HAL Id: hal-02385072
https://hal.archives-ouvertes.fr/hal-02385072
Submitted on 11 Feb 2021HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Species: A Small-Molecule Mimic of Amine Oxidases
Martine Largeron, Anne Neudörffer, Maurice-Bernard Fleury
To cite this version:
Martine Largeron, Anne Neudörffer, Maurice-Bernard Fleury. Oxidation of Unactivated Primary Aliphatic Amines Catalyzed by an Electrogenerated 3,4-Azaquinone Species: A Small-Molecule Mimic of Amine Oxidases. Angewandte Chemie International Edition, Wiley-VCH Verlag, 2003, 42 (9), pp.1026-1029. �10.1002/anie.200390263�. �hal-02385072�
Oxidation of Unactivated Primary Aliphatic Amines
Catalyzed by an Electrogenerated 3,4-Azaquinone
Species: a Small Molecule Mimic of Amine Oxidases.
Martine Largeron*, Anne Neudorffer, and Maurice-Bernard Fleury.
[*]Prof. M.-B. Fleury, Dr. M. Largeron, Dr. A. Neudorffer Laboratoire de Chimie Analytique et Electrochimie, UMR 8638 CNRS-Université René Descartes
Faculté des Sciences Pharmaceutiques et Biologiques
4, Avenue de l’Observatoire, 75270 Paris Cedex 06 (France) Fax : 01 44 07 35 88
E-mail : largeron@pharmacie.univ-paris5.fr
The copper amine oxidases are ubiquitous enzymes that occur in bacteria, yeast, plants, and mammals. These enzymes catalyze the two-electron oxidative deamination of a primary amine to produce the corresponding aldehyde and ammonia, with subsequent two-electron reduction of dioxygen to hydrogen peroxide. To carry out this reaction, copper amine oxidases, which belong to the new class of proteins designated quinoproteins, contain two cofactors: an organic cofactor, 2,4,5-trihydroxyphenylalanine quinone (TPQ,
Figure 1), and a cupric ion.[1] This class also includes
bacterial methylamine oxidases which use a peptide-bound tryptophan tryptophylquinone (TTQ)[2] and lysyl oxidase, a
mammalian copper amine oxidase, whose active site has been identified as lysine tyrosylquinone (LTQ).[3]
It is well established that TPQ catalyzes the conversion of an amine substrate to an aldehyde through a pyridoxal-like transamination mechanism, which results in the reduction of TPQ to an aminoquinol form.[4] Although
there is no question regarding the crucial role of copper in the biogenesis of TPQ from its tyrosine precursor, the role of the copper cofactor during amine oxidation is, however, less well understood. Recent intensive studies have shown that an electron transfer from the cofactor to the metal is not required during the catalytic turnover, so that the metal essentially stabilizes a reduced oxygen intermediate.[5] These results are supported by the fact
that the mature form of bovine lysyl oxidase retains its catalytic function in the absence of copper.[6]
Substantial mechanistic information has been obtained through catalytic aerobic deamination of benzylamine mediated by synthetic models of natural quinones, which confirm the occurrence of the transamination mechanism.[7]
However, these models failed to oxidize unactivated primary aliphatic amines under the same experimental conditions,
with the exception of a recent study showing that the metal ion complex of TTQ model compound was able to oxidize aliphatic amines, in anhydrous organic media, whereas no reaction took place in the absence of the metal ion.[8]
We recently showed that electrogenerated 3,4-iminoquinone 1ox (Figure 1) acts as an efficient catalyst for the autorecycling oxidation of benzylamine through the transamination process reported for amine oxidase cofactors. The catalytic cycle produced the reduced catalyst 1red and N-benzylidene-benzylamine as the product of benzylamine oxidation in 3200% yield (based on the initial concentration of 1red.[9] In contrast to other
existing amine oxidase model cofactors, 1ox was also very active toward aliphatic amines. Herein, we report for the first time the catalytic oxidation of unactivated primary aliphatic amines by a small molecule enzyme mimic, in the absence of metal ion.
Unstable 3,4-iminoquinone 1ox was electrogenerated from 3,4-aminophenol 1red by using anodic-controlled potential electrolysis at a platinum electrode in deaerated methanol that contained an excess of primary aliphatic amine. At a potential at which the 3,4-aminophenol 1red could be oxidized to the iminoquinone form 1ox (see Experimental Section),[10] the anodic current remained
unchanged for a long time, consistent with steady-state catalytic behaviour. Accordingly, a high value was found for the total number of electrons (n) transferred per molecule of 1red in the catalytic process. These results indicated that the 1red/1ox system behaved as a redox mediator for the indirect electrochemical oxidation of unactivated primary aliphatic amines to the corresponding alkylimines, according to the transamination mechanism reported in Scheme 1.[9] After exhaustive electrolysis, the
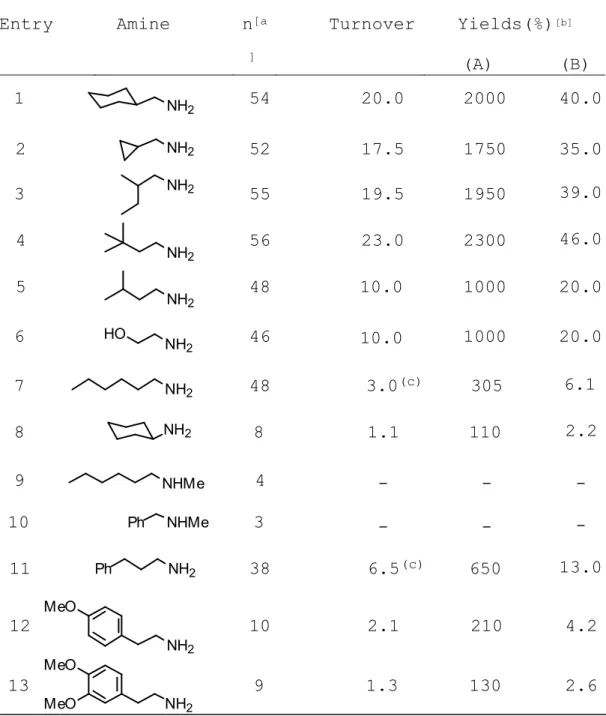
catalyst 1ox was lost irreversibly as corroborated by the anodic current, which remained negligible upon further addition of amine substrate. Unstable alkylimine was isolated in terms of the corresponding aldehyde 2,4-dinitrophenylhydrazone (DNPH) obtained upon workup of the oxidized solution with 2,4-dinitrophenylhydrazine. Table 1 shows the effects of varying the structure of the amine substrate.[11] The most efficient substrates for 1ox were
primary alkyl amines (Table 1, entries 1-6), with high yields of isolated DNPH ranging from 1000 to 2300% (relative to the mediator). With n-hexylamine (Table 1, entry 7), although a high value of 48 was found for n, the catalytic oxidation of more hydrophobic longer-chain amine produced DNPH in only 305% yield (based on 1red), along with 255% of a second product identified as the osazone
(1,2-bis-DNPH). Interestingly, as for the enzymes themselves, -branched amines like cyclohexylamine (Table 1,entry 8) were found to be poor substrates for the model catalyst 1ox, whereas secondary amines (Table 1, entries 9 and 10) were not reactive at all.
Primary amines bearing aromatic substituents also underwent deamination, though the turnover was much lower on account of a more rapid deterioration of the catalyst. With phenylpropylamine (Table 1, entry 11), a value of 38 was found for n. Similarly to n-hexylamine (Table 1, entry 7), both DNPH (650%) and 1,2-bis-DNPH (100%) could be isolated at the end of the catalytic process. Further investigations would be necessary to justify the formation of the osazone which seems favored when starting amines are not sterically encumbered by and branching.
With ring-substituted phenylethylamines, the catalytic process ceased after roughly 2 turnovers (Table 1, entries 12 and 13). Close inspection of the exhaustively oxidized solution revealed that electrogenerated 3,4-iminoquinone
1ox was trapped with the tautomeric enamine form of the
alkylimine extruded during the catalytic process (Scheme 2, step 7) through an inverse-electron-demand Diels-Alder reaction,[12] affording unstable 1,4-benzoxazine derivatives
isolated as stable products 2 and 3 in 50 and 65% yields, respectively, after a subsequent two-electron oxidation reaction (Scheme 2, step 9). Interestingly, this multi-step one-pot electrochemical procedure could lead to novel neuroprotective agents, due to the structural similarity of
2 and 3 with earlier reported 1,4-benzoxazine derivatives.[13]
In summary, we have demonstrated for the first time that unactivated primary aliphatic amines can be efficiently oxidized by a synthetic model cofactor of amine oxidases in the absence of metal ion. The catalyst 1ox exhibits the same substrate specificity as the copper amine oxidases themselves, that is, poor reactivity with -branched primary amines and no reactivity toward secondary amines. The reaction displays two features that are most often associated with enzymatic systems: a) the reaction is likely enhanced through the participation of neighboring substituents, as they prevented the competing formation of Michael adducts (in this case, the benzoyl and 2-hydroxy groups); b) the presence of an active hydroxyl proton very probably constitutes an essential component of the catalytic activity (in this case, the 2-hydroxy proton, analogous to that of TPQ).[14] We are currently developing
2-hydroxy group to convert a catalytic inert species into a highly effective enzymatic prosthetic group.
Experimental Section
General procedure - Controlled-potential electrolysis
was carried out in a cylindrical, three-electrode, divided cell (9 cm diameter), using an electronic potentiostat. In the main compartment, a platinum grid (60 cm2 area) served as the anode (working electrode). A platinum sheet was placed in the concentric cathodic compartment (counter electrode), which was separated from the main compartment with a glass frit. The reference electrode was an aqueous saturated calomel electrode (SCE), which was isolated from the bulk solution in a glass tube with a fine-porosity frit. The electrolyte solution [0.04 mol.L-1 lithium perchlorate or tetraethylammonium perchlorate indifferently, in methanol] was poured into the anodic and the cathodic compartments, as well as into the glass tube that contained the SCE electrode. 3,4-Aminophenol[15] 1red
(0.1 mmol) and an excess of primary aliphatic amine (5 mmol) were added to the solution in the main compartment (250 mL), and the resulting solution was then oxidized under nitrogen, at room temperature, at + 600 mV vs SCE
(initial current 30-40 mA), i.e. at a potential following the anodic peak observed in cyclic voltammetry, characteristic of the two-electron oxidation of 1red to
1ox. After exhaustive electrolysis (6-10h, n = 8 to 56),
i.e., when a negligible value of the current was recorded
(0.5-1.0 mA), the solution was worked up by addition of 2.5 mmol of 2,4-dinitrophenylhydrazine reagent (in 5 mL of H2SO4, 15 mL of EtOH and 5 mL of water) because 5 mmol of
primary amine gave 2.5 mmol of alkylimine (scheme 1). After 1h, the resulting solution was concentrated to 40 mL. The solid was collected by filtration, washed with water, and dried in a vaccuum dessicator. The identity and purity of DNPH was confirmed by TLC and 1H NMR, after comparison with
an authentical sample. In the case of n-hexylamine or phenylpropylamine, the solid was chromatographed on silica gel to separate DNPH from 1,2-bis-DNPH. Control studies indicated that the amount of aldehyde produced either from simple autoxidation or from electrochemical oxidation of the starting amine in the absence of catalyst was negligible.
Isolation of 1,4-benzoxazines 2 and 3 - After
exhaustive electrolysis (6h, n = 9 or 10), the electrolysis solution was poured into a molar acetic acid buffered aqueous solution of pH 4.8 (50 mL). The resulting hydroalcoholic solution was concentrated to remove
methanol. The yellow solid, identified as the 1,4-benzoxazine derivative, was collected by filtration, washed with water, and dried in a vacuum dessicator.
2. M.p. 162°C; 1H NMR (300 MHz, CDCl3): = 2.25 (m, 1H), 2.75 (t, J = 6 Hz, 2H), 3.15(m, 2H), 3.75 (s, 3H), 3.90 (s, 3H), 5.90 (d, J = 8 Hz, 1H), 6.55 (d, J = 9 Hz, 1H), 6.75(d, J = 8 Hz, 2H), 6.95 (d, J = 8 Hz, 2H), 7.05 (d, J = 8 Hz, 2H), 7.45 (d, J = 9 Hz, 1H), 7.55 (m, 3H), 7.70 (d, J = 8 Hz, 2H), 8.00 (d, J = 8 Hz, 2H), 13.10 (s, 1H); 13C NMR (75 MHz, CDCl3): = 35.7, 46.2, 55.2, 55.3, 81.2, 108.3, 113.9, 114.2, 122.7, 127.9, 128.3, 128.9, 129.2, 129.6, 131.2, 131.6, 133.2, 138.2, 150.7, 154.5, 158.1, 160.3, 161.9, 202.2; MS-DCI: m/z : 509 MH+]. References
[1] a) J.P. Klinman, Chem. Rev. 1996, 96, 2541-2561; b) J.P. Klinman, J. Biol. Chem. 1996, 271, 27189-27192; c) J.A. Stubbe, W.A. Van der Donk, Chem. Rev. 1998, 98, 705-776; d) J.P. Klinman, Proc. Nat. Acad. Sci. U.S.A. 2001, 98, 14766-14768; e) M.A. Halcrow, Angew. Chem. 2001, 113, 358-362; Angew. Chem. Int. Ed. 2001, 40, 346-349.
[2] Y.-L. Hyun, V.L. Davidson, Biochemistry, 1995, 34, 816-823 and references therein.
[3] S.X. Wang, N. Nakamura, M. Mure, J.P. Klinman, J. Sanders-Loehr, J. Biol. Chem. 1997, 272, 28841-28844. [4] M. Mure, S.A. Mills, J.P. Klinman, Biochemistry 2002,
41, 9269-9278.
[5] a) S.A. Mills, J.P. Klinman, J. Am. Chem. Soc. 2000,
122, 9897-9904; b) B. Schwartz, A.K. Olgin, J.P.
Klinman, Biochemistry 2001, 40, 2954-2963; c) S.A. Mills, Y. Goto, Q. Su, J. Plastino, J.P. Klinman,
Biochemistry 2002, 41, 10577-10584; E.I. Solomon, P.
Chen, M. Metz, S.-K. Lee, A.E. Palmer, Angew. Chem.
2001, 113, 4702-4724; Angew. Chem. Int. Ed. 2001, 40,
4570-4590.
[6] C. Tang, J.P. Klinman, J. Biol. Chem. 2001, 276, 30575-30578.
[7] a) E.J. Rodriguez, T.C. Bruice, J. Am. Chem. Soc. 1989,
111, 7947-7956; b) S. Itoh, M. Mure, M. Ogino, Y.
Ohshiro, J. Org. Chem. 1991, 56, 6857-6865; c) M. Mure, J.P. Klinman, J. Am. Chem. Soc. 1995, 117, 8707-8718 ; d) Y. Lee, L.M. Sayre, J. Am. Chem. Soc. 1995, 117, 11823-11828 and references therein; e) S. Itoh, N. Takada, S. Haranou, T. Ando, M. Komatsu, Y. Ohshiro, S. Fukuzumi, J. Org. Chem. 1996, 61, 8967-8974 and references therein; f) K.Q. Ling, J. Kim, L.M. Sayre, J.
[8] S. Itoh, M. Tanaguchi, N. Takada, S. Nagatomo, T. Kitagawa, S. Fukuzumi, J. Am. Chem. Soc. 2000, 122, 12087-12097.
[9] M. Largeron, M.-B. Fleury, J. Org. Chem. 2000, 65, 8874-8881.
[10] The cyclic voltammogram of 3,4-aminophenol 1red (0.1 mmol), recorded under our experimental conditions (see Experimental Section), showed an oxidation peak Pa at + 500 mV vs SCE, the sweep rate being 0.5 V s-1. The
addition of 5 mmol of primary amine had two effects: first, a shift of the peak Pa to 0.0 mV vs SCE, due to the ionization of the 4-hydroxyl group; second, an increase of the anodic peak intensity suggesting that 3,4-iminoquinone 1ox can act as a catalyst even at the time scale of cyclic voltammetry, competitively with the succeeding oligomerization. For similar behaviour of quinonoid species see : K. Kano, M. Nakagawa, K. Takagi, T. Ikeda, J. Chem. Soc. Perkin Trans 2 1997, 1111-1119 and references therein.
[11] In some cases, yield of DNPH was lower than that expected from the value of n due to the partial conversion of unstable alkylimine into volatile aldehyde, at the time scale of anodic electrolysis.
[12] Recently, we have evidenced similar inverse electron demand Diels-Alder reactions of 1ox with R1R2CHCH2NH2
amines, but under different experimental conditions which have been defined as optimal for the synthesis of 1,4-benzoxazine derivatives; see: M. Largeron, A. Neudorffer, M. Vuilhorgne, E. Blattes, M.-B. Fleury,
Angew. Chem. 2002, 114, 852-855; Angew. Chem. Int. Ed.
2002, 41, 824-827.
[13] M. Largeron, B. Lockhart, B. Pfeiffer, M.-B. Fleury, J.
Med. Chem. 1999, 42, 5043-5052.
[14] M. Mure, J.P. Klinman, J. Am. Chem. Soc. 1993, 115, 7117-7127.
[15] Reduced catalyst 1red was synthesized from commercially available 2-nitroresorcinol through a four-step sequence previously reported; see: R. Larget, B. Lockhart, B. Pfeiffer, A. Neudorffer, M.-B. Fleury, M. Largeron,
Legends for Figure and Schemes
Figure 1 - Quino-cofactors derived from protein-bound
tyrosine (TPQ, LTQ), tryptophan (TTQ), together with 3,4-iminoquinone model cofactor 1ox.
Scheme 1 - Mechanism of catalytic oxidation of primary
aliphatic amines mediated by electrogenerated 3,4-iminoquinone model cofactor 1ox.
Scheme 2 - Inactivation of model cofactor 1ox by
ring-substituted phenylethylamine substrates, leading to 1,4-benzoxazine derivatives 2 and 3.
Table 1. Catalytic oxidation of primary aliphatic amines
mediated by the electrogenerated 3,4-iminoquinone 1ox model
cofactor.
Entry Amine n[a ]
Turnover Yields(%)[b] (A) (B)
[a] total number of electrons transferred per molecule of mediator; [b] determined by weight of the aldehyde derivative DNPH, relative to
the mediator (A) and to the amine substrate (B), respectively; [c] the
1,2-bis-DNPH derivative was formed also; turnover: 2.5 (entry 7); 1.0 (entry 11). NH2 NH2 NH2 NH2 NH2 NH2 HO NH2 NHMe Ph NH2 NH2 MeO NHMe NH2 MeO MeO NH2 1 2 3 4 5 6 7 8 9 10 11 12 13 54 52 55 56 48 46 48 8 4 3 38 10 9 2000 1750 1950 2300 1000 1000 305 110 -650 210 130 Ph 20.0 17.5 19.5 23.0 10.0 10.0 3.0(c) 1.1 -6.5(c) 2.1 1.3 40.0 35.0 39.0 46.0 20.0 20.0 6.1 2.2 -13.0 4.2 2.6
O O HN O N H H N O (CH2)4 LTQ O O N N H TTQ O HO HN O O TPQ HN O OH O 1ox Figure 1 O HN NH O H
Scheme 1 Ph O OH HN O Ph O OH H2N O NH NH3 Ph O OH N O R H+ Ph O OH N O R Ph O OH H2N O -2e - H+ 1red 1ox Ph O OH H N O R HN R R N R R R NH2 alkylimine 1) 2) 3) 4) 5) 6) R NH2
-Alkylimine 7) Ar N H Ar HN O O H O H N O O H O Ar HN N O O H O Ar HN 8) 9) -2e-,-2H+ 2: Ar = 4-MeO-C6H4 3: Ar = 3,4-MeO-C6H3 Scheme 2 Ar Ar
Short text for the table of contents
Mimic of amine oxidases, electrogenerated 3,4-iminoquinone 1ox exhibits, under metal-free conditions, high catalytic
performance in the chemoselective oxidation reaction of unactivated primary aliphatic amines.
Keywords : amino compounds . chemoselectivity .
electrochemical oxidation . enzyme mimics . quinones Ph O OH HN O Ph O OH H2N O Pt anode 1red 1ox
Indirect 1ox - mediated oxidation
-(cat.)
![Figure 1), and a cupric ion. [1] This class also includes bacterial methylamine oxidases which use a peptide-bound tryptophan tryptophylquinone (TTQ) [2] and lysyl oxidase, a mammalian copper amine oxidase, whose active site has bee](https://thumb-eu.123doks.com/thumbv2/123doknet/14584471.541340/3.892.124.769.121.1174/figure-includes-bacterial-methylamine-oxidases-tryptophan-tryptophylquinone-mammalian.webp)