrement, sont respectivement l'épidémiologiste et le pathologiste qui collaborent avec nous dans le cadre de notre étude sur le cancer du sein, ce qui a permis d'étudier des échantillons mammaires.
Quant à moi, j'ai fait le génotypage des gènes étudiés dans une cohorte de 258 Caucasiens Français et j'ai calculé les fréquences allèliques. Louis Pérusse est l'un des investigateurs impliqués dans la Quebec Family Study qui nous a donné accès aux échantillons d'ADN des Caucasiens Français. Alain Bélanger, chercheur au Centre de recherche du CHUQ, a conjointement participé à cette étude avec ma directrice de recherche Chantai Guillemette. Elle a également fait le design et la conceptualisation de l'étude, l'analyse des données de même que la rédaction et la révision de l'article. Le manuscrit inséré dans ce mémoire est identique à celui publié.
Le présent mémoire comprend une introduction générale englobant les concepts abordés dans le cadre de mon projet de maîtrise. Le manuscrit inséré suit cette introduction et constitue un chapitre distinct. Ce manuscrit a été rédigé en anglais et selon les critères exigés par le périodique. Cependant, il est ici accompagné d'un résumé en français et comporte les sections suivantes: résumé, introduction, hypothèse et objectifs, méthodologie, résultats, discussion et conclusion, bibliographie, tableaux et figures. Dans la dernière section de ce mémoire de maîtrise, une discussion générale résume l'ensemble des résultats obtenus et propose également de futures avenues de recherche envisagées pour la poursuite du projet. Aussi, une liste des références citées dans l'introduction et la discussion termine ce mémoire. Enfin, j'ai joint, en annexe, trois autres manuscrits auquels j'ai contribué durant ma maîtrise.
Remerciements
Tout d'abord, je tiens à remercier mes parents, Ranko et Tatjana, pour m'avoir appuyée tout au long de mes études. Ma sœur Aleksandra est également pour moi une source de motivation et de divertissement. Je remercie mon plus grand complice, Alexandre, pour son amour, sa générosité, sa patience, son humour et sa compréhension. Je ne serais pas rendue là aujourd'hui sans vous!
Ensuite, je remercie sincèrement ma directrice de recherche, Dr. Chantai Guillemette, pour m'avoir si bien formée et pour m'avoir donnée le privilège d'effectuer mes études graduées dans son équipe de recherche. Merci d'avoir cru en moi et de m'avoir accordée autant de confiance et de responsabilités. J'ai beaucoup apprécié ton support constant dans tous mes projets. Je suis fière d'avoir pu bénéficier d'un aussi bon modèle dans le domaine de la science. J'admire énormément ta rigueur, ton dynamisme et ta détermination et pendant ma maîtrise j'ai compris que c'est un cheminement vers la quasi perfection, ce qui va certainement m'être très utile tout au long de ma vie.
Évidemment, remerciements à mes anciens et récents collègues au Centre de Recherche du CHUL. Je débuterais par Lyne Villeneuve qui est une source de référence indispensable. Merci d'avoir endurer mes nombreuses questions. J'espère que tu vas avoir des jumeaux! Kim Journault, maintenant en congé de maternité, été la première à m'initier au monde des UGT. Merci de m'avoir si bien expliquer comment maniper avec la rigueur, surtout pour les Westerns. On se voit sur le terrin de golf l'année prochaine! Un très gros merci à Hugo Girard pour son aide au niveau informatique, par exemple EndNote. Même si tu m'as écoeurée à maintes reprises, tu es une personne très sympatique et généreuse. Vincent Ménard, merci de m'avoir encouragée dans mes projets. Appelles-moi pour être la représentante de ta future compagnie! Anne-Sophie Bélanger, ma coéquipière pour le projet méthylation, j'ai beaucoup apprécié à travailler avec toi. Merci pour ta compréhension et ta simplicité. On se reprendra pour le magazinage de Nôel avec Viny! Marie-Odile Benoit-Biancamano est un vrai dictionnaire des animaux et des plantes. Je te remercie énormément pour avoir corriger ce mémoire. Également merci pour tous les
conseils que tu m'as donnée pour mon chien! Merci à Johanie Lépine pour sa franchise, son sens d'humour et son divertissement avec son talent de chanteuse. Je suis très heureuse de t'avoir côtoyée pendant ces trois années. Judith Bellemare est une fille travaillante et sympatique. Merci de m'avoir faite découvrir les « amourettes d'agneau »! Un gros merci à Patrick Caron pour tout le temps passé à analyser mes essais enzymatiques. Tu es un chimiste hors-pair! Merci à Jean-Philippe Adam pour nous avoir fait rire si souvent avec tes histoires possibles et impossibles. Eric Lévesque, je te remercie pour tes précieux conseils quant aux différentes manières de faire une même manip, souvent plus pratiques. Merci à Mario Harvey, un nouveau membre de l'équipe CG, pour sa gentillesse et pour avoir corriger mon article. Olivier Bernard, ancien étudiant à la maîtrise et pharmacien, même si j'ai été la cible de tes nombreuses manigances, je suis très contente de t'avoir connu. Merci à Sarah Chouinard, ta douceur et ta gentillesse font de toi une personne exeptionnelle. Remerciements à toute l'équipe du Dr. Olivier Barbier, soit Mélanie Verreault (bientôt maman de deux enfants), Jocelyn Trottier (si serviable) et Jenny Kaeding (très expressive), pour avoir été des collègues tant appréciés. Merci à Nicolas Léveillé, NTB (Nick The Brain) ou anti-UGT, dont la maturité dépasse l'âge. Je suis certaine que tu vas avoir beaucoup de succèes dans ton post-doc en Europe. J'apprécie sincèrement tout le temps que tu as investi pour nos conversations. Tu m'as fait beaucoup réfléchir tant au niveau professionnel que personnel. Merci encore ! Profonds remerciements à Dr. Olivier Barbier et Dr. Alain Bélanger pour leurs remarques et suggestions très utiles lors de nos réunions d'éguipes hebdomadaires. Je veux également remercier à Ronal Maheux pour toutes ces conversations sur l'heure du dîner ou bien dans la salle HPLC pendant que j'écrivais ce mémoire. Tu a été si professionnel, serviable et sympatique et tu es une très bonne personne ressource. On ira voir une game des Canadiens un moment donné ! Merci aussi à Alain Fortier pour les heures et les heures de plaisir à parler de tout et de rien. C'était très amusant d'être la principale cible de tes écœurements. Comme on dit, on écœure ceux qu'on aime ! Je passerai te voir pour ne pas que tu t'ennuies trop. Merci à toutes les personne suivantes avec qui j'ai eu la chance de travailler à un moment ou un autre et qui m'ont particulèrement marquée : Line Cantain, Andréa Fournier, Martin Perreault, Olivier Larouche, Julie Lessard, Jean-François Gagnon et Frédéric Guénard.
Finalement, je tiens à remercier les Fonds de la Recherche en Santé du Québec (FRSQ) pour m'avoir décernée une bourse de recherche à la maîtrise. De plus, j'aimerais tout spécialement remercier la Faculté de Pharmacie de l'Université Laval et tout son personnel pour m'avoir guidée à travers mes études et pour l'obtention d'une bourse du Fonds d'Enseignement et de Recherche (FER). Je me sens très privilégiée d'avoir effectué mes études de deuxième cycle au sein de cette faculté.
et vous n 'aurez pas à travailler un seul jour de votre vie.
Table des matières
Résumé i Abstract ii Avant-Propos iii
Remerciements vii Table des matières xi Liste des tableaux xiii Liste des figures xiv CHAPITRE I : Introduction 1
1. Dyslipidémie 2 1.1. Hypercholestérolérnie 5
1.1.1. Hypercholestérolérnie familiale 8
1.2. Hypertriglycéridémie 9 2. Traitement médical de la dyslipidémie 10
2.1.Statines 14 2.1.1. Métabolisme des statines 18
2.1.2. Effets indésirables et pléitropes des statines 19
2.2. Fibrates 21 2.2.1. Interactions médicamenteuses des fibrates 22
2.2.2. Fénofibrate 23 3. UDP-Glucuronosyltransférases (UGT) 25
3.1. Réaction de glucuronidation 26 3.2. Profil d'expression des UGT 29 3.3. Superfamille multigénique des UGT 29
3.3.1. Famille UGT 1A 32 3.3.2. Nouvelle classe de protéines UGT1A 34
3.3.3. Famille UGT2B 35 CHAPITREE : Hypothèse et objectifs 37
Hypothèse générale du projet de recherche 38
Objectifs du projet de recherche 39
CHAPITRE III: Résultats 40 Identification des UGT humaines impliquées dans le métabolisme du fénofibrate et
l'influence des variants génétiqiues 40 CHAPITRE FV : Discussion et conclusion 66
Discussion 67 Conclusion 73 BIBLIOGRAPHIE 74 ANNEXE 1 : CHAPITRE V 94
Étude pharmacogénomique de la glucuronosyltransférase UGT1A3 humaine conjuguant
les estrogènes 94 ANNEXE 2 : CHAPITRE VI 135
Association de polymorphismes des gènes UGT1A8 et UGT2B7 à une formation
variable des glucuronide s phénolique et acyl de l'acide mycophénolique 135
Caractérisation des variants communs des UGT1A8, UGT1A9 et UGT2B7 présentant des capacités altérées pour l'inactivation des metabolites 4-hydroxylés mutagéniques de
Liste des tableaux
Table 1 : La composition des lipoprotéines 4 Table 2 : Phénotypes de Frederickson 4 Table 3 : Classification d'ATP III pour LDL-C, cholestérol total et HDL-C 6
Table 4 : Classification des triglycérides 9 Table 5 : Effets des différents hypolipémiants sur les lipides dans le sang 11
Table 6 : Études cliniques démontrant les effets bénéfiques des statines et des fibrates 13
Liste des figures
Figure 1 : Corrélation entre le niveau sanguin du cholestérol et le risque
cardiovasculaire. Étude MRFIT, 361 662 hommes âgés de 35 à 57 ans 3 Figure 2: Structure des statines (A) et structure de l'HMG-CoA (B). Les statines
possèdent un élément de structure commun qui ressemble à l'HMG-CoA 15 Figure 3: Comparaison de l'efficacité des statines. La résuvastatine est la statine la plus
efficace pour réduire le LDL-C 16 Figure 4 : Structure des fibrates. Les fibrates les plus utilisés sont le gemfibrozil et le
fénofibrate 21 Figure 5 : Enzymes retrouvées dans les phases I et II du métabolisme des
médicaments. La phase I est principalement catalysée par les cytochromes P450 tandis que la phase II regroupe diverses classes d'enzymes de conjugaison dont les
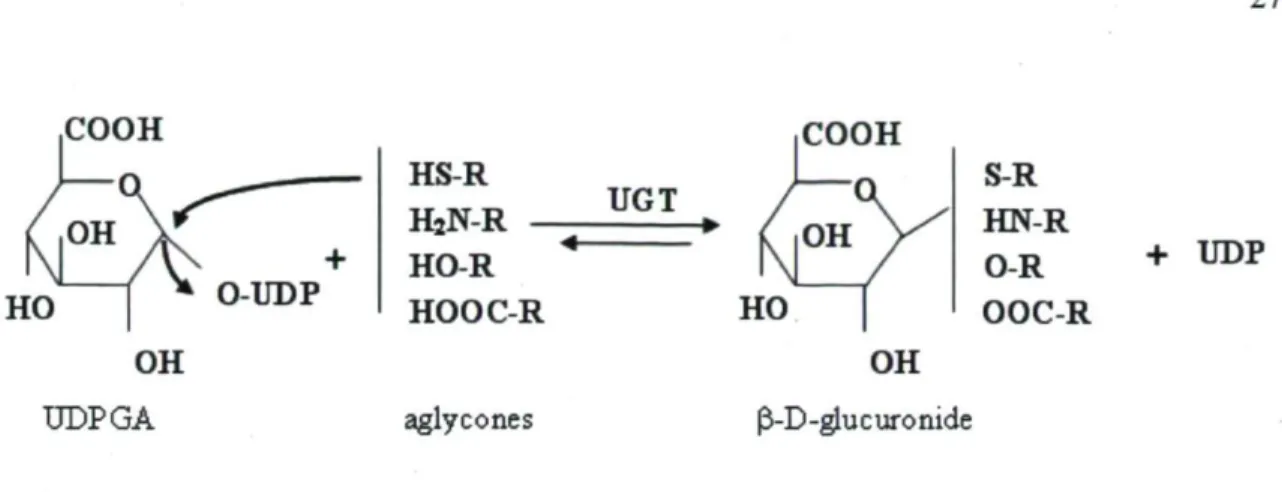
UGT représentent la plus forte proportion 26 Figure 6 : Réaction de glucuronidation catalysée par les enzymes UGT. La réaction
enzymatique implique deux molécules : l'acide glucuronique (UDPGA) qui agit comme co-facteur et le substrat (aglycone). La réaction forme le glucuronide et libère
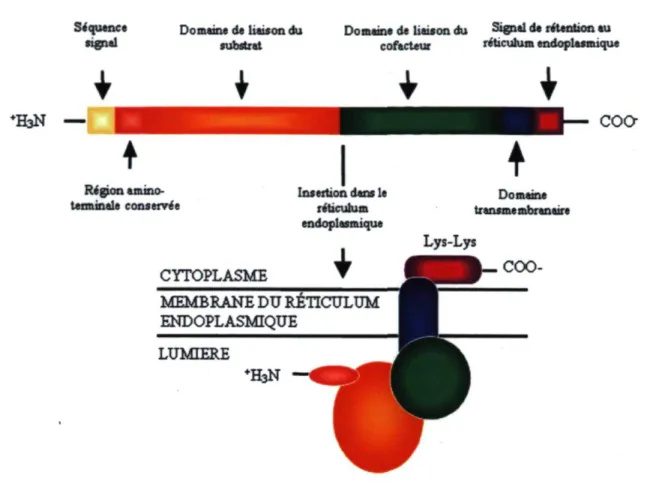
l'uridine diphosphate (UDP) 27 Figure 7 : Localisation et structure protéique schématique des UGT dans le reticulum
endoplasmique. La structure primaire des UGT est constituée de deux domaines structuraux, soit une région amino-terminale responsable de la liaison au substrat et
une région carboxy-terminale qui se fixe au co-facteur UDPGA 28 Figure 8 : Profil d'expression des UGT chez l'homme. Les UGT sont localisées aux
portes d'entrée des agents chimiques de même que dans plusieurs organes
périphériques. Une isoenzyme peut être exprimée dans plusieurs tissus et à l'inverse,
un tissu peut exprimer plusieurs types d'isoformes 30 Figure 9 : Classification des enzymes UGT humaines. Les familles UGT1 et UGT2
partagent plus de 40% d'homologie. À l'intérieur de la famille 1A et 2, les différentes
isoformes partagent au moins 66% et 59% d'homologie, respectivement 31 Figure 10 : Organisation génomique du gène UGT1A. Un seul gène code pour les
enzymes de la famille UGT1 A. Les UGT1A possèdent un exon 1 distinct et partagent les exons 2 à 5. Le tout couvre une longueur de 200 kb dans la région chromosomique
2q37... 33 Figure 11 : Épissage alternatif des exons 5a et 5b génère les protéines UGT1A il et i2,
respectivement. Les isoformes UGT1 Ai2 sont enzymatiquement inactives, contrairement aux isoformes UGTIAil, mais agissent comme modulateurs de la
glucuronidation pour plusieurs substrats 35 Figure 12 : Organisation génomique du gène UGT2B. Les gènes de la famille UGT2B
sont regroupés en cluster dans la région chromosomique 4ql3 d'une longueur de 30 kb. Les UGT2B sont encodées par des gènes différents mais tous composées de six
La mortalité due aux troubles cardiovasculaires (CVD) a décliné depuis les années 1960, mais les CVD restent la cause principale de décès tant chez les hommes que chez les femmes de toutes les races aux États-Unis. Une grande proportion d'individus âgés souffrira de maladies cardiovasculaires. Par exemple, chez les hommes âgés de 55 ans et plus, presque la moitié de tous les décès sont attribués aux CVD en comparaison avec moins de 25 % pour tous les cancers et moins de 2 % pour toutes les infections. Une proportion encore plus forte de mortalités (56%) est due aux CVD chez les femmes plus âgées (65 ans et plus) avec moins de 20 % due aux cancers (CHD, 1998). En 1998, 29.9 % des décès au Canada ont été provoqués par les CVD et l'organisation mondiale de la santé prédit que les maladies cardiovasculaires seront la principale cause de mortalité et de morbidité dans le monde d'ici 2010 (WHO, 2005). Ainsi, elles constituent la maladie fatale la plus prévalente et la plus coûteuse, soulignant le besoin de sa prévention et sa guérison.
Les facteurs les plus importants qui favorisent le développement des CVD, tels que le tabagisme, la dyslipidémie, l'hypertension, le diabète, l'obésité, l'alcool, le manque d'activité physique régulière, l'absence de consommation régulière de fruits et légumes et les facteurs psycho-sociaux contribuent à plus de 90 % du risque attribuable de la population, comme le démontre l'étude INTERHEART, réalisée à travers 52 pays (Yusuf et al., 2004).
La dyslipidémie, qui se définit comme une concentration anormale de lipoprotéines ou de lipides (cholestérol et/ou triglycérides) dans le sang, est un facteur de risque d'importance particulière (NCEP, 2002). La fréquence de la dyslipidémie varie avec la population étudiée et avec sa définition. L'incidence est la plus élevée chez les patients avec le CVD prématuré, qui est défini comme une maladie cliniquement évidente survenant avant 55 ans chez les hommes et avant 65 ans chez les femmes. Également, la fréquence de dyslipidémie est très élevée chez les personnes contrôles de même âge sans maladies cardiovasculaires : de l'ordre de 80 à 88 % en comparaison avec approximativement 40 à 48 % (NCEP, 1993). La concentration moyenne du cholestérol sérique total chez les adultes aux États-Unis a
à 1994. Au cours de la même période, le pourcentage d'adultes américains avec un niveau de cholestérol total de 240 mg/dL (6.22 mmol/L) ou plus a décliné de 20 à 17 % (Carroll et al., 2005). Des études épidémiologiques ont démontré une corrélation positive entre le niveau de cholestérol total et le risque de troubles cardiovasculaires (Stamler et al., 1986; NCEP, 1993) (Figure 1). C'est également valable pour des hommes plus jeunes (âgés de 40 ans est moins) (Stamler et al., 2000).
s n JS *
1
3Î 2
sr i — ■ — i — ■ — i — ■ — i — i — i — i — iÎOO ISO 200 2SO 300 330 Niveau sanguin du cholestérol (mg/dL)
Figure 1 : Corrélation entre le niveau sanguin du cholestérol et le risque cardiovasculaire. Étude MRFIT, 361 662 hommes âgés de 35 à 57 ans (Stamler et al.,
1986(Stamleretal., 1986)).
Les classes majeures de dyslipidémie sont regroupées selon le phénotype de Frederickson (Fredrickson, 1971) (Tableau 2 et Tableau 1 pour la composition des lipoprotéines). Une variété de facteurs, dont certains sont familiaux, peuvent produire ce désordre lipidique.
Lipoprotéine Taille
(nm) L : P Composition Rôle métabolique Chylomicrons 100-1000 99: 1 4% VLDL IDL LDL HDL 30-70 25-35 15-25 7.5-10
Transport des triglycerides alimentaires.
Transport des triglycérides du foie vers les tissus extrahépatiques. Résidus du catabolisme des VLDL et stade préliminaire des LDL.
Transport du cholestérol du foie vers les tissus extrahépatiques.
50 : 50 35-41% Transport du cholestérol des tissus extrahépatiques vers le foie. 9 2 : 8 89: 11 79:21 23% 43% 58%
L : lipides; P : protéines; C : cholestérol; VLDL : lipoprotéines de très faible densité; IDL : lipoprotéines de densité intermédiaire; LDL : lipoprotéines de faible densité; HDL : lipoprotéines de haute densité
(Adapté de Lôffler, 1998 (Loffler, 1998); Biggerstaff et al., 2004 (Biggerstaff and Wooten, 2004))
Table 2 : Phénotypes de Frederickson Phénotype
de Frederickson
Lipoprotéines dont la concentration est
élevée dans le sang
Niveaux typiques de lipides I Chylomicrons Triglycérides > 99e centile
Ha LDL Cholestérol total > 90e centile; triglycérides et/ou apo-B peuvent être > 90e centile.
lib LDL et VLDL Cholestérol total et/ou triglycérides peuvent être > 90e centile; apo-B > 90e centile
III Résidus de VLDL et de chylomicrons
Cholestérol total et triglycérides > 90e centile
rv
VLDL Cholestérol total > 90e centile; il peut aussi y avoir triglycérides > 90e centile ou faible HDLV Chylomicrons
et VLDL
lipoprotéines de haute densité (HDL-C) sont plus particulièrement positivement corrélés avec les CVD (LaRosa et al., 1990). De nombreuses études cliniques et épidémiologiques, ainsi que des études animales et fondamentales, ont identifié le LDL-C comme la meilleure cible pour une intervention. Pour les patients qui n'ont pas d'autres facteurs de risque, quelques changements de mode de vie, tels qu'une diète faible en cholestérol et en lipides et une augmentation de l'activité physique par exemple, normalement suffisants pour diminuer le risque de CVD. Cependant, une thérapie médicamenteuse est recommandée pour les patients qui ont déjà souffert d'un événement cardiovasculaire, qui sont porteurs de plusieurs facteurs de risque ou pour lesquels les niveaux de LDL-C sont au-dessus de 190 mg/dl (NCEP, 2002).
Il existe plusieurs troubles dyslipidémiques dont l'hypercholestérolérnie et l'hypertriglycéridémie.
1.1. Hypercholestérolérnie
Par hypercholestérolérnie, on entend un taux élevé de cholestérol sanguin. Ce n'est pas une maladie en soi mais un marqueur métabolique qui est considéré comme un facteur de risque de maladies cardiovasculaires. L'hypercholestérolérnie est un des facteurs de risque modifiables majeurs pour les CVD, même chez les personnes de 65 ans et plus (Manolio et al., 1992).
Le Third Report of the Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III, ou ATP III) a récapitulé les recommandations actuelles pour la gestion du cholestérol élevé dans le sang (NCEP, 2002). Les directives de diverses organisations diffèrent dans leur applicabilité, l'estimation des risques et les recommandations (Broedl et al., 2003). Les directives d'ATP III sont basées sur des observations épidémiologiques qui ont démontré une corrélation positive entre le niveau de cholestérol total et le risque de troubles cardiovasculaires (Wallis et al., 2000). Ces directives tiennent compte de l'absence (la prévention primaire) ou de la présence (la
Des essais cliniques ont démontré que la baisse ciblée de cholestérol chez les patients avec hypercholestérolérnie réduit la morbidité due aux CVD. Une méta-analyse de 38 tests de préventions primaires et secondaires, par exemple, a constaté que pour chaque réduction de 10 % du cholestérol sérique, la mortalité due aux CVD serait réduite de 15 %, et le risque de mortalité total de 11 %. Cependant, aucune augmentation de la mortalité n'a été observée chez les sujets sans maladie cardiovasculaire (Gould et al., 1998).
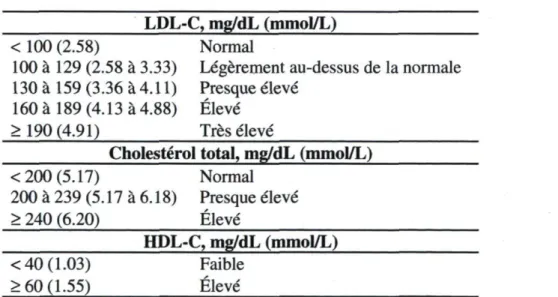
Les recommandations d'ATP III pour le traitement de l'hypercholestérolérnie sont basées sur les fractions de LDL-C et tiennent compte de la coexistence des troubles cardiovasculaires (Ballantyne et al., 2000; NCEP, 2002). Il y a cinq étapes majeures dans la détermination de la catégorie de risque d'un individu, qui servent de base pour les directives du traitement (NCEP, 2002).
Étape 1: L'obtention du profil lipidique. Les résultats sont classifies tel qu'indiqué dans le tableau ci-dessous (Tableau 3).
Table 3 : Classification d'ATP III pour LDL-C, cholestérol total et HDL-C LDL-C, mg/dL (mmol/L)
< 100 (2.58) Normal
100 à 129 (2.58 à 3.33) Légèrement au-dessus de la normale 130 à 159 (3.36 à 4.11) Presque élevé
160 à 189 (4.13 à 4.88) Élevé > 190 (4.91) Très élevé
Cholestérol total, mg/dL (mmol/L) < 200 (5.17) Normal 200 à 239 (5.17 à 6.18) Presque élevé > 240 (6.20) Élevé HDL-C, mg/dL (mmol/L) < 40 (1.03) Faible > 60 (1.55) Élevé
placent le patient dans des conditions semblables à celles qui provoquent des maladies cardiovasculaires : le diabète, les maladies symptomatiques de l'artère carotide, la maladie artérielle périphérique, l'anévrisme aortique abdominal et les facteurs de risque multiples qui confèrent une probabilité de plus de 20 % de développer les CVD dans les dix prochaines années (voir l'étape 4 ci-dessous).
Étape 3 : L'identification des facteurs majeurs des CVD autres que ceux liés aux LDL : le tabagisme, l'hypertension (pression artérielle > 140/90 ou médication antihypertensive), le HDL-C bas (< 40 mg/dL [1.03 mmol/L]), l'histoire familiale de CVD prématuré et l'âge (hommes 45 ans, femmes 55 ans). Il est à noter qu'une concentration de HDL-C de 60 mg/dL (1.55 mmol/L) est un facteur de risque "négatif, c'est-à-dire que sa présence enlève un facteur de risque au compte total.
Étape 4 : Si deux facteurs ou plus ont été identifiés à l'étape 3 chez les patients sans CVD ou avec leurs équivalents (voir l'étape 2), le risque de développer une CVD dans les dix prochaines années est évalué en utilisant les modifications de l'ATP III des tables de risque de Framingham (voir http://www.nhlbi.nih.gov/.). Une étude de validation a constaté que la prédiction de Framingham pour les CVD est valable chez les hommes et femmes blancs ainsi que chez les hommes et femmes noirs. Cependant, cette prédiction surestime le risque chez les hommes américains de descendance japonaise et hispaniques et chez les femmes amérindiennes (D'Agostino et al., 2001). Également, plusieurs études ont suggéré que les critères de Framingham surestiment aussi le risque dans les populations européennes et asiatiques (Bastuji-Garin et al., 2002; Brindle et al., 2003; Liu et al., 2004). Il n'est pas clair si ces différences sont réelles ou si elles sont dues aux divergences dans les méthodes de recherche et/ou les procédures de jugement.
Étape 5 : L'évaluation de la catégorie de risque du patient. Cela permet de décider quand il faut amorcer des changements de style de vie et quand il faut considérer des traitements médicamenteux (voir les tables au http://www.nhlbi.nih.gov/.).
Les directives d'ATP IE considèrent les risques absolus et les niveaux de LDL-C pour évaluer l'admissibilité aux thérapies hypolipémiantes. Son impact a été examiné sur la population ayant droit au traitement en analysant des données de 13 589 sujets dans le cadre de l'étude NHANES HI (Fedder et al., 2002). En général, l'admissibilité pour le traitement a augmenté de 157 et 122 % pour les hommes et les femmes, respectivement, et de 131 et 201 % pour les 65 ans et plus et 45 ans et moins, respectivement (tout sexe confondu). La perturbation dans le métabolisme des lipoprotéines est souvent familiale. Dans une étude, par exemple, 54 % de tous les patients avec une CVD prématurée (et 70 % avec une anomalie lipidique) avaient des antécédents familiaux (Genest et al., 1992). Un de ces désordres est l'hypercholestérolérnie familiale.
1.1.1. Hypercholestérolérnie familiale
L'hypercholestérolérnie familiale est un désordre monogénique autosomal causé par des défauts dans le gène qui code pour le récepteur d'apo B/E (LDL) (Hobbs et al., 1990; Ishibashi et al., 1993; Grossman et al., 1995; Austin et al., 2004). Le dommage dans la fonction de ces récepteurs aboutit à une clairance réduite des particules LDL de la circulation et une élévation du LDL-C plasmatique. Elle est aussi caractérisée par l'assimilation accrue de LDL par les MSR (macrophage scavenger receptor), aboutissant à la production augmentée de LDL oxydés qui semblent avoir une plus forte activité athérogénique (Steinberg et al., 1989). On peut voir une présentation clinique semblable à l'hypercholestérolérnie familiale sous la forme d'une maladie autosomale récessive rare dans laquelle le défaut semble impliquer une protéine adaptatrice pour le récepteur des LDL et des mutations dans l'apo B, un ligand pour le récepteur des LDL (Garcia et al., 2001).
L'hypercholestérolérnie familiale est caractérisée par un effet de dosage du gène, où les homozygotes sont plus défavorisés que les hétérozygotes. Par exemple, dans un rapport, la clairance de LDL-C était réduite de 27 % chez les hétérozygotes et de 53 % chez les homozygotes. L'excès de LDL-C peut se déposer dans les artères pour former un athérome ou dans la peau et les tendons et causer les xanthomatose et xanthelasma. La fréquence de
al., 1993).
1.2. Hypertriglycéridémie
L'hypertriglycéridémie désigne l'élévation du taux de triglycérides dans le sang. La concentration de triglycérides dans le sérum peut être stratifiée comme suit (Tableau 4) (NCEP, 2002) :
Table 4 : Classification des triglycérides LDL-C, mg/dL (mmol/L)
< 150 (1.7) Normal 150 à 199 (1.7 à 2.2) Presque élevé 200 à 499 (2.2 à 5.6) Élevé > 500 (5.6) Très élevé
(Adapté de Adult Treatement Panel III au http://www.nhlbi.nih.gov/.)
La lipoprotéine lipase (LPL) hydrolyse les triglycérides contenus dans les chylomicrons et les VLDL. Également, elle facilite le transfert du cholestérol de ces lipoprotéines aux HDL (Brewer, 2004). L'hydrolyse des triglycérides libère les acides gras qui sont alors utilisés comme source d'énergie ; ils peuvent être reconvertis en triglycérides ou stockés dans les tissu adipeux. Les mutations dans le gène LPL affectent variablement l'activité de l'enzyme et donc la dégradation des chylomicrons et des VLDL qui transportent les triglycérides endogènes. Les mutations en N-terminal (ex : Glyl88Glu, Asp9Asn et Asn291Ser) réduisent l'activité de la LPL et sont associées à une augmentation des triglycérides dans le sang et une diminution du HDL-C (Sprecher et al., 1996; Wittrup et al., 1999). Au contraire, la mutation en C-terminal (Ser447Ter) peut augmenter l'affinité de liaison des LDL, ce qui se traduit par une augmentation de l'activité de l'enzyme de même que par une légère diminution des triglycérides sanguins (Wittrup et al., 1999).
L'hypertriglycéridémie est associée à un risque accru de maladies cardiovasculaires. La force de cette relation a été complètement confirmée dans un rapport en 2007 avec deux
larges études prospectives impliquant plus de 3 500 cas de CVD et 44 000 hommes et femmes d'âge moyen, en plus d'une méta-analyse de 27 études prospectives populationnelles englobant un total de plus de 10 000 cas de CVD et 260 000 participants. Cette analyse a rapporté que le risque de CVD attribuable à l'hypertriglycéridémie était équivalent chez les femmes et les hommes (Sarwar et al., 2007). Cependant, par d'autres études, l'hypertriglycéridémie a été soulignée comme étant la plus prédictive quant au risque de CVD chez les personnes plus jeunes et chez les femmes (Hokanson and Austin, 1996; Nordestgaard et al., 2007). Dans les deux cas, l'hypertriglycéridémie est liée à un risque augmenté d'événements cardiovasculaires.
L'hypertriglycéridémie est aussi associée à une mortalité accrue chez les patients avec des maladies cardiovasculaires connues et peut aussi réduire la survie après la chirurgie de greffe de contournement de l'artère coronaire (Haim et al., 1999; Sprecher et al., 2000). Dans l'étude Bezafîbrate Infarction Prevention (BIP), chez les patients avec les CVD, on a observé une augmentation de la mortalité ainsi que des niveaux de triglycérides dans le sang. Le risque le plus élevé a été noté principalement dans les sous-groupes de patients avec un cholestérol total et un LDL-C élevés et chez les femmes avec des niveaux de HDL-C supérieurs à 45 mg/dL (1.16 mmol/L) (Haim et al., 1999).
2. Traitement médical de la dyslipidémie
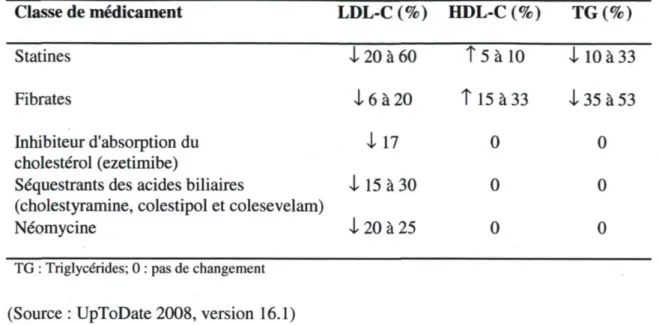
Les hypolipémiants englobent plusieurs classes des médicaments qui incluent les statines, les fibrates, l'ezetimibe, les séquestrants des acides biliaires et la néomycine. Ces médicaments diffèrent quant à leur mécanisme d'action et aux degré et type de modifications du profil lipidique. Ils sont bénéfiques chez les personnes dyslipidémiques pour la prévention primaire, mais aussi pour la prévention secondaire des maladies cardiaques. Voici un résumé de leurs effets (Tableau 5):
i 20 à 60
î
5à 10 4 10 à 33 4 6 à 20î
15 à 33 i 3 5 à 5 31 17 0 0
i 15 à 30 0 0
i 20 à 25 0 0
Table 5 : Effets des différents hypolipémiants sur les lipides dans le sang
Classe de médicament LDL-C (%) HDL-C (%) TG(%) Statines
Fibrates
Inhibiteur d'absorption du cholestérol (ezetimibe)
Séquestrants des acides biliaires
(cholestyramine, colestipol et colesevelam) Néomycine
TG : Triglycérides; 0 : pas de changement
(Source : UpToDate 2008, version 16.1)
Les statines et les fibrates sont les plus importants agents hypolipémiants (Wierzbicki, 2004). Les statines sont des inhibiteurs sélectifs de la synthèse du cholestérol (Rosenson, 2004b). La lovastatine était le premier représentant des statines en 1987. Par contre, les fibrates constituaient la première classe de drogues approuvées pour le traitement de dyslipoprotéinémie. Le clofibrate était leur premier représentant en 1967 aux États-Unis (Fazio and Linton, 2004). En activant le récepteur nucléaire PPARa (Peroxisome Proliferator Activated Receptor de type alpha) les fibrates modulent l'expression d'un grand nombre de gènes cibles du métabolisme des triglycérides et du cholestérol (Chapman, 2003). Depuis, plusieurs analogues des deux classes de médicaments ont été conçus et leur efficacité a été confirmée par plusieurs études cliniques dans la prévention primaire et secondaire des maladies cardiaques (Tableau 6).
D'autre part, rezetimibe est une drogue qui inhibe l'absorption du cholestérol alimentaire (van Heek et al., 2001). Comme le cholestérol exogène contribue pour 25 à 50 % des niveaux sanguins de cholestérol, le traitement avec ce dernier seul ou en combinaison avec des statines ou des fibrates peut significativement aider à la thérapie hypocholestérolémiante (Wierzbicki, 2004). Quant aux séquestrants des acides biliaires et la néomycine, ils sont principalement employés pour abaisser la réabsorption des acides
biliaires et ainsi réduire les taux de cholestérol total. Cependant, l'administration de séquestrants des acides biliaires est souvent limitée par les effets indésirables, tels que la constipation, les nausées et la diarrhée (Davidson and Toth, 2004; Rosenson, 2005).
Les médicaments hypolipémiants constituent la catégorie thérapeutique dont la croissance est la plus rapide au Canada, avec une augmentation de 15,5% en 2004. Par exemple, l'atorvastatine (LipitrV40) est le médicament qui a été le plus prescrit au Canada en 2004 (EMS, 2005). De plus, différentes études ont démontré une corrélation positive entre une réduction des niveaux de cholestérol et la survenue d'événements cardiovasculaires (Wierzbicki, 2004), ce qui accentue la valeur de l'intervention médicamenteuse au niveau de la modulation des lipoprotéines. Aussi, on a révélé des effets désirables non lipidiques ou effets pléiotropes liés à ces médicaments. Ces faits soulignent une grande importance pharmacologique des drogues hypolipémiantes et d'autres recherches sont nécessaires afin de mieux comprendre tant leurs effets bénéfiques que néfastes.
Table 6 : Etudes cliniques démontrant les effets bénéfiques des statines et des fibrates
Drogue Patients Prév. Référence Etudes sur les statines
Scandinavian Simvastatin Survival Study (4S)
Anglo-Scandinavian Cardiac Outcomes Trial-Lipid
Lowering Arm (ASCOT-LLA) Air Force/Texas Coronary Atherosclerosis Prevention study (AFCAPS/TexCAPS) Cholesterol and Recurrent Events (CARE) Simvastatine 4 444 2° Atorvastatine 10 305 Ie Lovastatine 6 605 Ie Pravastatine 4 159 Pedersen et al., 2004 Sever et al., 2003 Gotto et al., 2000 Plehn et al., 1999 Etudes sur les fibrates
Diabetes Atherosclerosis Intervention Study (DAIS) Veterans Affairs High-Density Lipoprotein Cholesterol Inter-vention Trial Study (VA-HIT)
Fénofibrate 731
Gemfibrozil 2531
St. Mary's, Ealing, Northwick
Park Diabetes Cardiovascular Bezafibrate
Disease Prevention(SENDCAP) 164
Stockholm Ischaemic Heart Disease Secondary Prevention
Study Clofibrate 555 2° Steiner, 2001 2° Rubins et al., 1999 2° Elkeles etal., 1998 2° Carlson et al., 1988 Prév : prévention 1°: primaire 2°: secondaire
2.1. Statines
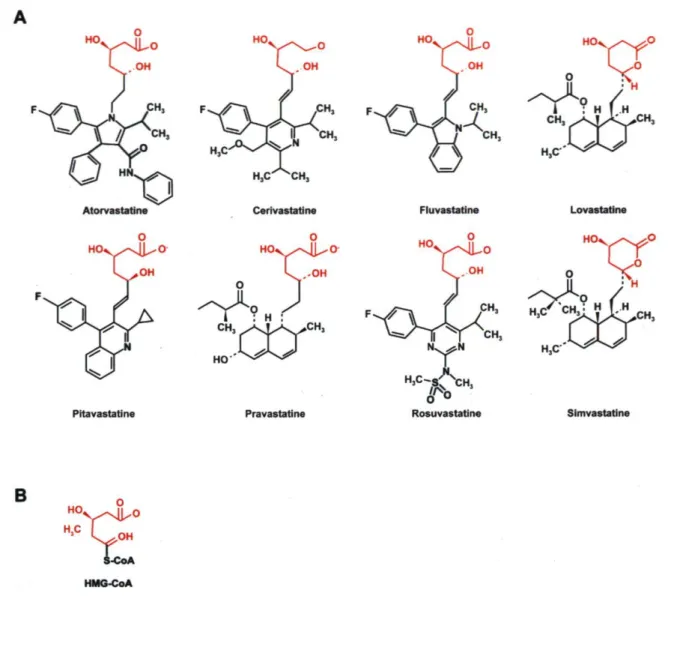
Les statines actuellement disponibles sur le marché sont les lovastatine, pravastatine, simvastatine, fluvastatine, atorvastatine et rosuvastatine. Ces agents sont des inhibiteurs compétitifs de l'enzyme limitante de la synthèse hépatique du cholestérol, la 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase. Les statines occupent une partie du site de liaison du HMG-CoA, bloquant ainsi l'accès de son substrat au site actif de l'enzyme (Istvan and Deisenhofer, 2001). En diminuant la synthèse endogène de cette molécule, les statines provoquent une augmentation de l'expression hépatique des récepteurs aux LDL, résultant en une clairance élevée du LDL-C du plasma (Ness et al., 1996). De plus, l'atorvastatine réduit la production de VLDL, via un effet médié par la sécrétion hépatique de l'apo B (Conde et al., 1996), et le traitment médicamenteux est associé à une réduction de l'activité de l'enzyme HMG-CoA reductase (Ness et al., 1998). Étant d'origine synthétique, les statines possèdent un élément de structure commun qui ressemble à l'HMG-CoA mais qui leur procure une affinité trois fois plus élevée que son substrat naturel (Williams and Feely, 2002) (Figure 2).
Pitavastatine Pravastatine Rosuvastatine Simvastatine
B
HO
V^-°
S-CoA HMG-CoA
Figure 2: Structure des statines (A) et structure de l'HMG-CoA (B). Les statines possèdent un élément de structure commun qui ressemble à l'HMG-CoA.
_ J S 1
I
Q. ro5
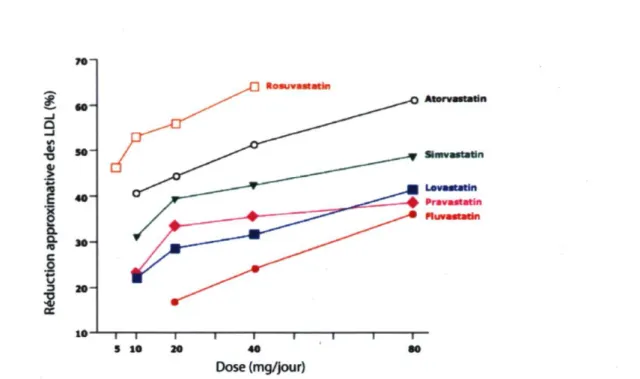
CC l » - l 5 0 " 3 0 1 0 -10 A t o r v a s t a t i n T 1 -3 10 —r-20 Dose (mg/jour)Figure 3: Comparaison de l'efficacité des statines. La résuvastatine est la statine la plus efficace pour réduire le LDL-C.
Effets sur les LDL : Les statines sont les médicaments les plus puissantes pour réduire le LDL-C, résultant en des diminutions de 30 à 63 % (Levy et al., 1993; Illingworth et al., 1994; Larsen and Illingworth, 1994; Jones et al., 1998; Rosenson, 2003) (Figure 3). Dans une cohorte de patients avec athérosclérose documentée et un niveau de LDL-C > 130 mg/dL (3.4 mmol/L), le pourcentage de patients atteignant l'objectif de réduire le niveau de LDL-C sous 100 mg/dL (2.6 mmol/L) était beaucoup plus haut avec l'atorvastatine qu'avec la fluvastatine, la lovastatine ou la simvastatine (32 % contre 1, 10 et 22 %, respectivement) (Brown et al., 1998). Cependant, la rosuvastatine semble être encore plus puissante que l'atorvastatine (Jones et al., 2003; Rosenson, 2003). Dans plusieurs études, la rosuvastatine (20 à 40 mg/jour) a réduit le LDL-C de plus de 63 %. Par exemple, les effets de plusieurs statines, dont la rosuvastatine (10 à 40 mg/jour), l'atorvastatine (10 à 80 mg/jour), la simvastatine (10 à 80 mg/jour) et la pravastatine (10 à 40 mg/jour) ont été comparés chez 2 431 adultes avec l'hypercholestérolérnie (LDL-C entre 160 et 240 mg/dL) (Jones et al., 2003). Après six semaines de traitement, plus de patients prenant la rosuvastatine ont réussi à atteindre le niveau cible de LDL-C (< 100 mg/dL [2.6 mmol/L]); 82, 89 et 89 % de patients ont atteint le niveau cible de LDL-C avec 10, 20 et 40 mg de rosuvastatine respectivement, tandis que les pourcentages les plus hauts obtenus avec
1 "atorvastatine et la simvastatine étaient de 85 et 82 %, respectivement. La rosuvastatine a été approuvée par la Food and Drug Administration aux États-Unis à une de dose de 5 à 40 mg/jour (www.fda.gov); on a toutefois observé, quoique rarement, une insuffisance rénale chez les patients traités avec 80 mg/jour de rosuvastatine (Jones et al., 2003). En débutant le traitement, la dose maximale recommandée de rosuvastatine est de 20 mg/jour pour des patients avec des niveaux de LDL-C au-dessus de 190 mg/dL (4.9 mmol/L). En 2005, il a été conseillé de recommander une dose de départ de 5 mg/jour pour les patients exigeant une baisse de LDL-C moins agressive et pour ceux présentant des facteurs de prédisposition pour la myopathie, incluant les patients asiatiques, les patients prenant de la cyclosporine et les patients souffrant d'insuffisance rénale sévère (www.fda.gov/cder/drug/InfoSheets/ HCP/RosuvastatinHCP.pdf). Si un patient est tolérant aux doses élevées de rosuvastatine et a besoin d'un traitement lipidique plus efficace, la dose peut être augmentée jusquà un maximum de 40 mg/jour. Cependant, cette dose serait prescrite seulement aux patients qui en ont vraiment besoin. Jusqu'à 40 mg/jour, la fluvastatine est la statine la moins puissante (voir figure 3 ci-haut). Par contre, à 80 mg/jour, la fluvastatine est aussi efficace pour diminuer le niveau de LDL-C que la plupart des statines autres que la rosuvastatine et l'atorvastatine (Eidelman et al., 2002). La fluvastatine présente moins d'interactions médicamenteuses et de toxicité musculaire que certaines autres statines. Il y a un effet hypolipémiant additif lorsque n'importe laquelle des statines est utilisée en combinaison avec un séquestrant d'acides biliaires (Brown et al., 1990; Pan et al., 1990; Sprecher et al., 1994).
Effets sur les HDL : La simvastatine (40 à 80 mg/jour) semble être plus efficace que l'atorvastatine (20 à 40 mg/jour) pour augmenter le niveau de HDL-C dans le sang et les concentrations d'apolipoprotéine A-I (Kastelein et al., 2000). Cependant, c'est encore la rosuvastatine qui est la plus efficace, augmentant le niveau de HDL-C de plus de 10 % (Jones et al., 2003).
Effet sur les triglycérides : L'atorvastatine et la rosuvastatine sont les statines les plus efficaces pour baisser le niveau de triglycérides (14 à 33 %) chez les patients avec hypercholestérolérnie (Bakker-Arkema et al., 1996; Dart et al., 1997; Davidson et al., 1997; Jones et al., 2003). Ces données ont été confirmées dans l'étude mentionnée précédemment
chez 2 431 adultes avec hypercholestérolérnie (Jones et al., 2003). Après six semaines de traitement, ceux prenant l'atorvastatine et la rosuvastatine avaient une réduction plus forte des triglycérides que ceux prenant la simvastatine et la pravastatine; par exemple, pour la dose 40 mg, les réductions étaient de 26.8, 26.1, 14.8 et 13.2 %, respectivement. Des résultats semblables ont été observés quand l'atorvastatine a été comparée avec la lovastatine (Davidson et al., 1997). Les effets de l'atorvastatine et de la rosuvastatine sur le niveau des triglycérides dans le sang sont dépendants de la dose (Bakker-Arkema et al., 1996; Jones et al., 2003). Par exemple, dans une série de 56 patients avec hypertriglycéridémie dont la concentration de triglycérides moyenne était de 600 mg/dL et la concentration de LDL-C était de 120 mg/dL, l'administration d'atorvastatine aux doses de 5, 20, ou 80 mg/jour a permis de réduire le niveau de triglycérides de 27, 32 et 46 %, respectivement et le niveau de LDL-C de 17, 33 et 41 %, respectivement (Bakker-Arkema et al., 1996).
2.1.1. Métabolisme des statines
Les statines, surtout celles hydrophobes, sont métabolisées principalement via l'oxydation par des cytochromes P450 (CYP), en particulier par CYP3A4 pour la majorité des statines et par CYP2C9 pour la fluvastatine et la pitavastatine; 50 % des médicaments disponibles sont également métabolisés par cette voie (Corsini et al., 1999). Il a été démontré que la prise simultanée de deux drogues métabolisés par CYP3A4 entraînait leur compétition pour la même voie, résultant ainsi en un niveau élevé des deux médicaments (Davidson and Toth, 2004). Pour éviter des effets néfastes, une des deux drogues co-administrées peut être remplacée par un autre agent ayant le même effet, mais qui n'est pas métabolisé par CYP3A4. Également, plusieurs études ont rapporté la glucuronidation comme voie catabolique des statines, surtout par les enzymes UGT1A1 et 1A3 (Prueksaritanont et al., 2002a; Fujino et al., 2003). Le métabolisme des statines produit des dérivés plus hydrophiles, permettant leur élimination biliaire, de même que rénale, quoiqu'en moindre proportion (Davidson and Toth, 2004).
La variabilité dans la réponse et les effets secondaires des hypolipémiants peut être expliquée par une combinaison de facteurs, dont les variations dans les gènes impliqués dans le métabolisme des médicaments. Par exemple, CYP2D6, qui est un membre de la
famille des cytochromes P450, est fonctionnellement absent chez 7 % des Caucasiens et des Afro-américains, tandis que cette déficience est plutôt rare chez les Asiatiques. Le phénotype de CYP2D6 semble être important chez les patients traités avec la simvastatine, car il peut affecter l'efficacité et la tolérance au traitement (Mulder et al., 2001). Également, les polymorphismes dans le gène codant pour l'HMG-CoA reductase semblent aussi affecter la réponse aux statines. Dans une étude sur la pravastatine (40 mg/jour), les individus portant deux variants génétiques communs (la fréquence des hétérozygotes est de 6.7 %) fortement liés avaient une réduction plus faible du cholestérol total ainsi que du LDL-C en comparaison avec les individus homozygotes non variants (-32.8 contre -42.0 mg/dL 0.85 contre-1.09 mmol/L] pour le cholestérol total et -27.7 contre -34.1 mg/dL [-0.72 contre-0.88 mmol/L] pour le LDL-C) (Chasman et al., 2004). Ce polymorphisme n'avait aucun effet sur le changement du niveau de HDL-C suite au traitement. Il a été souligné que des doses plus faibles de statines peuvent être plus efficaces chez les Asiatiques que chez les Caucasiens (Liao, 2007). Les informations pour la prescription de la rosuvastatine recommandent une dose initiale inférieure pour les Asiatiques à celle suggérée pour les autres groupes ethniques, étant donné des différences observées dans leurs pharmacocinétiques (www.astrazeneca-us.com/pi/crestor.pdf). Il n'y a toutefois aucune recommandation de cet ordre pour les autres statines.
2.1.2. Effets indésirables et pléitropes des statines
Des effets indésirables arrivent moins fréquemment avec les statines qu'avec les autres classes d'agents hypolipémiants. Bien que les effets indésirables de la monothérapie sont rares, leur incidence augmente si certains médicaments sont co-administrés (Williams and Feely, 2002) et on doit en tenir compte, car un tiers des prescriptions des statines est en combinaison avec des médicaments avec lesquelles elles peuvent interagir (Bellosta et al., 2004). Le dysfonctionnement hépatique a été rapporté, cependant le risque réel semble être très faible. La myopathie reste un effet secondaire important, bien que relativement rare. La simvastatine est la statine causant le plus fréquemment la rhabdomyolyse (Omar and Wilson, 2002). Il a été observé que les statines lipophiles (simvastatine, lovastatine, atorvastatine et fluvastatine) sont davantage associées aux événements défavorables en comparaison avec les statines hydrophiles (pravastatine et rosuvastatine) (Brewer, 2003;
Rosenson, 2004a). Ceci peut s'expliquer par le fait que les substances hydrophiles peuvent être éliminées directement, sans nécessiter leur métabolisme via les gènes spécifiques. Dans certaines études, les statines semblent causer seulement une légère augmentation des effets secondaires en comparaison avec le placebo (Kashani et al., 2006; Armitage, 2007). Cependant, l'expérience dans la pratique clinique suggère que les effets secondaires musculaires sont relativement communs, y compris les effets secondaires exigeant l'arrêt de la thérapie aux statines. L'explication de cette différence est incertaine, mais peut être liée aux critères de sélection des études cliniques, qui limitent la capacité de généraliser les résultats pour établir les profils des effets secondaires, ce qui a été observé dans une population plus large de patients.
D'autre part, les statines ont montré de nombreux effets pléitropes. En effet, des rapports attribuent des propriétés anti-inflammatoires aux statines, qui réduiraient l'adhésion des monocytes à l'endothélium (Weber et al., 1997), en produisant des cytokines (Ferro et al., 2000), en exprimant le MHC II inductible qui mène à la répression de l'activité des lymphocytes T (Kwak et al., 2000) et en empêchant l'activation des voies inflammatoires importantes impliquant NF-KB (Ortego et al., 1999) et STAT-1 (Cignarella et al., 2003).
D'autres études ont remarqué des effets thrombotiques directs (en plus des effets anti-inflammatoires), réduisant l'hyperactivité des plaquettes, la formation de la thromoxane et de la fibrine (Schieffer and Drexler, 2003). De plus, il existe des évidences que les statines pourraient abaisser la pression artérielle (Sposito et al., 1999; Tonolo et al., 2000). Finalement, une découverte surprenante de l'étude WOSCOPS (West of Scotland Coronary Prevention Study), où les hommes avec hypercholestérolérnie étaient traités avec la pravastatine en prévention primaire, a révélé une réduction de 30 % du risque de développer le diabète, défini comme un niveau de glucose sanguin > 126 mg/dL (7 mmol/L) (Freeman et al., 2001). Les mécanismes ne sont toutefois pas encore connus. Puisque la cerivastatine n'est plus disponible sur le marché à cause des risques potentiels de sécurité tels que la myopathie et la rhabdomyolyse (Chang et al., 2004), la rosuvastatine est de toute évidence considérée comme la drogue la plus efficace pour le traitement de dyslipidémie (Schachter, 2005).
2.2. Fibrates
Trois fibrates sont actuellement disponibles aux États-Unis soit: gemfibrozil, fénofibrate et clofibrate. Cependant, ce dernier n'est plus utilisé puisqu'il a été associé à la chondrocalcinose et à des cancers gastro-intestinaux (WHO, 1984). D'autres fibrates sont disponibles, tels que le bézafibrate et le ciprofibrate (Figure 4).
o irv-°v/
c o>
Ht J
H—
*ijy°x
c
°" ,jry°xr
Bézafibrate Cipofibrate Clofibrate
0
f Y ° ^ ^ ^ C 0
2H
Fénofibrate Gemfibrozil
Figure 4 : Structure des fibrates. Les fibrates les plus utilisés sont le gemfibrozil et le fénofibrate.
Les effets majeurs des fibrates sont la réduction des triglycérides sanguins (de 35 à 50 %) et l'augmentation du HDL-C (de 15 à 25 %) (Fruchart et al., 1998; Staels et al., 1998). Ces effets sont obtenus par l'activation sélective du récepteur nucléaire PPARa (Vu-Dac et al., 1995; Staels et al., 1998), qui est surtout exprimé dans les tissus catabolisant des acides gras (Barbier et al., 2002).
Deux facteurs contribuent à la diminution des triglycérides suite au traitement avec les fibrates (Staels et al., 1998). Tout d'abord, la sécrétion hépatique de VLDL est réduite. Aussi, l'élimination des triglycérides est permise par une augmentation de l'expression de la lipoprotéine lipase (LPL), qui peut être expliquée en partie par la diminution de l'expression d'apoC-III, une apolipoprotéine qui inhibe la lipoprotéine lipase, favorisant ainsi la libération des acides gras. De plus, la réduction de la concentration d'apoC-III dans les VLDL favorise le catabolisme de ces lipoprotéines (Chapman, 2003).
Trois mécanismes contribuent à l'augmentation du HDL-C après le traitement aux fibrates (Staels et al., 1998). Dans un premier lieu, il y a une stimulation de la synthèse des apolipoprotéines A-I et A-Il (Vu-Dac et al., 1995; Berthou et al., 1996; Jin et al., 1996). L'effet direct sur l'apo A-I résulte en la stabilisation des transcrits de l'ARNm, menant à la traduction et la sécrétion plus forte d'apo A-I contenant les particules d'HDL (Berthou et al., 1996; Jin et al., 1996). Ainsi, cet effet, de même que l'induction des transporteurs transmembranaires, tel que ABCA1, favorisent le transport inverse du cholestérol et son élimination dans la bile (Barbier et al., 2002). Des effets semblables ont été démontrés pour l'ARNm d'apo A-II et pour sa sécrétion (Vu-Dac et al., 1995). Ensuite, il y a un transfert accru d'apo A-I et d'autres composants de surface, ce qui diminue le transfert de HDL en VLDL. Finalement, il y a moins d'inhibition par les VLDL (en raison de la réduction de leur concentration) sur la synthèse hépatique d'apo A-I.
À la différence des statines, qui manifestent leur efficacité clinique à travers une large gamme de niveaux de LDL-C, les fibrates ont principalement montré leur efficacité à réduire le nombre d'événements cardiovasculaires chez les patients avec un niveau élevé de triglycérides (au-dessus de 200 mg/dL [2.2 mmol/L]) (Frick et al., 1987; BIPstudy, 2000) ou avec un HDL-C bas (au-dessous de 40 mg/dL [1.0 mmol/L]). Également, les fibrates peuvent diminuer plusieurs caractéristiques du syndrome métabolique (Rubins et al., 1999).
2.2.1. Interactions médicamenteuses des fibrates
Les interactions médicamenteuses entre les fibrates et les statines ont souvent été rapportées et ont une importance prédominante, sachant que leur co-administration est fréquente lors du traitement de la dyslipidémie. Les fibrates ont été associés à la toxicité musculaire (Magarian et al., 1991), un effet qui est plus prononcé lorsque ces derniers sont administrés en combinaison avec les statines (Pierce et al., 1990; Miller and Spence, 1998). Cet effet peut être empêché par l'inhibition compétitive de CYP3A4, menant à une réduction du métabolisme des statines. La pravastatine et la fluvastatine n'étant pas majoritairement métabolisées par le CYP3A4, elles seraient de meilleurs candidats lorsque la combinaison d'une statine avec un fibrate est nécessaire. Cependant, cette observation est incertaine (Athyros et al., 1997). La glucuronidation, qui est un sentier métabolique important pour
l'excrétion rénale des statines lipophiles, semble être significativement inhibée par le gemfibrozil, mais pas par le fénofibrate (Ballantyne and Davidson, 2003). Dans des études cliniques, le niveau de statines dans le sang augmente de 1.9 à 5.7 fois lorsque les sujets sont traités avec le gemfibrozil, alors qu'il reste inchangé avec le fénofibrate. Ceci pourrait s'expliquer par le fait que les enzymes UGT1 Al et 1A3 sont inhibées par la compétition du gemfibrozil, alors que le fénofibrate est le substrat d'autres enzymes UGT. Ainsi, le fénofibrate a été démontré comme n'ayant aucun effet inhibiteur in vitro (Prueksaritanont et al., 2002b). De plus, dans les études avec le fénofibrate, l'incidence de la myopathie est faible, que le patient prenne aussi une statine ou non (Keech et al., 2005). Ainsi, le fénofibrate est le fibrate de choix pour les patients qui nécessitent une thérapie combinée avec une statine (Rosenson, 2004a).
Les fibrates interfèrent avec le métabolisme de la warfarine, un anti-coagulant (Miller and Spence, 1998). Une interaction médicamenteuse est considérée comme cliniquement importante si elle est présente entre deux médicaments qui sont co-administrées et si l'effet résultant amène à un ajustement de la dose ou à une autre intervention clinique (Williams and Feely, 2002). Ainsi, étant donné que cette interaction est cliniquement importante, la dose de warfarine doit être réduite de 30% chez des patients traités avec un fibrate et le warfarin.
2.2.2. Fénofibrate
Le fénofibrate peut être prescrit de deux manières différentes : en formulation nanocristalline (145 mg pris quotidiennement sans obligation de prendre aux repas) ou en capsules micronisées (200 mg pris quotidiennement mais avec les repas) (Adkins and Faulds, 1997). Dans les deux cas, une réduction de dose est nécessaire chez les patients atteints de maladies rénales. Le fénofibrate est surtout approuvé pour la baisse de triglycérides chez les sujets avec une hyperlipoprotéinémie de type IV et V (Tableau 2). Dans une étude chez 84 patients avec hyperlipidémie combinée où le fénofibrate ou l'atorvastatine (10 mg/jour) ont été administrés aléatoirement, le fénofibrate s'est avéré plus efficace dans la réduction des triglycérides et du VLDL-C sanguins, tandis que
l'atorvastatine était plus efficace dans la diminution du LDL-C et du cholestérol total (Ooi et al., 1997).
Dans la récente étude FIELD, 9 795 patients atteints de diabète de type II ont été traités avec le fénofibrate (200 mg par jour) et aucun résultat significatif quant aux événements cardiovasculaires n'a été constaté (Keech et al., 2005). Cependant, dans une plus petite étude (731 patients avec le diabète de type H), une diminution de la progression angiographique des maladies cardiovasculaires a été observée avec la prise de fénofibrate (200 mg/jour) (DAISstudy, 2001).
Une interaction médicamenteuse importante du fénofibrate est son augmentation de la clairance de la cyclosporine. Par exemple, chez 43 individus ayant subi une transplantation cardiaque, le fénofibrate a mené à une réduction de 30 % des niveaux de cyclosporine (Boissonnat et al., 1994). De plus, cinq de ces patients ont eu un rejet aigu associé à la diminution des niveaux de cyclosporine.
Jusqu'à présent, très peu d'investigations ont porté sur l'étude de la voie principale du métabolisme du fénofibrate. Prueksaritanont et ses collaborateurs ont démontré que les enzymes UGT 1 Al, UGT 1 A3, UGT1A9 et UGT2B7 présentent la plus grande réactivité pour la conjugaison du fénofibrate (Prueksaritanont et al., 2002b). Toutefois, peu d'enzymes UGT avaient été incluses dans l'étude.
Le métabolisme du fénofibrate est complexe même s'il n'inclut pas les enzymes CYP. En effet, plusieurs enzymes UGT le métabolisent, ce qui peut engendrer une grande variété d'interactions médicamenteuses possibles. Donc, les effets réels chez un individu restent toujours difficiles à prévoir et dépendent de facteurs génétiques et environnementaux, des habitudes de vie ainsi que de l'état de santé.
3. UDP-Glucuronosyltransférases (UGT)
Le processus de detoxification chez l'homme se divise en trois phases: la phase I, ou réaction de fonctionnalisation, la phase II, ou réaction de conjugaison, et la phase III qui correspond à l'élimination. La phase I implique l'ajout d'un groupement fonctionnel réactif sur un composé hydrophobe (Friedberg, 1998). Les cytochromes P450 (CYP450) sont les enzymes principalement impliquées dans la catalyse de cette réaction. La molécule produite, par une réaction d'oxydation, de réduction ou d'hydrolyse, possède un groupement polaire de type hydroxy (OH), amino (NH2), thiol (SH) ou carboxy (COOH). Les produits résultants de la phase I sont généralement moins liposolubles et donc plus accessibles pour les enzymes de conjugaison (enzymes de phase II) telles que les UGT (Dutton, 1980). En plus de ces dernières, la phase II regroupe diverses classes d'enzymes de conjugaison telles que les sulfotransférases, les glutathiones-S-transférases, les méthyltransférases et les arylamines N-acétyltransférases (Figure 5) (Evans and Relling,
1999). Ces enzymes conjuguent divers groupements polaires sur des produits de la phase précédente, ce qui augmente la polarité des molécules. Elles deviendront donc encore plus polaires qu'après la phase de fonctionnalisation et seront plus facilement excrétées via la bile et l'urine (Liston et al., 2001; Guillemette, 2003). Finalement, la phase III correspond à l'élimination des produits formés par les phases I et IL
Chez l'homme, les UGT sont impliquées dans le processus de detoxification de substances endogènes, tels que les hormones stéroïdiennes (corticostéroïdes, minéralocorticoïdes, progestatifs, androgènes et oestrogènes) et thyroïdiennes, les acides biliaires, les acides gras de même que la bilirubine (Liu et al., 1995; Bélanger et al., 1998; Hum et al., 1999; Jude et al., 2001; Lépine et al., 2004) et de substances exogènes, comme les médicaments, les vitamines liposolubles, les polluants environnementaux ainsi que plusieurs carcinogènes retrouvés, entre autres, dans le tabac (Bartsch et al., 1992; Jin et al., 1993a; Nowell et al.,
1999; Stillwell et al., 1999; Bernard and Guillemette, 2004; Carlini et al., 2005; Mori et al., 2005). Étant donné que l'accumulation des substances endogènes et des substances exogènes peut être nocive pour l'organisme, leur métabolisme par ces trois phases est très important car il empêche cette accumulation et prévient ainsi les effets néfastes potentiels.
Phase I
CYP1A1/2 CYP1B1 CYP2A6Phase II
CYP2C19 CYP3A4/5/7 NAT2Figure 5 : Enzymes retrouvées dans les phases I et II du métabolisme des médicaments. La phase I est principalement catalysée par les cytochromes P450 tandis que la phase II regroupe diverses classes d'enzymes de conjugaison dont les UGT représentent la plus forte proportion.
3.1. Réaction de glucuronidation
La glucuronidation est catalysée par la famille des UGT et cette réaction est considérée comme un mécanisme de detoxification qui modifie l'activité biologique et pharmacologique de différents composés en augmentant l'hydrophilicité de la molécule et ainsi facilitant son élimination de l'organisme (Mackenzie et al., 1997; Bélanger et al., 1998; Hum et al., 1999). La glucuronidation implique deux molécules : l'acide glucuronique (UDPGA) qui agit comme co-facteur et le substrat (Figure 6). Au cours de la réaction, les UGT transfèrent un groupement acide glucuronique sur le groupement fonctionnel du substrat, formé lors de la première phase de detoxification, ou sur une molécule déjà fonctionnalisée (Tukey and Strassburg, 2000; Guillemette, 2003). L'ajout du groupement glucuronide provoque un encombrement stérique, empêchant la liaison aux récepteurs, mais aussi une plus grande polarité, ce qui facilite l'élimination.
,COOH iCOOH HSR H2NR -HO-R HOOC-R aglycones UGT + UDP
Figure 6 : Réaction de glucuronidation catalysée par les enzymes UGT. La réaction enzymatique implique deux molécules : l'acide glucuronique (UDPGA) qui agit comme co-facteur et le substrat (aglycone). La réaction forme le glucuronide et libère l'uridine diphosphate (UDP).
Les UGT sont des protéines transmembranaires retrouvées au niveau du reticulum endoplasmique de la cellule (Figure 7) (Mackenzie et al., 1997). Ce sont des protéines de 527 à 530 acides aminés, d'un poids moléculaire entre 50 et 57 kDa, qui peuvent être divisées en deux domaines structuraux : une région amino-terminale responsable de la liaison au substrat et une région carboxy-terminale qui se fixe au co-facteur UDPGA (Hum et al., 1999). La partie amino-terminale est moins conservée d'une isoenzyme à l'autre et cette portion variable semble conférer la spécificité enzymatique des UGT. Une séquence signal, qui dirige la protéine dans le reticulum endoplasmique, est également présente dans cette région de la protéine, à l'exception d'UGTIAlO (Kinosaki et al., 1993; Strassburg et al., 2000). Cette séquence signal est clivée à la suite de l'insertion de la protéine dans le reticulum endoplasmique (Mackenzie et al., 1984). Quant au domaine carboxy-terminal, il est responsable de la rétention de l'enzyme à la membrane. De plus, il contient un domaine transmembranaire qui divise la protéine en deux parties : une petite portion située au niveau du cytoplasme et la majeure partie de la protéine située dans la lumière du reticulum endoplasmique.
Séquence Domaine de liaison du
signai substrat
Domame de liaison du Signal de rétention au cofecteur reticulum endoplasmique
♦H3N
coo-Région amino-terminale conservée CYTOPLASME Insertion dans le reticulum endoplasmique MEMBRANE DU RETICULUM ENDOPLASMIQUE LUMIERE ♦H3N Domaine tr ansme mbr an air e Lys-Lys
COO-Figure 7 : Localisation et structure protéique schématique des UGT dans le reticulum endoplasmique. La structure primaire des UGT est constituée de deux domaines structuraux, soit une région amino-terminale responsable de la liaison au substrat et une région carboxy-terminale qui se fixe au co-facteur UDPGA.
La glucuronidation est quantitativement la plus importante réaction de la phase II de detoxification (Jansen et al., 1992) (Figure 5). Elle est reconnue comme étant un processus d'inactivation et d'excrétion des substrats endogènes et exogènes. Par exemple, pour ce qui est du 2-hydroxyamino-l-méthyl-6-phenylimidazo[4,5-b]pyridine (N-OH-PhIP), un composé hautement carcinogène retrouvé dans la viande bien cuite et qui est impliqué dans l'étiologie du cancer du côlon, la conjugaison par les UGT est considérée comme étant sa principale voie d'élimination (Nowell et al., 1999; Malfatti and Felton, 2001). Cependant, dans le cas de certains médicaments, en acquérant de nouvelles propriétés biologiques suite à leur glucuronidation, il arrive que les produits glucuronidés soient plus actifs que les produits originaux. C'est le cas notamment de la morphine-6-glucuronide qui est un