IDENTIFICATION DES PROTEINES RECONNUES PAR
LES ANTICORPS H36 ET H72 CHEZ L'AMIBE
DICTYOSTELIUM DISCOÏDE UM
Mémoire présenté
à la Faculté des études supérieures et postdoctorales de l'Université Laval dans le cadre du programme de maîtrise en microbiologie
pour l'obtention du grade de Maître es sciences (M. Sc.)
DEPARTEMENT DE BIOCHIMIE, DE MICROBIOLOGIE ET DE BIO-INFORMATIQUE
FACULTÉ DES SCIENCES ET DE GÉNIE UNIVERSITÉ LAVAL
QUÉBEC
2012
L'amibe Dictyostelium discoideum sécrète des corps multilamellaires (CMLs) à partir de sa voie endocytique lorsqu'elle est cultivée en présence de bactéries. L'objectif de la présente étude consistait à comprendre le rôle des CMLs dans la physiologie de D. discoideum en caractérisant l'antigène de l'anticorps H36 présent sur les CMLs et celui de l'anticorps H72 qui se retrouve dans la voie endocytique. L'identification des antigènes a été réalisée par une immunoprécipitation suivie par une analyse en spectrométrie de masse. L'identité de l'antigène H36 a été déterminée et a été confirmée par des analyses en Western blot pour être la protease cprG aussi nommée CP7. Dans le cas de l'anticorps H72, l'identification n'a pas été fructueuse. Cette étude démontre que les CMLs sécrétés contiennent donc la protease cprG. Il s'agit de la deuxième description de l'association d'une protease avec des structures membranaires sécrétées par l'amibe D. discoideum.
Abstract
The amoeba Dictyostelium discoideum secretes multilamellar bodies (MLBs) from its endocytic pathway when grown in the presence of bacteria. The objective of this study was to understand the role of MLBs in the physiology of D. discoideum by characterizing the antigens recognized H36 and H72 antibodies. These antigens are found on MLBs or in the cell's endocytic pathway. The identification of antigens was performed by immunoprécipitation followed by mass spectrometry analyses. The identity of the H36 antigen was determined and was confirmed by
Western blot analyses as the cprG protease, which is also called CP7. In the case of the H72 antibody, the identification has been unsuccessful. This study demonstrates that secreted MLBs therefore contain the cprG protease. This is the second description of the association of a protease with secreted membrane structures by D. discoideum.
Avant-Propos
Tout d'abord, j'exprime mes profonds remerciements à mon directeur Steve Charette pour m'avoir fait confiance et accueilli dans son laboratoire. Qu'il soit aussi remercié de m'avoir dirigé et encouragé à réussir ma maîtrise.
J'aimerais remercier toute l'équipe de Dr Charette surtout Geneviève Filion et Valérie Paquet pour leur collaboration pendant ce projet. J'ai eu la chance de passer ma maitrise dans une merveilleuse équipe.
Je tiens à remercier les membres de mon comité d'encadrement, Dre Caroline Duchaine et Dr David Marsolais, pour m'avoir donné la chance de continuer ma maîtrise et pour leurs conseils à propos de mon projet.
Je souhaite également remercier Dre Louise Brisson, directrice du programme maîtrise et doctorat en biochimie et microbiologie pour avoir accepté ma demande d'admission.
Je tiens aussi à remercier la Chaire de la fondation J.-D. Bégin et du fond Alphonse L'Espérance de la fondation de l'IUCPQ ainsi que le FRQS (Fonds de recherche du Québec - Santé) pour avoir financé ma maitrise.
Un remerciement spécial à Christian Ortis et Bahijeh qui m'ont beaucoup aidé pour la rédaction de mon mémoire.
Merci à mes amis: Alireza, Ramtin et Ali pour tous les bons moments.
Table des matières
Résumé ii Abstract iii Avant-Propos iv Table des matières vi Liste des tableaux viii Liste des figures ix Liste des abréviations x Chapitre 1: Introduction 11
1.1. Les protozoaires 12 1.2. La phagocytose 13 1.3. Les interactions protozoaires-bactéries et la production des corps multilamellaires (CMLs) 16
1.3.1. Le phénomène d'enrobage bactérien chez l'amibe 17 1.3.2. Le phénomène d'enrobage bactérien chez les ciliés 20
1.4. L'amibe Dictyostelium discoideum 21 1.5. H36 et H72, des marqueurs de la voie endocytique chez D. discoideum 25
Chapitre 2: Hypothèses et objectifs 30
2.1. Hypothèse 31 2.2. Les objectifs de ce projet 31
Chapitre 3: Matériel et méthodes 32
3.1. Réactifs divers 33 3.2. Anticorps monoclonaux 33
3.3. Culture d'amibes 33 3.4. Immunofluorescence 34 3.5. Immunoprécipitation 36 3.6. Electrophorèse en gel de polyacrylamide 37
3.7. Coloration du gel de polyacrylamide 38
3.8. Spectrométrie de masse 39 3.9. Analyse informatique des protéines identifiées 39
3.11. Préparation de l'ADN génomique 41 3.12. Réaction de polymerase en chaine (PCR) 41
3.13. Electrophorèse sur gel d'agarose 43
3.14. Clonage 44 3.15. Électroporation 47
Chapitre 4: Résultats 49 4.1. Localisation cellulaire des protéines reconnues par H36 et H72 50
4.2. Isolement des protéines 52 4.3. Identification et analyse des protéines immunoprécipitées 53
4.4. Confirmation de l'identification de la protéine reconnue par H36 57 4.5. Confirmation de l'identification de la protéine reconnue par H72 59
Chapitre 5: Discussion 65 5.1. Isolement des protéines 66 5.2. L'identification de l'antigène de l'anticorps H72 67
5.3. La présence de la protéine cprG sur les CMLs 68
Chapitre 6: Conclusion et perspectives 73
6.1. Conclusion générale 74
6.2. Perspectives 75 Chapitre 7: Bibliographie 76
Liste des tableaux
Tableau 1. Composants du gel de résolution 38 Tableau 2. Composants du gel de concentration 38 Tableau 3. Oligonucleotides utilisés pour faire l'amplification des gènes codants pour les
protéines p35 et p20 par PCR 42 Tableau 4. Réactifs pour les réactions d'amplification PCR par échantillon de 20 pi 43
Liste des figures
Figure 1. La phagocytose 15 Figure 2. Image en microscope électronique à transmission montrant une vésicule expulsée de A.
castellanii contenant des bactéries L. pneumophila 18 Figure 3. Distribution des tailles des CMLs produits par A. polyphaga et A. castellanii après avoir
ingurgité!, pneumophila 19 Figure 4. La survie de L. pneumophila associée à A. castellanii en milieu liquide est augmentée
comparativement aux bactéries libres 20 Figure 5. Processus de la libération d'une vésicule contenant Salmonella (flèches) à partir de
Tetrahymena 21 Figure 6. Le cycle de vie chez D. discoideum 23
Figure 7. Les cellules D. discoideum sont capables de produire des CMLs qui contiennent des
billes de polystyrène 24 Figure 8. Modèle de la voie phago-endocytique chez D. discoideum 27
Figure 9. La production des CMLs H36-positifs par l'amibe en présence de bactéries Klebsiella
digestibles 29 Figure 10. Décompte des cellules en utilisant un hématocytomètre 35
Figure 11. Carte génétique du plasmide pDM 323 44 Figure 12. Les étapes nécessaires de processus du clonage d'un fragment d'ADN dans un
plasmide 45 Figure 13. Carte génétique du plasmide pSP73 47
Figure 14. Analyse par immunofluorescence de CMLs pour la présence des protéines reconnues
par les anticorps H36, H72 et H161 en co-culture avec la bactérie K. aerogenes 51 Figure 15. Gel SDS-PAGE suite à une immunoprécipitation et montrant la protéine reconnue par
l'anticorps H36 52 Figure 16. Gel SDS-PAGE suite à une immunoprécipitation et montrant la protéine reconnue par
l'anticorps H72 53 Figure 17. Le gène codant pour la protéine cprG 54
Figure 18. Domaines fonctionnels de la protéine cprG 54 Figure 19. Le gène codant pour la protéine DDBG0278831 (p20) 55
Figure 20. Domaines fonctionnels de la protéine DDBG0278831 (p20) 56 Figure 21. Le gène codant pour la protéine DDBG0270018 (p35) 56 Figure 22. Analyse des domaines fonctionnels de la protéine DDBG0270018 (p35) 57
Figure 23. Western Blot avec les anticorps H36 et AD-7.5 sur des protéines n'ayant pas été
exposées au B-merchapthoéthanol 58 Figure 24. Western Blot avec les anticorps H36 et AD-7.5 sur des protéines ayant été exposées au
P-merchapthoéthanol 59 Figure 25. Clonage du gène p20 dans le plasmide pDM323 60
Figure 26. Clonage du gène p35 dans le plasmide pDM323 61 Figure 27. Western blot pour confirmer l'expression de GFP-p20 et GFP-p35 62
Figure 28. Western Mot par l'anticorps H72 63 Figure 29. La vérification de la localisation des protéines p20-GFP et p35-GFP dans des cellules
de D. discoideum 64 Figure 30. L'anticorps H36 marque partiellement des CMLs purifiés sécrétés 71
AMP : Adenosine monophosphate
APS : Ammonium persulfate (persulfate d'ammonium) ARNr : Acide ribonucléique ribosomique
°C : Degré Celsius
CFU : colony-forming units (unité formatrice de colonie) CHUL : Centre Hospitalier de l'Université Laval
CML : Corps multi lamellaire
DAPI : 4',6'-diamidino-2-phénylindole dNTP : Désoxyribonucléotides
ECL : Enhanced chemiluminescence
EDTA : Ethylenediaminetetraacetic acid (acide ethylene diamine tétra acétique) ESI : Electrospray Ionization (Ionisation par électronébuliseur)
GFP : green fluorescent protein (protéine fluorescente verte) GPI : Glycosylphosphatidylinositol
IBIS : Institut biologie integrative et des systèmes IgG : Immunoglobuline G
m : Mètre
MALDI : Matrix-assisted laser desorption/ionization (désorption-ionisation laser assistée par matrice)
min : Minute ml : Millilitre pi : Microlitre
NADPH : Nicotinamide adenine dinucléotide phosphate ng : Nanogramme
pb : Paire de base
PCR : Polymerase chain reaction (réaction de polymerase en chaîne) PMSF : Phenylmethylsulfonyl fluoride (fluor de phénylméthylsulfonyle,)
SDS-PAGE : Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (electrophorèse sur gel de polyacrylamide en présence de dodécylsulfate de sodium)
TEMED : Tetraméthyléthylènediamine U : Unité
Chapitre 1
1.1. Les protozoaires
Les protozoaires sont des organismes eucaryotes, unicellulaires et hétérotrophes appartenant à la famille des protistes (Parry 2004). Ils ont été découverts pour la première fois par Antonie Van Leeuwenhoek (1632 - 1723). La plupart des protozoaires sont mobiles et peuvent être isolés d'une très grande variété de niches écologiques, y compris l'eau, les sols humides de même qu'à l'intérieur d'autres organismes (Thomas et al. 2008; Norf et Foissner 2010). Certains d'entre eux peuvent être des agents pathogènes humains et peuvent provoquer des maladies graves (Jones et al. 1975; Landfear 2011). Les protozoaires sont des cellules microscopiques et peuvent varier en taille de 2 à 2000 pm (Corliss 2002).
Pendant de nombreuses années, les protozoaires ont été divisés en 4 sous-groupes selon leur mode de locomotion : les flagellés, les amibes, les sporozoaires et les ciliés (Corliss 2002; Parry 2004). Les flagellés sont parmi les principaux consommateurs de bactéries dans les écosystèmes aquatiques (Sherr et Sherr 2002). Ils sont identifiables par la présence de flagelles qu'ils utilisent pour se propulser et chasser dans un environnement liquide (Silflow et Lefebvre 2001). Les flagellés sont généralement de petite taille (2 à 30 pm) (Krometis et al. 2009). Pour leur part, les amibes peuvent prendre n'importe quelle forme. Elles utilisent des pseudopodes pour se déplacer et aussi pour piéger et enfermer leurs proies dans des vacuoles alimentaires. Ils sont dans une gamme de taille allant de 2 à 20 pm (Rogers et al. 2008), mais certaines espèces comme Amoeba proteus peuvent atteindre près d'un millimètre. Les sporozoaires sont généralement des parasites intracellulaires dont la plupart sont des pathogènes de mammifères. Ils sont immobiles et manquent d'organes locomoteurs comme des flagelles ou des pseudopodes (Lim et McFadden 2010). Les ciliés comme le nom l'indique, constituent un groupe de protistes se caractérisant par la possession de cils (des organites ressemblant à des poils) dont la longueur varie entre 2 pm et 15 pm (Dopheide et al. 2008). Les cils sont des structures similaires aux flagelles, mais ils sont plus courts et plus nombreux que les flagelles. La plupart des ciliés utilisent leurs cils pour la locomotion, l'attachement et l'alimentation (Adl et Berger 1996).
La nouvelle méthode de la classification des protozoaires, est basée sur une combinaison de données moléculaires, y compris la séquence des ARNr 18S et de certaines protéines, combinées à des informations phénotypiques et moléculaires (Smirnov et al. 2007). De nombreux protozoaires ont au moins deux étapes principales dans leur cycle de vie: le trophozoite (l'étape métaboliquement active) et le kyste (une forme dormante). L'enkystement est particulièrement fréquent et se produit aux fins de protection lorsque les conditions environnementales deviennent défavorables. (Ben Salah et Drancourt 2010).
1.2. La phagocytose
Les protozoaires sont d'importants consommateurs de bactéries et d'autres matières organiques dans l'environnement. Ils ingurgitent les particules par le processus de la phagocytose (Barker et Brown 1994). Les protozoaires brouteurs sont capable de se nourrir de bactéries avec des taux entre 0,2 et 1465 bactéries par heure (Parry 2004). Le processus de la phagocytose (Fig. 1) a été découvert pour la première fois par Metchnikoff en 1883 et permet aux protozoaires d'acquérir les nutriments nécessaires à leur croissance (Desjardins et al. 2005). La phagocytose n'est pas observée seulement chez les protozoaires; les macrophages et les neutrophiles du système immunitaire utilisent aussi ce processus pour éliminer les corps étrangers dans l'organisme (Underhill et Ozinsky 2002). La phagocytose permet l'internalisation des larges particules (typiquement > 500 nm de diamètre) comme les bactéries. Ce processus est dépendant du cytosquelette d'actine et est initialise par la liaison de la particule au phagocyte via des récepteurs extracellulaires (Sateriale et Huston 2011). L'attachement des particules sur la surface est accompagné par l'activation de nombreuses voies de signalisation qui induit la polymérisation de l'actine au site de liaison. Cela permet un mouvement membranaire qui mène à la formation de la coupe phagocytique et à l'internalisation des particules (Meza et al. 2006). Ensuite les phagosomes commencent le processus de maturation alors qu'ils fusionnent avec des lysosomes pour devenir des phagolysosomes (Fratti et al. 2001). Plusieurs protéines membranaires jouent un rôle crucial dans la maturation du
phagosome (Vieira et al. 2002). Par la suite, différentes enzymes telles que les enzymes hydrolytiques et la NADPH oxydase sont activées amenant une diminution du pH du phagosome qui devient alors un lysosome (Steinberg et al. 2007). La fonction du lysosome est d'agir comme un compartiment où se fait la dégradation des bactéries et autres particules internalisées. Ensuite, les lysosomes retournent à un pH neutre et se transforment alors en post-lysosomes. Finalement les déchets non digérés sont dirigés vers l'extérieur du protozoaire via le processus d'exocytose (Bucci et al. 2000). Comparativement aux protozoaires, les cellules du système immunitaire ne font habituellement pas l'exocytose des déchets non digérés.
1____H
' _____^_T I _______ _____ ^__ ____»L_> J2_____fl
_______ V a
jf 1
_L'
B
Microbe orother particle p i a ^ n . Q Chemotaxis and adherence of microbe to phagocyte Q Ingestion ot microbe
bye
Formation ol a phagosome Q Fusion ol the phagosome
with a lysosome to form a phagolysosome O Ngestlon ol Ingested
microbe by enzymes Formation of residual body containing indigestible material
Q Discharge of waste
Phases of phagocytosis
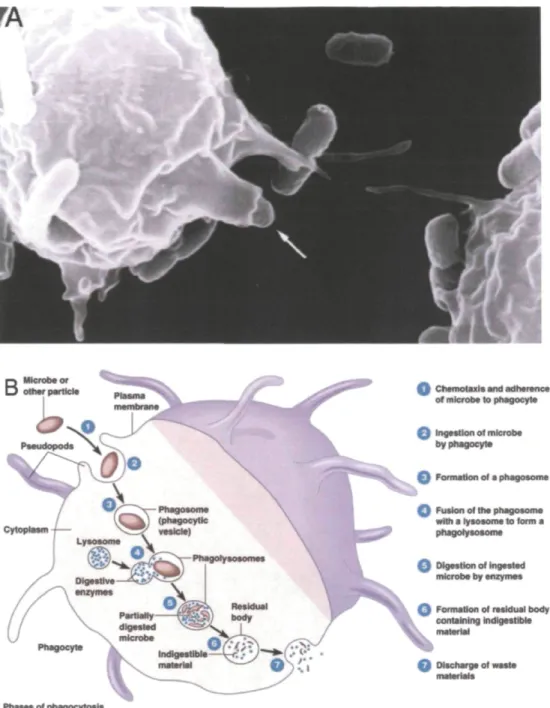
Figure 1. La phagocytose A) La phagocytose chez l'amibe Image en microscopie électronique à balayage. La flèche blanche montre une bactérie phagocytée par l'amibe. Figure tirée de Bozzaro et al. 2008. B) Le schéma du processus de phagocytose. Ce processus permet l'internalisation des bactéries et de larges particules dans les protozoaires. Source de l'image B: (httpy/classes.rnidlandstech.edu/Q.
1.3. Les interactions protozoaires-bactéries et la
production des corps multilamellaires (CMLs)
Les protozoaires sont parmi les plus grands prédateurs pour les bactéries. Ainsi certaines d'entre elles possèdent des stratégies défensives pré-ingestion telles que la formation de microcolonies, de cellules surdimensionnées ou la sécrétion de divers facteurs de virulence. Certaines bactéries ont aussi des moyens de défense post-ingestion telles que la croissance intracellulaire, la libération de toxines et la résistance à la digestion (Hahn et Hofle 1998). Nâgler a démontré en 1910 que les protozoaires libres et surtout les amibes peuvent être un réservoir pour des bactéries à Gram négatif. D'autres études ont démontré la présence de bactéries pathogènes dans les amibes du genre Acanthamoeba (Proca-Ciobanu et al. 1975).
Il est aujourd'hui établi qu'un grand nombre d'espèces bactériennes (plus de 23 espèces) appartiennent au groupe des bactéries résistantes aux amibes (amoeba resisting bacteria) comme Pseudomonas aeruginosa, Listeria monocytogenes, Legionella spp.. Vibrio spp. et Escherichia coli OI57:H7 (Greub et Raoult 2004; Thomas et al. 2010). Ces bactéries résistent à la prédation par les amibes et peuvent persister et se multiplier à l'intérieur des protozoaires (Barker et Brown 1994; Cirillo et al. 1997). Les diverses études suggèrent que les protozoaires pourraient agir comme des chevaux de Troie dans la propagation de ces bactéries pathogènes (Barker et Brown 1994; Greub et Raoult 2004). L'hypothèse est que les protozoaires peuvent servir de vecteur de transmission pour les bactéries dans l'environnement ou dans des structures artificielles (système de climatisation, chauffe-eau, unité dentaire, etc.) (Barbaree et al. 1986; Greub et Raoult 2004). Cependant, en plus de constituer un réservoir de bactéries, les protozoaires offrent aussi une protection à ces dernières en les isolant de certaines conditions environnementales hostiles par la formation des kystes contenant des bactéries ou par la production de vésicules extracellulaires formant un enrobage protecteur (Molmeret et al. 2005; Ben Salah et Drancourt 2010). Comparativement aux bactéries non pathogènes qui sont digérées par les amibes et autres protozoaires brouteurs, les bactéries résistantes à la prédation (souvent des bactéries pathogènes) ne sont pas digérées dans les lysosomes et peuvent se faire enrober dans du matériel protéolipidique (déchet métabolique du protozoaire) avant
d'être sécrétées hors de la cellule dans des structures multilamellaires (Rowbotham 1980; Bouyer et al. 2007). Ces corps multilamellaires (CMLs) sont formés de plusieurs couches concentriques de membranes lipidiques qui peuvent contenir des bactéries vivantes (Bouyer et cd. 2007; Berk et al. 2008). Les CMLs sécrétés peuvent aussi porter le nom de vésicules extracellulaires.
La viabilité des bactéries enrobées dans des CMLs est fortement augmentée, car ces structures leur offrent une protection contre des conditions environnementales défavorables. Le phénomène d'enrobage a été principalement étudié avec L. pneumophila (Greub et Raoult 2002; Bouyer et al. 2007). Il semble que ce phénomène soit généralisé, car outre L. pneumophila, de nombreuses bactéries comme Mycobacterium spp, Escherichia coli 0157: H7, Listeria monocytogenes, Salmonella enterica, et Parachlamydia peuvent être emballées dans des CMLs par des protozoaires (Gourabathini et al. 2008; Adekambi et al. 2006; Brandi et al. 2005; Greub et Raoult 2002). Des membres de deux principaux groupes des protozoaires sont connus pour produire des CMLs contenant des bactéries, y compris les amibes comme Acanthamoeba et des ciliés tels que Tetrahymena spp. et Glaucoma spp. (Rowbotham 1980; Brandi et al. 2005; Gourabathini étal. 2008).
1.3.1. Le phénomène d'enrobage bactérien chez l'amibe
Le phénomène d'enrobage a d'abord été observé pour L. pneumophila co-cultivé avec des Acanthamoeba (Rowbotham 1980). Ce genre d'amibe est connu pour sa grande capacité à phagocyter des particules en milieu de culture, ce qui facilite l'étude de ce phénomène. Deux espèces de ce genre d'amibes, Acanthamoeba polyphaga et castellanii ont été étudiées pour leur capacité à phagocyter L. pneumophila. À la suite de l'internalisation de la bactérie par l'amibe, la bactérie commence à se multiplier rapidement dans le phagosome. Enfin les amibes infectées produisent des CMLs contenant de nombreuses bactéries (Fig. 2) (Anand et al. 1983; Rowbotham 1986; Berk étal. 1998).
la H
i
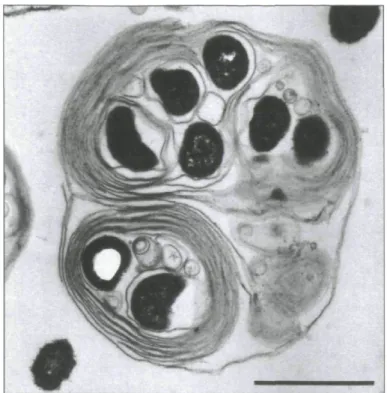
Figure 2. Image en microscope électronique à transmission montrant une vésicule expulsée de A. castellanii contenant des bactéries L. pneumophila. Figure tirée de Berk era/. 1998.
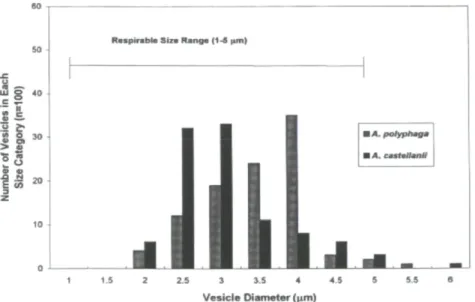
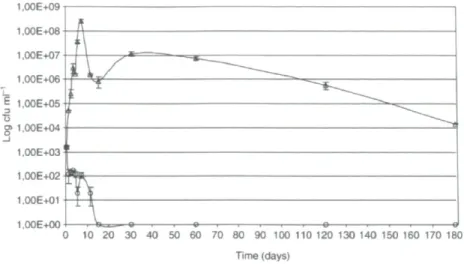
La taille des CMLs produits par les Acanthamoeba varie entre 2,1 à 6,4 pm (Fig. 3). Cela représente la taille théorique de particules respirables (c'est-à-dire les particules avec une dimension inférieure à 7 pm peuvent pénétrer et se déposer dans le système respiratoire). Chaque amibe peut produire jusqu'à 25 CMLs en 24h (Berk et al. 1998). Les bactéries L. pneumophila libres ne peuvent pas survivre dans le milieu pauvre (milieu d'enkystement) pour une longue période, mais les bactéries associées à A. castellanii peuvent survivre pour une période d'au moins 6 mois (Bouyer et al. 2007) (Fig. 4). L. pneumophila cultivée avec A. castellanii devient 100 fois plus virulente
comparativement à la même souche cultivée sur gélose (Cirillo et al. 1994). De plus, les légionnelles enrobées sont plus résistantes à des conditions défavorables telles que la congélation, la sonication et les biocides comparativement aux bactéries libres (Berk et al. 1998). La maladie du légionnaire est transmise via les aérosols (Philippe et al. 2006). Les CMLs produits par les amibes et contenant L. pneumophila participent probablement à la transmission de la maladie à cause de leur taille. Cela permet d'émettre l'hypothèse
À part L. pneumophila d'autres bactéries pathogènes comme Mycobacterium avium, Helicobacter pylori, Francicella tularensis et Chlamydia pneumoniae sont aussi capables de se multiplier dans A. castellanii ou A. polyphaga (Greub et Raoult 2002; Adekambi et al. 2006; El-Etr et al. 2009). Jusqu'à présent, il a été confirmé que les amibes sont capables de produire des CMLs contenant les souches de Mycobacterium avium, Burkholderia cepacia et Chlamydia, mais pour les autres souches cela n'a pas encore été confirmé (Cirillo et al. 1997; Marolda et al. 1999; Greub et Raoult 2002). Les résultats démontrent que les bactéries ingurgitées dans l'amibe sont plus résistantes à la chloration et au traitement antimicrobien que les bactéries libres (Winiecka-Krusnell et al. 2002). Les autres espèces d'amibe comme Naegleria, Vahlkampfia, Platyamoeba, Saccamoeba, Vexilliferia, Echinamoeba et Willaertia peuvent supporter la multiplication des légionelles, mais la capacité de la production des CMLs par ces amibes n'a pas encore été déterminée (Greub et Raoult 2004).
Figure 3. Distribution des tailles des CMLs produits par A. polyphaga et A. castellanii après avoir ingurgité L. pneumophila. Plus de 90% des CMLs produits par chaque espèce d'amibes possède une taille entre 1 à 5 pm. Figure tirée de Berk étal.
e 1.00E+05 1.00E+03 1.0OE+O2 1.0OE+O1 1.00E+00 0 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 Time (days)
Figure 4. La survie de L. pneumophila associée à A. castellanii en milieu liquide est augmentée comparativement aux bactéries libres. Dans l'échantillon d'amibes infectées par L. pneumophila, la viabilité des bactéries a été détectée par décompte d'unité formatrice de colonie (cfu), même après 180 jours. En revanche, dans l'échantillon contrôle (bactéries libres sans amibe), aucune bactérie viable n'a pas été détectée après 11 jours. Les bactéries viables dans les échantillons de 11 jours et plus étaient enrobées. Figure tirée de Bouyer et al. 2007.
1.3.2. Le phénomène d'enrobage bactérien chez les
ciliés
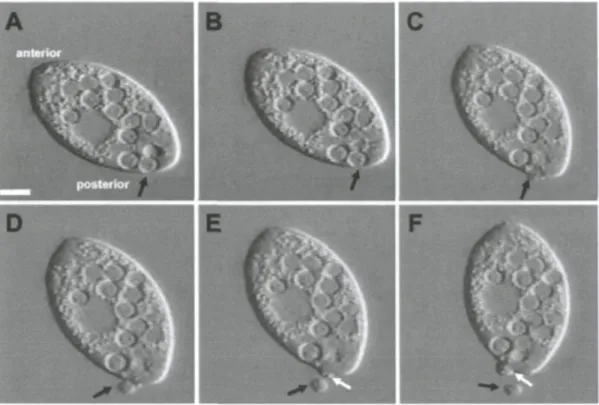
Les ciliés sont aussi capables de libérer un grand nombre de CMLs. Par exemple, Tetrahymena sp. infecté par Salmonella enterica stéréotype Thompson produit des CMLs contenant une forte densité de cette bactérie (Fig. 5) (Brandi et al. 2005; Rehfuss et al. 2011). Cette bactérie a la capacité d'échapper à la digestion dans les vacuoles alimentaires de ce protozoaire. Les cellules Tetrahymena commencent à expulser des vésicules environ lh après l'ingurgitation des bactéries. La taille de ces vésicules varie entre 3,1 à 8,0 pm (Brandi et al. 2005). Les résultats montrent qu'une proportion significative des Salmonella enrobées par les ciliés, est très résistante aux détergents et à la chloration en comparaison aux bactéries libres.
Figure 5. Processus de la libération d'une vésicule contenant Salmonella (flèches) à partir de Tetrahymena. Les flèches noires montrent la libération d'une vésicule contenant S. enterica. Les flèches blanches montrent la protrusion de la membrane de Tetrahymena au moment de la libération des vésicules. Figure tirée de Brandi et al. 2005.
Glaucoma sp. est un autre exemple des ciliés qui peut expulser des CMLs contenant des bactéries vivantes. Les bactéries E. coli 0157:H7, Salmonella enterica et L. monocytogenes peuvent être aussi enrobées dans des CMLs par le cilié Glaucoma (Gourabathini et al. 2008).
1.4. L'amibe Dictyostelium discoideum
Afin d'étudier le processus phagocytique et l'enrobage bactérien, Dictyostelium discoideum a été utilisé comme organisme modèle de protozoaire dans le présent travail
de maîtrise. D. discoideum est un eucaryote unicellulaire faisant partie du groupe des amibes dites sociales (Fey et al. 2009). La phylogénie moléculaire indique qu'il s'agit d'une espèce d'amibe appartenant à l'embranchement des Mycétozoaires se retrouvant dans le sol et la litière de feuilles humides (Baldauf et al. 2000). C'est en 1935 que K. Râper a décrit, pour la première fois, cette amibe (Râper et Smith 1939). Le génome de D. discoideum, est le premier génome de protozoaire libre (non pathogène) à avoir été séquence. Ce génome de 34Mb est haploïde et réparti sur six chromosomes. Il possède un haut pourcentage de nucleotides A/T (78.8%) et contient approximativement 12 500 gènes (Eichinger et al. 2005). En raison de son cycle de vie, qui implique une phase unicellulaire et une autre sous forme multicellulaire (Fig. 6), et de son comportement, D. discoideum est utilisé comme organisme modèle pour étudier la différenciation cellulaire, la phagocytose, la voie endocytique, l'exocytose, la motilite cellulaire, la transduction du signal, le chimiotactisme et la mort cellulaire programmée (Steinert et Heuner 2005; Annesley et Fisher 2009). Un des principaux avantages d'utiliser D. discoideum comme modèle est la facilitée de créer des mutants et d'en comparer les phénotypes avec la souche parentale afin de déduire la fonction du gène muté (Annesley et Fisher 2009).
D. discoideum se nourrit de bactéries par le processus de phagocytose et vit sous forme unicellulaire en présence de nutriments (Baldauf et al. 2000). Cette amibe est aussi un hôte modèle pour certaines bactéries pathogènes pour les humains, telles que la légionellose, les mycobactéries, Pseudomonas aeruginosa et les salmonelles (Steinert et Heuner 2005). Cette propriété de D. discoideum favorise son utilisation comme modèle pour étudier les interactions hôte-pathogène (Steinert et Heuner 2005; Bozzaro et al. 2008; Clarke 2010; Dallaire-Dufrasne et al. 2011).
Certains résultats ont démontré que D. discoideum est aussi capable de produire et de sécréter des vésicules lorsque les cellules sont cultivées en présence de bactéries (Gezelius 1959; Mercer et Shaffer 1960; Barondes et al. 1985; Cooper et al. 1986; Fukuzawa et Ochiai 1993; Emslie et al. 1998; Marchetti et cd. 2004). Ces vésicules sont similaires aux CMLs produites par les Acanthamoeba et les ciliés et contiennent des
composés bactériens non digérés tels que des glycoconjugués et des protéines amibiennes. Néanmoins, l'enrobage de bactéries vivantes dans des CMLs produit par D. discoideum n'a jamais été confirmé jusqu'à maintenant.
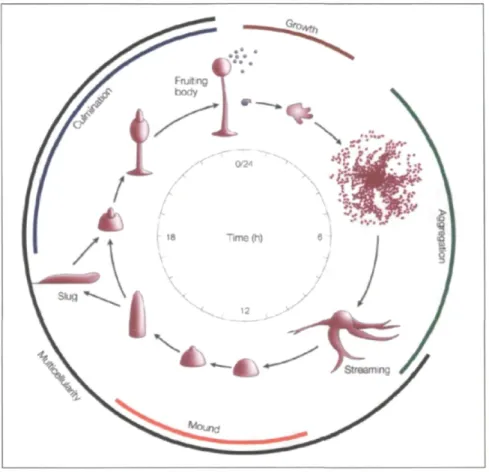
Figure 6. Le cycle de vie chez D. discoideum. Les amibes se nourrissent de bactéries, jusqu'à ce que la nourriture soit épuisée. Puis les cellules commencent un processus de signalisation cellule-cellule via la sécrétion d'AMPc. Ensuite les amibes s'assemblent pour former des agrégats. Ces derniers se constituent d'un pseudo plasmodium multicellulaire nommé "limace" (slug sur la figure). Lorsque les conditions sont favorables, la limace forme un corps fructifère qui contient des spores. Chaque spore produit une amibe haploïde unique pour relancer le cycle lorsqu'elle se retrouve en condition favorable (Henderson 1975). L'image présentée ici a été tirée du site internet http://dictybase.org.
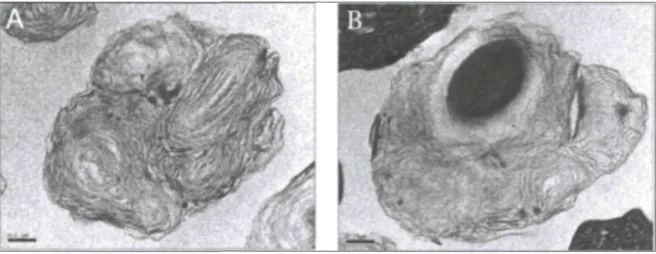
Au cours des deux dernières années, l'équipe du Dr. S. Charette a effectué plusieurs expériences pour confirmer que D. discoideum peut produire des CMLs et que ceux-ci peuvent contenir des bactéries enrobées. Afin de démontrer la présence de ce processus chez D. discoideum, Mycobacterium smegmatis a été choisie comme modèle bactérien et l'enrobage de cette bactérie a été observé avec deux souches différentes de D. discoideum, DH1-10 et AX2 (Charette, communication personnelle). Les résultats confirment que D. discoideum peut aussi expulser les bactéries emballées après leur ingestion. Le même test d'emballage a été effectué en utilisant des billes de polystyrène (1 pm de diamètre). Ces billes ne sont pas dégradées dans la voie d'endocytose des amibes après leur phagocytose (Charette et al. 2006). Comme les bactéries, les billes incubées avec les amibes sont emballées et sécrétées dans des CML (Paquet, communication personnelle) (Fig. 7).
_Sta
■V
Figure 7. Les cellules D. discoideum sont capables de produire des CMLs qui contiennent des billes de polystyrène. A) CML sécrété par les amibes en absence de bille. B) CML sécrété en présence de billes de polystyrène. Après avoir été phagocyté par l'amibe, la bille est incluse dans un CML puis relarguée dans le milieu. Cette figure a été préparée par V. Paquet.
1.5. H36 et H72, des marqueurs de la voie endocytique
chez _D. discoideum
Afin d'avoir une meilleure compréhension des mécanismes moléculaires impliqués dans le processus d'enrobage bactérien et la phagocytose chez l'amibe, le laboratoire de S. Charette dispose de plusieurs anticorps monoclonaux (H161, H36 et H72) dirigés contre diverses protéines membranaires de D. discoideum (Fig. 8). Ces anticorps se sont montrés très utiles pour l'étude de la voie endocytique de cette amibe. Ils ont été produits via l'immunisation de souris avec des extraits de membranes cellulaires de D. discoideum (Ravanel et al. 2001; Charette et al. 2006; Mercanti et al. 2006).
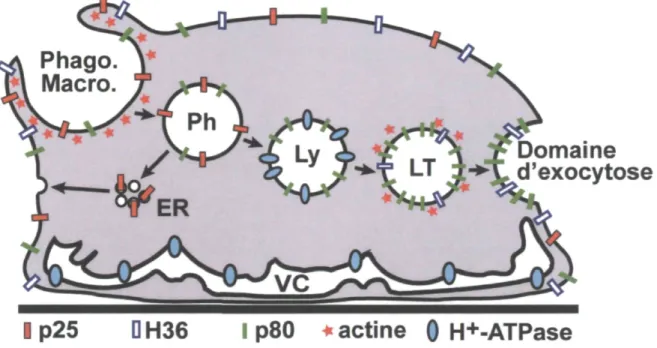
L'anticorps H161 reconnaît une protéine de 80 kDa (p80) avec trois domaines transmembranaires qui présente une homologie avec une protéine transporteur de zinc chez l'humain (CTR1) (Ravanel étal. 2001). Par une analyse en immunofluorescence, il a été montré que la protéine p80 est localisée à la surface cellulaire et sur les compartiments phagocytiques. La concentration de cette protéine augmente dans les endosomes matures. En effet, plus un endosome progresse dans sa maturation, plus sa surface sera occupée par cette protéine. La protéine p80 s'accumule aussi sur les membranes des lysosomes tardifs et le domaine d'exocytose situé sur la membrane plasmique de l'amibe (Ravanel et al. 2001).
L'anticorps H72 détecte une protéine de 25 kDa (p25) localisée principalement à la surface cellulaire et dans les endosomes de recyclage (Charette et al. 2006). Il a également suggéré que p25 est constamment recyclée à partir de la voie phagocytique vers la membrane plasmique via justement les endosomes de recyclages. La protéine p25, reconnue par l'anticorps H72, a permis de mettre en lumière le triage protéique dans les compartiments endosomaux (Charette et Cosson 2008). L'antigène de l'anticorps H72 n'est pas encore connu.
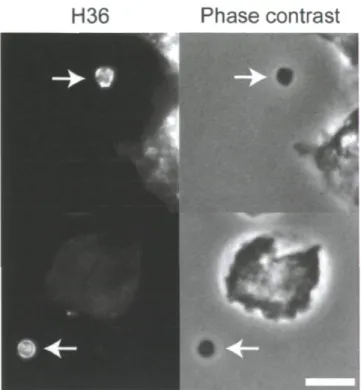
L'anticorps H36 marque les CMLs produits par D. discoideum (Fig. 9). Cet anticorps détecte une protéine avec un poids moléculaire d'environ 46 kDa, mais la caractérisation de l'antigène H36 n'est pas encore été réalisée. (Charette et al. 2006). Les analyses d'immunofluorescence ont montré qu'il existe un lien évident entre la protéine détecté par H36 et la membrane plasmique ainsi que les compartiments d'endocytose chez D. discoideum (Mercanti et al. 2006).
Pour confirmer que les protéines H72 et H161 trouvées dans les phagosomes nouvellement formés proviennent de la surface cellulaire et non d'un autre compartiment intracellulaire, la surface des cellules seulement a été marquée avec les anticorps H72 et H161. Ensuite, les cellules ont été autorisées à phagocyter les particules de levure pendant 3 minutes avant de les fixer et de les incuber avec un anticorps secondaire fluorescent. Les protéines H72 et H161 ont été détectées à des niveaux similaires dans les phagosomes nouvellement formés et à la surface cellulaire. Cela indique que les protéines H72 et H161 observées dans les coupes phagocytiques et dans les phagosomes nouvellement formés proviennent effectivement de la membrane plasmique. Ces protéines ont été capturées lors de la formation des phagosomes et ne proviennent pas de compartiments intracellulaires pendant la formation du phagosome (Mercanti et al. 2006). Le même processus a été répété en utilisant l'anticorps H36 comme marqueur. Cet anticorps aussi est présent à la surface des cellules Dictyostelium. L'anticorps H36 a été exclus de coupes phagocytiques et a été retrouvé seulement à des niveaux très faibles dans les phagosomes nouvellement formés (Mercanti et al. 2006).
Phago.
Macro, j / * " * X
ER
LT i V l
d'exocytose
Domaine
VC
lp25
DH36
p80
*
actine
0
H+-ATPase
Figure 8. Modèle de la voie phago-endocytique chez D. discoideum. L'anticorps H72 qui reconnaît la protéine p25 est localisé principalement à la surface cellulaire, sur les coupes phagocytiques ainsi que les endosomes de recyclages. L'anticorps H161 dont l'antigène est la protéine p80 est capable de marquer la membrane plasmique et tous les compartiments de la voie endocytique tout en s'accumulant dans les lysosomes tardifs (LT). L'anticorps H36 détecte une protéine principalement présente à la surface de la cellule. La protéine du cytosquelette d'actine est située sur les compartiments à l'entrée et à la sortie de la voie endocytique. La vacuole contractile (VC) est un organite impliqué dans l'osmorégulation qui contient la protéine H-ATPase. Figure fournie par S. Charette.
Dans une tentative de caractérisation de l'antigène de l'anticorps H36, un extrait cellulaire de D. discoideum a été traité avec l'enzyme PGNase F (Peptide N-Glycosidase F). Cette enzyme est capable d'hydrolyser presque tous les types de chaînes de glycopeptides par clivage du GlcNAc liée aux résidus d'asparagine des oligosaccharides de mannose, hybrides et complexes des glycoprotéines N-liées. Tel que démontré par un l'utilisation de cette enzyme, la protéine reconnue par H36 est glycosylée puisque le poids moléculaire de la protéine diminue après traitement (S. Charette, communication personnelle).
Étant donné que les protéines p25 et p46 sont membranaires (confirmé par les analyses immunofluorescence) (Charette et al. 2006; Mercanti et al. 2006), on peut supposer qu'elles ont au moins un domaine transmembranaire ou un point d'ancrage lipidique tels qu'un glycosylphosphatidylinositol (GPI ancrage).
L'identification des protéines reconnues par les anticorps H36 et H72 permettra de déterminer les protéines impliquées dans la voie endocytique et la formation des CMLs.
H36
Phase contrast
Figure 9. La production des CMLs H36-positifs par l'amibe en présence de bactéries Klebsiella digestibles. Les matériaux à partir de la plaque phagocytaire des cellules D. discoideum cultivés sur un tapis de K. aerogenes ont été déposés sur une lame de verre. Elles ont été traitées par l'immunofluorescence (IF) en utilisant l'anticorps H36. Les images ont été obtenues par microscopie à épifluorescence. Les vésicules extracellulaires sont indiquées par des flèches. La barre d'échelle: 5 pm. Cette figure a été préparée par V. Paquet.
Chapitre 2
2.1. Hypothèse
Nous pensons que l'identification des protéines reconnues par les anticorps H36 et H72 nous aidera à mieux comprendre le mécanisme de la voie phagocytique et la formation des CMLs chez D. discoideum et à augmenter encore plus l'utilité de ces anticorps comme marqueurs de la voie phago-endocytique.
2.2. Les objectifs de ce projet
1) Vérification de la présence de la protéine p25 reconnue par l'anticorps H72 sur les CMLs
2) Isolement des protéines reconnues par les anticorps H36 et H72 par une approche utilisant l'immunoprécipitation
3) Identification des protéines isolées par spectrométrie de masse
4) Analyse informatique des séquences des gènes codant pour les protéines identifiées
5) Confirmation de l'identité des protéines reconnues par H36 et H72 soit par analyse en Western blot ou par l'expression d'une version GFP des protéines identifiées.
Chapitre 3
3.1. Réactifs divers
Une partie du matériel nécessaire pour ce projet a été fourni par diverses compagnies. Les billes de protéine G sépharose, l'anticorps Anti-IgG de souris (Al 1029) et
l'anticorps Anti-GFP ont été fournis par Invitrogen (Camarillo, CA). Le cocktail d'inhibiteurs de proteases, l'albumine de sérum bovin (bovine serum albumin, BSA), les
billes de protéine A sépharose, le DAPI (4'6-diamidino-2-phenylindole), le détergent NP40 et la paraformaldehyde ont été obtenus chez Sigma-AIdrich (St. Louis, MO). L'ampicilline, l'acrylamide et l'agarose ont été achetées chez Roche (Basel, Suisse). La Taq ADN polymerase, la Pfu ADN polymerase, la protéinase-K, la T4 ADN ligase, ainsi que des enzymes de restriction et le marqueur de poids moléculaire pour l'électrophorèse d'ADN ont été procurés par New England Biolabs (Burlington, Canada). Les détergents Triton-X 100 et Tween 20 ont été fournis par VWR (Pennsylvania, USA). Le LB Agar et le N,N,N,N-7efra Méthyle thylene Diamine (TEMED) ont été fournis par EMD (Darmstadt, Germany). La trousse de réactifs pour chimioluminescence (ECL) a été
acquise chez Thermo Scientific (Rockford, IL USA). Les trousses de purification d'ADN
ont été obtenues chez Qiagen (Québec, Canada)
3.2. Anticorps monoclonaux
Les anticorps monoclonaux H36, H72 et H61 reconnaissant des protéines membranaires chez D. discoideum (Ravanel et al. 2001; Mercanti et al. 2006) proviennent du laboratoire du Pr Pierre Cosson (Université de Genève, Genève). L'anticorps monoclonal anti-cprG (AD7.5) a été fourni par le laboratoire du Dr Hudson Freeze (Burnham Institut, La Jolla, California) (Ord et al. 1997).
3.3. Culture d'amibes
La souche DH1-10 de l'amibe D. discoideum qui a été utilisée pour cette étude provient du Stock center de Dictybase (http://dictybase.org/). Il s'agit d'une souche axénique qui peut être facilement cultivée en laboratoire dans du milieu liquide. Les amibes ont été cultivée dans des boîtes de Pétri contenant 12 ml de milieu HL5 composé de 1,43% de bactopeptone, 0,72% d'extrait de levure, 50 mM de maltose, 3,6 mM de Na2HP04 et 3,6
mM KH2PO4. Afin d'éviter la contamination bactérienne, 15 pg/ml de l'antibiotique tetracycline a été ajouté dans le milieu de culture. Les cellules de DH1-10 ont été changées de milieux de culture deux fois par semaine et elles ont été incubées à une température de 21 °C.
3.4. Immunofluorescence
L'immunofluorescence est une méthode permettant de visualiser et de localiser un antigène spécifique au sein de la cellule par l'interaction entre l'anticorps et l'antigène et par l'utilisation d'anticorps couplés à des fluorochromes et détectables par microscopie à épi-fluorescence (Coons et al. 1941 ).
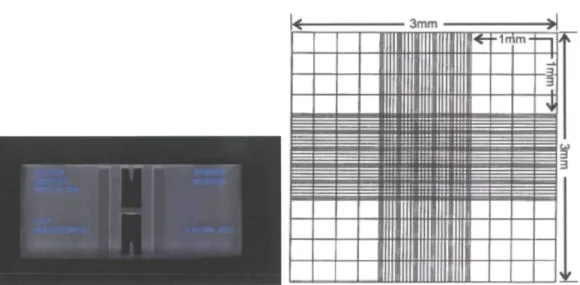
Le matériel analysé en immunofluorescence provenait soit de co-culture d'amibes avec des bactéries sur un milieu solide ou d'amibe seules en culture liquide. Dans le cas des co-cultures, du matériel provenant de plage de phagocytose a été resuspendu dans du milieu HL5 et déposé sur une lamelle pour lh30. Dans le cas des cellules en culture liquide, le milieu HL5 a été retiré des Pétris contenant les amibes pour être remplacé par 8 ml de milieu HL5 frais, les cellules ont été resuspendues dans le milieu en utilisant une pipette. Cette suspension a été transférée dans un tube de 15 ml. Une aliquote de la suspension de la cellule était placée sur un hématocytomètre pour permettre de déterminer la concentration de cellules dans la suspension (Fig. 10). Une quantité de 5xl05 cellules de D. discoideum a été déposée sur des lamelles dans des plaques de six puits et par la suite, le volume de chacun des puits a été complété à 500 pi de milieu HL5 frais. Les cellules ont adhéré pendant lh30, puis ont été fixées pendant 30 minutes dans une solution de paraformaldehyde 4% diluée dans le milieu HL5. Ensuite, les cellules ont été rincées trois fois avec 2 ml de tampon PBS-NH4CI (40mM) suivi d'un second rinçage avec 2 ml de tampon PBS IX (2 mM Na2HP04, 14,7 mM KH2P04, pH 6,0). Les cellules ont été perméabiUsées en incubant celles-ci pendant 2 minutes dans une solution de methanol à -20°C (Mercanti et al. 2006). Les lamelles ont ensuite été transférées dans une solution de PBS IX. Un blocage non spécifique a été effectué en déposant les lamelles dans une solution PBS IX contenant 0,2% d'albumine de sérum bovin (PBS-BSA) pendant 5 minutes à température ambiante. Les cellules sur les lamelles ont été
incubées avec l'anticorps H36 ou H72 dilué 1:1000 dans du PBS-BSA pendant 45 minutes. Les lamelles ont été lavées 3 fois pendant 5 minutes avec du tampon PBS et ensuite l'anticorps secondaire fluorescent (Anti-IgG de souris) couplé à Alexa -Fluor 488 a été dilué 1:400 dans du tampon PBS-BSA et a été déposé sur les cellules pour une période de 30 minutes à température ambiante. Le dernier rinçage a été fait par le tampon PBS et les lamelles ont été déposées sur des lames de verre en ajoutant 10 pi de Prolong Gold (Invitrogen, Camarillo), une solution de montage, puis scellées avec du vernis à ongles. Afin d'éviter l'exposition à la lumière entre les observations au microscope, les lames ont été emballées dans du papier d'aluminium. L'observation et les photos des cellules ont été prises avec un microscope à épifluorescence équipé d'une caméra numérique (Zeiss, Oberkochen).
_
n_*t
irT i m -r
v i ■II+-
4
^ ^ 3 3min
HUM
i
tFigure 10. Décompte des cellules en utilisant un hématocytomètre Après la préparation de la suspension de cellules de D. discoideum, 15 pi de celle-ci ont été déposée sur des lames de verre et le décompte fait par observation des amibes visibles au microscope dans chacun des quatre carrés. Chaque grand carré qui correspond à un volume de 0,1 pi est divisé en 16 petits carrés. Les images de la figure ont été tirées du site internet http://www.tpub.com/corpsman/232.htm.
3.5. Immunoprécipitation
L'immunoprécipitation est une méthode qui est basée sur la précipitation d'un antigène spécifique dans un complexe de protéines en utilisant la protéine A ou la protéine G qui ont une haute affinité pour la région Fc de l'anticorps. Les protéines A ou G sont fixées à des billes de sépharose. L'antigène d'intérêt peut par la suite êtres séparé par migration sur un gel de polyacrylamide SDS-PAGE (Bonifacino et al. 2001).
Le décompte des cellules a été fait en utilisant un hématocytomètre et 1,5-2,0 x IO7 cellules de Dictyostelium ont été prélevées à partir de six Pétris contenant des amibes en culture liquide. Les cellules ont été centrifugées pendant 5 minutes à 1 400 g. Les cellules ont été suspendues dans 5 ml de tampon de lyse (1% glycerol, 1% de NP40 et 1 mM d'EDTA) contenant 50 pg/ml de Phenylmethanesulfonylfluoride (PMSF), un inhibiteur de proteases, et incubées pendant 15 minutes à 4°C. Les échantillons ont été centrifugés pendant 15 minutes à 13 000 g (4°C) et ensuite le surnageant a été transféré dans un nouveau tube. En même temps, les billes de sépharose ont été préparées comme suit: 200 pi de la protéine A sépharose ou de protéine G sépharose ont été lavées trois fois avec 1 ml de tampon de lyse. Les billes ont été remises en suspension dans 300 pi de tampon de lyse. Ensuite 100 pi de ces billes ont été ajoutés à 400 pi de tampon de lyse comme contrôle et 200 pi ont été ajoutées à 800 pi de tampon de lyse contenant 50 pi de l'anticorps H36 ou H72. Les tubes contenant les billes ont été déposées sur une plaque agitatrice pendant une heure à 4°C pour permettre aux anticorps de se lier aux billes. Les billes ont ensuite été lavées deux fois avec 500 pi de tampon de lyse et resuspendues dans un volume total de 300 pi. Par la suite, 50 pi des billes contenant des anticorps H36 ou H72 ont été mélangés avec 630 pi du surnageant. Ce mélange a été incubé pendant 16 heures à 4°C pour être ensuite lavé trois fois avec 500 pi de tampon de lyse. Les protéines immunoprécipitées attachées aux anticorps H36 et H72 ont été séparées par electrophorèse dans un gel de polyacrylamide contenant 0,4% de SDS (SDS-PAGE).
3.6. Electrophorèse en gel de polyacrylamide
L'électrophorèse est une technique permettant de séparer des molécules en faisant migrer celles-ci dans une matrice d'agarose ou de polyacrylamide sous l'action d'un champ électrique. La séparation de molécules comme les protéines et de l'ADN est l'une des principales utilisations de cette technique. L'électrophorèse des protéines en condition dénaturante s'effectue en présence d'un détergent anionique, le sodium dodécylsulfate (SDS), qui se fixe à tous les types de molécules protéiques et provoque la denaturation des protéines sous une forme linéaire. La séparation des protéines par SDS-PAGE se base donc sur une seule propriété à savoir leur masse moléculaire (Laemmli 1970).
Le système Laemmli (Laemmli 1970) a été utilisé pour la séparation des protéines immunoprécipitées par les anticorps. Le gel de polyacrylamide a été coulé entre deux plaques de verre séparées de 1 mm par deux entretoises. Ce gel est constitué de deux sections distinctes: la partie inférieure du gel s'appelle le gel de résolution (Tableau 1) et la partie supérieure du gel est nommée le gel de concentration (Tableau 2). Celui-ci (pH 6,8) a une concentration d'acrylamide plus faible tandis que le gel de résolution possède une concentration plus élevée et un pH plus basique de 8,6. Le gel de résolution est coulé en premier et polymérisé avant que soit coulé et polymérisé le gel de concentration. Un peigne a été utilisé pour créer des puits pendant la polymérisation. Les plaques de verre contenant le gel ont été installées verticalement dans une chambre à electrophorèse contenant un tampon (196 mM glycine, 0,1% SDS, 50 mM Tris-HCl pH 8,3). Les protéines immunoprecipitiées ont été mélangées avec le tampon Tex (2% SDS, 5% p-mercaptoéthanol, 10% glycerol, 62,5 mM Tris-HCl pH 6,8) et elles ont été chargées dans les puits du gel. Les protéines ont été migrées à 120V pendant lh45 afin de les séparer selon leur poids moléculaire.
Tableau 1. Composants du gel de résolution Ge de résolution ( 2 X 1 mm) Produits 7% 7,5% 8% 8,5% 10% 12% 12,5% 13% 14% 15% 20 % Eau (ml) 5,2 5,0 4,8 4,7 4,2 3,5 3,3 3,2 2,8 2,5 0,8 Buffer A (ml) 2,5 2,5 2,5 2,5 2,5 2,5 2,5 2,5 2,5 2,5 2,5 Acrylamide 30%(ml) 2,3 2,5 2,7 2,8 3,3 4,0 4,2 4,3 4,7 5,0 6,7 APS 10% (ul) 60 60 60 60 60 60 60 60 60 60 60 TEMED (ul) 6 6 6 6 6 6 6 6 6 6 6 Volume final (ml) 10 10 10 10 10 10 10 10 10 10 10
Buffer A : 18,45g de TRIS-HCL, 77,0g de TRIS-Base, 2,0g de SDS, pH : 8,8, Filtrer millipore : 0,22 pm
Tableau 2. Composants du gel de concentration
Gel de concentration 2 gels 4 gels
Eau (ml) 6,5 9,75 Buffer C (ml) 2,5 3,75 Acrylamide 30% (ml) 1 1,5 APS 10% (ul) 60 90 TEMED (ul) 20 30 Volume final (ml) 10 15 Buffer C : 30g de TRIS-Base (0,5M), 2g SDS (0,4%)
3.7. Coloration du gel de polyacrylamide
Une fois l'électrophorèse terminée, les gels ont été transférés dans des cassettes de plastique et rincés trois fois avec de l'eau ultrafiltrée. La coloration s'obtient en ajoutant 25 ml d'une solution de bleu de Coomassie (500 ml de methanol 100%, 100 ml d'acide acétique glacial, 400 ml d'eau, lg de bleu de Coomassie). Les cassettes contenant des gels ont été déposées sur une plaque agitatrice pendant 15 minutes à température ambiante et ensuite lavées trois fois avec de l'eau stérile. Pour enlever l'excédant de colorant sur les gels, 25 ml de solution décolorante (10% acide acétique glacial, 10% methanol) a été ajouté à la cassette avant d'être déposé sur une plaque agitatrice. Le tampon de décolorant a été renouvelé périodiquement jusqu'à ce que les bandes deviennent visibles sur les gels.
3.8. Spectrométrie de masse
La spectrométrie de masse est une technique souvent employée pour la caractérisation des protéines et pour déterminer leurs compositions (Aebersold et Mann 2003). Les protéines séparées via electrophorèse sont isolées du gel et digérées par des proteases
comme la trypsine ou la pepsine. Ensuite les protéines sont ionisées par les méthodes
Electrospray Ionization (ESI) ou Matrix-Assisted Laser Desorption-Ionization (MALDI). Enfin les échantillons sont analysés par le spectromètre pour obtenir un
spectre de la masse des peptides avec étalonnage externe (Aebersold et Mann 2003).
Après la coloration du gel SDS-PAGE avec le bleu de Comassie, les bandes désirées
ont été découpées avec une lame de scalpel stérile et ont été gardées dans 100 pi d'eau ultrafiltrée à 4°C. L'analyse des échantillons en spectrométrie de masse a été effectuée à la plateforme de protéomique du CHUL (Québec Canada).
3.9. Analyse informatique des protéines identifiées
Le logiciel Scaffold (http://proteomesoftware.com) a été utilisé pour l'analyse des
résultats de l'identification des protéines à partir des résultats produits en spectrométrie de masse. Les applications disponibles sur NCBI
(http://ncbi.nlm.nih.gov/protein) ont été utilisées pour déduire si les protéines identifiées sont retrouvées dans d'autres organismes ou si elles sont uniques et spécifiques à l'amibe. Le logiciel disponible sur SMART (http://smart.embl-heidelberg.de/) a été employé pour identifier les domaines fonctionnels des protéines identifiées comme la présence d'un peptide signal, les domaines transmembranaires et d'autres domaines d'interaction ou fonctionnels (enzyme). Les applications disponibles sur Expasy (www.expasy.ch) ont permis de déterminer toutes les caractéristiques physico-chimiques des protéines (hydrophobicité, structures secondaires [hélice alpha, feuillet bêta, région flexible], etc.).
3.10. Immunobuvardage (Western Blot)
La technique de l'immunobuvardage (western blot) permet de détecter une protéine spécifique en utilisant une sonde chémiluminescente dans un extrait cellulaire de protéines tout en donnant des informations sur leurs tailles. Cependant, cette méthode dépend de l'utilisation d'un anticorps de haute qualité dirigée contre la protéine désirée (Towbin et al. 1979).
Pour obtenir un extrait des protéines totales de l'amibe D. discoideum, lxlO6cellules ont été récoltées par centrifugation à 1 400 g pendant 5 minutes et mélangées avec 0,5 volume de tampon Tex 3x (18,8 ml de tampon C, 15 ml glycerol et 3,45 g SDS). Les extraits ont été dénaturés à 95°C pendant 5 minutes et puis les protéines ont été séparées sur des gels de polyacrylamide (SDS-PAGE) pour être ensuite transférées sur une membrane de nitrocellulose sous une tension de 100 V pendant 1 heure. Les membranes ont été immergées dans 50 ml de tampon TBS (10 mM de Tris pH 7,4 et 150 mM de NaCI) pendant 5 minutes puis elles ont été bloquées avec le tampon TBSM (10 mM de Tris pH 7,4, 150 mM de NaCI et 7% de lait écrémé) pendant 16 heures à 4°C. Les membranes ont été lavées à cinq reprises pendant 5 minutes avec une solution de TBST (10 mM de Tris pH 7,4, 150 mM de NaCI et 0,1% de Tween 20). Afin de détecter la protéine spécifique, les anticorps primaires, ceux détectant les protéines d'intérêt, ont été ajoutés à la solution contenant la membrane et incubés pendant 90 minutes à température ambiante. Pour les anticorps sous forme d'ascite comme H36, une dilution 1:20 000 dans 20 ml de TBST a été utilisée et pour les anticorps comme Anti-cprG, qui sont des surnageants de culture cellulaire, une dilution 1:50 dans 20 ml de TBST a été utilisée. Les membranes ont été lavées à trois reprises pendant 5 minutes avec du tampon TBST et ont été incubées avec l'anticorps secondaire (anti-IgG de souris conjugué à la peroxydase) à la concentration 1:20 000 dans 20 ml de TBST pendant une heure avec agitation à température ambiante. Les membranes ont été lavées six fois dans le tampon TBST (chaque lavage ayant une durée de 5 minutes). Pour observer les protéines ciblées par les anticorps primaires, 2 ml de la solution d'Enhanced Chemiluminescence (ECL) ont été ajoutés sur les
membranes pendant 5 minutes à la température ambiante. Celles-ci ont été transférées dans une chambre noire. Un film photosensible a été déposé sur chaque membrane et exposé pendant des périodes de 5 secondes, 30 secondes et 1 minute. Le développement de chaque film a permis de révéler la présence du signal lumineux produit par la peroxydase couplée à l'anticorps secondaire.
3.11. Préparation de l'ADN génomique
Les cellules D. discoideum ont été remises en suspension dans du milieu HL5 à une concentration d'au moins 1000 cellules/pl et un volume de cellule a été mélangé avec un volume de tampon de lyse [10 mM Tris, pH 8,3, 50 mM KC1, 2,5 mM MgCl., 0,45% Nonidet P-40 (NP40), et 0,45% Tween 20] contenant de la Proteinase K (20 pg/pl de PK pour chaque 25 pi de LysB) (Charette et Cosson 2004). Les mélanges de cellules ont été incubés à 95 °C pendant 1 minute pour inactivation.
3.12. Réaction de polymerase en chaîne (PCR)
La PCR est une méthode qui sert à amplifier de façon spécifique une région d'un ADN matriciel. Cette méthode est largement utilisée en biologie moléculaire. La PCR requiert les éléments suivants dans le milieu réactionnel: l'ADN polymerase, les désoxyribonucléotide triphosphate (dNTP), les amorces, le tampon de l'ADN polymerase contenant du MgCl? (Valasek et Repa 2005).
Les amorces utilisées pour amplifier le gène codant pour chacune des protéines d'intérêt identifiées par spectrométrie de masse ont été déterminées à partir des séquences publiées sur le National Center for Biotechnology Information (http://blast.ncbi.nlm.nih.gov) et sur Dictybase (http://dictybase.org/) (Tableau 3). Les gènes codants pour les protéines p20 (DDB0306889) et p35 (DDB0306604) ont été amplifiés par PCR en utilisant la Go Taq DNA Polymerase. Le Tableau 4 présente les concentrations requises pour chaque réaction. L'amplification de l'ADN pour les gènes DDB0306889 et DDB0306604 a été réalisée dans une appareil d'amplification
PCR (Master Cycler d'Eppendorf) sous les conditions de cycle suivantes: (1) 95°C pendant 2.30 minutes, (2) 95°C pendant 30 secondes, (3) 42,5°C pendant 30 secondes, (4) 68°C pendant 1-2 minutes, (5), 30 cycles des étapes 2 à 4, et 6) une étape d'extension terminale à 68°C pendant 10 minutes.
Tableau 3. Oligonucleotides utilisés pour faire l'amplification des gènes codants pour les protéines p35 et p20 par PCR
Nom de
l'amorce Séquence de l'amorce
P20-FWD1 GGATCCGGATCCAAAATGAACGGTCAAGGAAAGTATGTA P20-REV2 TCTAGA TCTAGA AAGGTTACCTTGATATGGTTCTTGGTA P35-FWD1 GGATCCGGATCCAAAATGGTATTAAGTTTATTAAATTAT
Tableau 4. Réactifs pour les réactions d'amplification PCR par échantillon de 20 pl Composante Volume (pl) 5X Tampon GoTaq 4 lOmMdNTP 1,6 20pM Amorce F WD 1,3 20pM Amorce REV 1,3 Eau 11 Go-taq ADN polymerase 0,1 ADN 1
3.13. Electrophorèse sur gel d'agarose
Les fragments d'ADN produits par les réactions PCR sont visualisés par electrophorèse sur gel d'agarose 1% qui permet la séparation des molécules d'ADN en fonction de leur taille. Ce gel est préparé par dissolution par chauffage de 0,5 g d'agarose dans 50 ml de tampon TBE IX. L'ajout de bromure d'éthidium (1 pg/ml) permet la visualisation des bandes pendant l'exposition du gel aux rayonnements ultraviolets. Le mélange est coulé dans une cassette de plastique et des peignes sont insérés dans le gel pour former des puits de chargement des échantillons. Après polymérisation, le gel est transféré à l'intérieur d'une cuve à electrophorèse contenant du tampon TBE IX et par la suite 5 pl de chaque échantillon est déposé dans chaque puits. La taille des fragments d'ADN est déterminée en ajoutant 5 pl de marqueur l'ADN (lkb à 10 kb) sur le gel. Une tension de 80 V est appliquée pour une durée de
45 minutes. Enfin, le gel est exposé aux rayons ultraviolets pour visualisation des bandes d'ADN amplifiés
3.14. Clonage
Pour étudier la fonction des protéines identifiées et pour, entre autres, s'en servir comme marqueur membranaire, il devient intéressant d'exprimer ces protéines dans l'amibe sous une forme fluorescente en produisant des protéines de fusion avec la protéine fluorescence verte (Green Fluorescent Protein, GFP). Plusieurs vecteurs sont disponibles pour permettre ce genre d'approche comme les plasmides de la famille pDM (Fig. 11) qui ont été développés récemment et qui permettent de faire des protéines de fusion avec la GFP dans un plasmide optimisé pour l'expression dans D. discoideum (Veltman et al. 2009).
Figure 11. Carte génétique du plasmide pDM323. Ce plasmide est optimisé pour l'expression les protéines avec la GFP dans D. discoideum. Figure tirée de Veltman et al. 2009.
gene for antibiotic resistance
Foreign DNA
region of interest DNA insertion actenalchromosome Bacteria platted on medium + antibiotic
L
Clone
Only bacteria containing recombinant DNA grow Culturel
P DNA
y w \ purification _^ Q
Cloning into a plasmid
Figure 12. Les étapes nécessaires de processus du clonage d'un fragment d'ADN dans un plasmide. L'image présentée ici a été tirée du site internet http://www.accessexcellence.org.
Après l'amplification des gènes d'intérêt par PCR, les amplicons ont été purifiés par la méthode de purification sur colonne (QIAGEN - QIAquick PCR Purification Kit). Les gènes codants pour les protéines p20 et p35 ainsi que les plasmides pSP73 (Fig. 13) ont été coupés par les enzymes de restriction pour ensuite être ligaturés ensemble. La ligature a été réalisée en mélangeant 100 ng de vecteur et d'insert dans un ratio 1:3 avec une unité d'ADN T4 ligase et son tampon (50 mM Tris-HCl, 10 mM MgC12, 1 mM ATP, 10 mM Dithiothreitol). Le mélange a été incubé à 14°C pendant 16 heures. Une aliquote de 10 pl du mélange des ligatures a été utilisée pour transformer 50 pl de cellules compétentes (E. coli MC106U en faisant un choc thermique (42°C pour 45 secondes). Les cellules transformées ont été étalées sur les pétris contenant une gélose Luria Bertani (LB) contenant 100 pg/ml d'ampicilline et celles-ci ont été incubées pendant 16 h à 37°C. Les colonies obtenues ont été transférées dans des tubes contenant 2 ml du milieu LB avec ampicilline (100 pg/ml) et incubées pendant 16 h à 37°C. Les plasmides ont été purifiés à l'aide de la trousse de purification QIAprep et ont été incubés en présence des enzymes de restriction afin de libérer l'insert. La migration sur gel d'agarose a permis de déterminer les plasmides possédant les inserts. L'analyse par séquençage a été exécutée sur les échantillons positifs par le service d'analyses biomoléculaires (IBIS, Québec). La vérification de la séquence a été faite afin de s'assurer de l'absence de mutations dans le gène. L'ADN des clones positifs identifiés a été ensuite coupé avec les enzymes de restrictions (BamHI et Xbal) pour libérer les inserts contenant les gènes pour être ensuite ligaturé dans le vecteur d'expression pDM323 (Veltman et al. 2009) en répétant la procédure décrite ci-dessus.
Xrryu IKK'-» Scat 1690 P w l 15». Fspl 143? 5.;' ' I i stan Bai i i stan EcoRV 14 19 FcoRi .4 Saci M Kpnl 40 Sf7!_ 4? P _ f | , 45 Xtwl 51 SM i 57 .Ace I 58 Psfl Stooi H..-U II 75 PWull 83 X ' . Î I ■ .
-Figure 13. Carte génétique du plasmide pSP73. Ce plasmide simple a été utilisé comme plasmide de clonage primaire. L'image de cette figure a été tirée du site internet http://www.promega.com.
3.15. Electroporation
L'électroporation est une méthode qui consiste à introduire de l'ADN étranger dans une cellule hôte à travers la membrane. Cette méthode est basée sur l'utilisation d'une impulsion électrique qui est capable de provoquer la rupture réversible de la membrane pour introduire l'ADN plasmique dans la cellule (Gunn et al. 1995).
Pour chaque electroporation entre lxlO7 et 2xl07 cellules de D. discoideum sont nécessaires. Les cellules sont préparées en ajoutant 10 ml du tampon d'électroporation (5 ml de tampon KH2PO4, 5 ml de tampon Na2HP04, 42,8 g de sucrose, 490 ml d'eau distillée) à une culture fraiche de cellules. Celles-ci ont ensuite été transférées dans un tube Falcon de 15 ml et ont été centrifugées à 1400 g pendant 5 minutes. Le surnageant a été éliminé et le culot cellulaire a été lavé dans le tampon d'électroporation et centrifugé une fois à 1400 g pendant 5 minutes. Le tampon a été retiré et le culot de cellules a été dilué avec du tampon d'électroporation. Un volume de 500 pl de tampon contenant les cellules et l'ADN plasmidique a été ajouté dans les cuvettes (#5520, 2 mm) d'électroporation. La cuvette a été laissée 5 minutes dans la glace et par la suite placée
dans l'appareil d'électroporation. Une impulsion électrique de 1,2 kV a été effectuée une fois sur les cellules. Immédiatement après, les cuvettes ont été mises sur la glace pendant
15 minutes. Pendant ce temps, une concentration de 1 pl de MgCL (lOOmM) et 1 pl de CaCL (lOOmM) a été ajouté au centre de grandes boîtes de Pétris. Le contenu de chaque cuvette a été transféré dans ces boîtes de Pétris et mélangé à ces deux solutions ioniques puis incubé pendant 15 minutes. Finalement, un volume de 35 ml de HL5 contenant de la tetracycline (15 pg/ml) a été ajouté aux boîtes de Pétri avant de les incuber à 21°C pendant 24 h. Afin de sélectionner les amibes ayant incorporé le plasmide, 5 ml de milieu HL5 contenant l'antibiotique G418 a été ajouté à chaque culture afin d'obtenir une concentration finale de 10pg/ml de l'antibiotique. Ensuite, les boîtes de Pétris ont été gardées pendant une semaine à 21°C jusqu'à l'apparition de colonies d'amibe au fond de la boîte de Pétri.
Chapitre 4
4.1. Localisation cellulaire des protéines reconnues par
H36 et H72
Afin de vérifier si la protéine reconnue par l'anticorps H72 est présente sur les CMLs, une analyse en immunofluorescence a été réalisée en utilisant la microscopie à épifluorescence. Nous savions déjà que la protéine reconnue par l'anticorps H36 est présente sur les CMLs (V. Paquet, communication personnelle). Nous avons donc utilisé cet anticorps comme contrôle positif (Fig. 14). De même, l'anticorps H161 a été utilisé comme contrôle négatif parce qu'il ne détecte pas les CML sécrétés (Fig. 14) (V. Paquet, communication personnelle). Les résultats de l'immunofluorescence montrent que l'antigène reconnu par l'anticorps H72 est présent sur la membrane de l'amibe et les endosomes de recyclage comme déjà décrit (Charette et al. 2006), mais il n'est pas présent sur les CMLs sécrétés dans le milieu (Fig. 14).
DIC
IF
DAPI
H36 à
___fc_.^^H
II
B
H 72
C
H161
Figure 14. Analyse par immunofluorescence de CMLs pour la présence des protéines reconnues par les anticorps H36, H72 et H161 en co-culture avec la bactérie À. aerogenes. La protéine reconnue par H36 (A) est trouvée à la membrane plasmique des cellules et sur les CML sécrétés. La flèche blanche montre l'anticorps associé à un CML. La protéine reconnue par H72 (B) est trouvée sur certaines membranes des cellules, mais n'est pas présente sur les CMLs. Il en est de même pour la protéine reconnue par H161 (C).
4.2. Isolement des protéines
La stratégie employée pour identifier les protéines reconnues par H36 et H72 s'appuie sur la méthode d'immunoprécipitation en utilisant les protéines A et G-sépharose. Les protéines associées aux anticorps H36 et H72 ont été séparées par electrophorèse dans un gel de polyacrylamide (SDS-PAGE) 12 %. Ces gels ont été colorés avec une solution de bleu de Coomassie. Une bande de 46 kDa spécifiquement précipitée par l'anticorps H36 (Fig. 15) et une bande de 25 kDa (Fig. 16) par l'anticorps H72 (étant donné que ces bandes n'étaient pas dans les échantillons contrôles) ont été découpées et envoyées pour analyse en spectrométrie de masse à la plateforme de protéomique du CHUL. Dans tous les gels, des bandes protéiques à environ 30 kDa ont été observées et le résultat de spectrométrie de masse montre que ces bandes correspondent aux différentes formes de la protéine Discoïdine. kDa 1 2 3 4 5 6 7
*«t "•■
« « » _■f.
Figure 15. Gel SDS-PAGE suite à une immunoprécipitation et montrant la protéine reconnue par l'anticorps H36. Identification des lignes sur le gel : 1) marqueur de poids moléculaire, 2) lysat cellulaire plus protéine G, 3) protéine G plus l'anticorps H36, 4) lysat cellulaire plus protéine G et l'anticorps H36, 5) lysat cellulaire plus protéine A, 6) protéine A plus l'anticorps H36, 7) lysat cellulaire plus protéine A et l'anticorps H36. L'antigène H36 est identifié par la flèche. * = contamination, ■ = protéines potentiellement intéressantes pour analyses spectrométriques futures.
Figure 16. Gel SDS-PAGE suite à une immunoprécipitation et montrant la protéine reconnue par l'anticorps H72. Identification des lignes sur le gel : 1) Marqueur de poids moléculaire, 2) lysat cellulaire plus protéine G, 3) protéine G plus l'anticorps H72. 4, 5 et 6) lysat cellulaire plus protéine G et l'anticorps H72. L'antigène de H72 est identifié par la flèche.
Une immunoprécipitation avec l'anticorps H72 a aussi été faite en utilisant la protéine A-sépharose, mais il n'y avait pas de bande correspondant à la protéine attendue sur le gel de polyacrylamide (SDS-PAGE).
4.3. Identification et analyse des protéines
immunoprécipitées
L'analyse des résultats obtenus à partir de la spectrométrie de masse indique que la protéine identifiée par l'anticorps H36 est la Cysteine Proteinase 7 (CP7) aussi nommée cprG. Le gène de cprG (Fig. 17) est situé entre les positions 1 740 048 pb et 1 741 532
sur le chromosome 3 de D. discoideum. La protéine codée par le gène cprG est une glycoprotéine de 47 kDa, de 460 aa qui est associée aux lysosomes (http://dictybase.org/). Cette protéine possède un peptide signal (Fig. 18) et sa structure quaternaire implique des ponts disulfures (Ord et al. 1997).
17 Genes 40k~ 1741k
+
cprG Gene Models DDB0215005 tRNfls ncRNflsFigure 17. Le gène codant pour la protéine cprG. Ce gène situé sur le chromosome 3 est d'une longueur de 1484 pb et contient un intron. L'image de cette figure a été tirée de http://dictybase.org/.
Figure 18. Domaines fonctionnels de la protéine cprG. Un peptide signal (trait rose) est situé dans la partie N-terminale de cette protéine. L'image de cette figure a été tirée de http://smart.embl-heidelberg.de.