HAL Id: tel-01526674
https://tel.archives-ouvertes.fr/tel-01526674
Submitted on 23 May 2017HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Caractérisation des carcinomes sarcomatoïdes primitifs
pulmonaires
Thibault Vieira
To cite this version:
Thibault Vieira. Caractérisation des carcinomes sarcomatoïdes primitifs pulmonaires. Cancer. Uni-versité Pierre et Marie Curie - Paris VI, 2016. Français. �NNT : 2016PA066552�. �tel-01526674�
Université Pierre et Marie Curie
ED 394
Sorbonne Universités, UPMC Univ Paris 06, GRC n°04, Theranoscan, F-75252, Paris, France
Caractérisation des carcinomes sarcomatoïdes primitifs
pulmonaires
Par Thibault Vieira
Thèse de doctorat de Physiologie physiopathologie
Dirigée par le Professeur Marie Wislez
Présentée et soutenue publiquement le 14 novembre 2016
Devant un jury composé de :
Joseph Gligorov, Professeur des Universités, Université Paris 6 (Président du jury) Marie Wislez, Professeur des Universités, Université Paris 6 (Directrice de thèse)
Claire Danel, Docteur, Maitre de conférences des universités Université Paris 7 (Rapporteur) Hélène Blons, Professeur des Universités, Université Paris Université Paris 5 (Rapporteur) Annette Larsen, Docteur, Directrice de recherche CNRS, Université Paris 6 (Examinateur) Nicolas Girard, Professeur des Universités, Université Claude Bernard Lyon 1 (Examinateur)
Remerciements
Je tiens tout d’abord à remercier chaleureusement Marie Wislez, qui a été ma directrice de thèse pendant ces quatre années. Merci Marie de m’avoir soutenu dans ce long chemin difficile. Avec toi j’ai appris la rigueur, la persévérance, l’écriture scientifique. Je retiens beaucoup d’enseignements à tes côtés.
Je remercie sincèrement Martine Antoine avec qui j’ai passé des heures à contempler au microscope ces tumeurs si particulières. Je suis toujours aussi impressionné par ton expertise. Je remercie avec beaucoup d’amitiés Nathalie Mathiot et Virginie Poulot pour leur aide précieuse dans la mise au point et l’accompagnement des techniques. Leurs soutiens et amitiés m’ont beaucoup aidé.
Merci à Anita Rodenas pour son aide dans la mise au point des nombreuses techniques d’immunohistochimie, de TMA.
Merci à mes compères du laboratoire Michael Duruisseaux et Vincent Fallet.
Je remercie Jacques Cadranel qui a m’a accueilli dans son service, son enseignement et son savoir m’ont été d’une aide précieuse.
Enfin, je remercie le Président du Jury (Joseph Gligorov), les rapporteurs de cette thèse (Claire Danel et Hélène Blons) et les examinateurs du jury (Nicolas Girard et Annette Larsen) pour leur temps consacré à cette soutenance.
Je dédicace cette thèse à Cloé ma tendre épouse, Anatole mon petit soleil Michelle et Roger mes parents Fabrice mon frère.
1
Table des matières
Abréviations ... 3
1 Introduction ... 4
1.1 Données épidémiologiques des cancers bronchopulmonaires ... 4
1.2 Classification anatomo-pathologique des cancers bronchopulmonaires ... 5
1.3 Spécificités des carcinomes sarcomatoïdes ... 6
1.3.1 Définition et caractéristiques pathologiques ... 6
1.3.2 Une particularité : la transition épithélio-mésenchymateuse ... 26
1.3.3 Caractéristiques cliniques ... 12
1.3.4 Caractéristiques moléculaires ... 12
1.3.5 Un modèle de cellule souche ? ... 31
1.3.6 Place de l’immunité ... 22
1.3.7 Approches thérapeutiques actuelles ... 26
2 Objectifs de thèse ... 33
3 Résumés des articles et manuscrits ... 35
3.1 Détermination de la réponse à la chimiothérapie ... 35
3.1.1 Article 1 : étude des caractéristiques cliniques et moléculaires des patients ayant un CBNPC de stade avancé et réfractaires à la chimiothérapie à base de platine ... 35
3.1.2 Article 2 : étude de l’efficacité de la chimiothérapie dans le sous type sarcomatoïde ... 43
3.1.3 Article 3 : étude de la résistance à la chimiothérapie de lignées de carcinomes sarcomatoïdes, implication de la voie SHH ... 49
3.2 Etude de la transitio-épithélio-mesenchymateuse et de l’invasion vasculaire ... 65
3.2.1 Article 4 : caractérisation pathologique, immunohistochimique et moléculaire des carcinomes sarcomatoïdes, implication de l’angio-invasion. ... 61
3.3 Immunité et carcinomes sarcomatoïdes ... 74
3.3.1 Article 5 : étude de l’infiltration immunitaire et de l’expression de PD-L1 au sein des carcinomes sarcomatoïdes ... 74
3.4 Caractérisation moléculaire ... 88
3.4.1 Article 6 : étude moléculaire à haut débit des carcinomes sarcomatoïdes ... 88
3.4.2 Article 7 : étude des mutations de l’exon 14 de MET dans une population de carcinomes sarcomatoïdes ... 97
3.4.3 Analyses par séquenceur de nouvelle génération ... 119
2
4.1 Projet immunologique ... 124
4.2 Projet cellules souches ... 126
5 Conclusion ... 123
6 Références bibliographiques ... 128
3
Abréviations
ALK Anaplastic lymphoma Kinase
CBNPC Carcinome Bronchique Non à Petites Cellules CBL Casitas B-Lineage Lymphoma
CSC Cellules Souches Cancéreuses EFGR Epidermal Growth Factor Receptor EGF Epidermal Growth Factor
FGFR1 Fibroblast Growth Factor Receptor 1 FISH Fluorescence In Situ Hybridization HGF Hepatocyte Growth Factor
IHC Immunohisthochimie
ITK inhibiteurs de tyrosine kinase
MET Mesenchymal-Epithelial Transition (gène) Shh Sonic Hedgehog
SNP Single Nucleotid Polymorphism
TEM Transition Epithélio-Mésenchymateuse TMA Tissue Micro-Array
4
1 Introduction
1.1 Données épidémiologiques des cancers broncho-pulmonaires
Le cancer bronchique se classe à la 4ième place des cancers en France, avec une incidence d’environ 39 495 nouveaux cas par an en 2012, soit 11,1% de l’ensemble des cancers (données Institut National du Cancer). Il s’agit de la première cause de mortalité par cancer, avec environ 29 949 décès estimés par an en 2012. Le pronostic est particulièrement sombre, avec une médiane de survie à 5 ans de 14% (tous stades confondus). Il touche préférentiellement les hommes (70%), et l’âge moyen au diagnostic est de 65 ans. Chez l’homme, le cancer bronchique est le 2ième cancer incident avec 28 211 nouveaux cas en 2012 (14,1% des cancers de l’homme), derrière le cancer de prostate, et reste la première cause de décès par cancer (21 326 décès en 2012, 25% des décès par cancer chez l’homme). Après une augmentation constante du nombre de cancers bronchiques chez l’homme jusqu’au début des années 2000, on assiste à une lente décroissance de son incidence (incidence standardisée de 52,9 pour 100 000 en 2000, et de 51,7 pour 100 000 en 2012). Chez la femme, le cancer bronchique est le 3ième cancer incident avec 11 284 nouveaux cas en 2012 (7,3% des cancers de la femme), derrière le cancer du sein et le cancer colorectal, et la 2ième cause de décès par cancer (8 623 décès en 2012, 13,7% des décès par cancer chez la femme). A l’inverse de la population masculine, on assiste actuellement à une augmentation de l’incidence et de la mortalité du cancer bronchique chez la femme (incidence standardisée de 9,7 pour 100 000 en 2000, et de 18,6 pour 100 000 en 2012). Cette hausse touche essentiellement les femmes jeunes, entre 45 et 60 ans (mortalité par cancer bronchique multipliée par 2,6 dans cette tranche d’âge chez la femme).
5
1.2 Classification anatomo-pathologique des cancers
broncho-pulmonaires
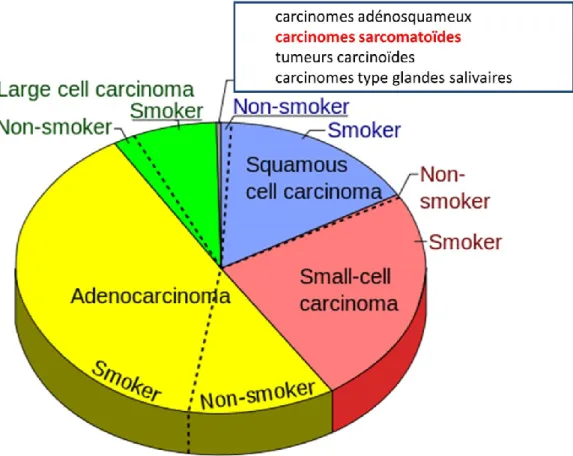
La grande majorité des cancers bronchiques correspondent au cancer bronchique non à petites cellules (CBNPC) (85%), le reste étant constitué des cancers bronchiques à petites cellules (15%). La classification actuelle des CBNPC repose sur la classification de l’OMS 2015 (Travis et al., 2015), dont les principales catégories sont : les adénocarcinomes, les carcinomes épidermoïdes et les carcinomes à grandes cellules. Les carcinomes sarcomatoïdes font partis du groupe des CBNPC (Figure 1). Ils représenteraient entre 0,4 à 3% des CBNPC (Yendamuri et al., 2012a).
6
1.3 Spécificités des carcinomes sarcomatoïdes
1.3.1 Définition et caractéristiques pathologiques
En 1958, Nash et Stout ont rapporté l’une des premières séries autopsiques concernant 5 cas de carcinomes à cellules géantes (NASH, STOUT, 1958). Ils décrivaient d’un point de vue macroscopique des tumeurs à prédominance périphérique, large, nécrotique à section gris-jaune, hémorragique avec une dissémination métastatique majeure. D’un point de vue microscopique ils notaient la présence de cellules géantes multinuclées, de taille très variable, pouvant présenter des noyaux hyperchromatiques et irréguliers pléomorphes, « bizarres », un cytoplasme éosinophilique et un haut rapport nucléo-cytoplasmique. Ils notaient une empéripolèse de polynucléaires neutrophiles (présence de polynucléaires neutrophiles à l’intérieur des cellules géantes). Dans certaines zones, la tumeur pouvait prendre un aspect de tumeur épithéliale, et dans d’autres un aspect de « sarcome », avec des cellules fusiformes organisée en travées et un aspect storiforme. Ils soulignaient déjà l’hétérogénéité de ces tumeurs et la nécessité d’individualiser ce sous-groupe. Ces tumeurs ont par la suite été rattachées au sein des CBNPC dans la classification OMS de 1999. Les descriptions pathologiques ne différent peu des classifications OMS successives 1999, 2004 et 2015.
Actuellement, la classification OMS 2015 regroupe ainsi sous le terme général de carcinomes sarcomatoïdes, tout carcinome comportant des cellules d’aspect sarcomateux c’est à dire ayant un aspect fusiforme ou un aspect de cellules géantes ou comportant un contingent sarcomateux (Figure 2). Cette classification définit cinq sous-types.
7 – des carcinomes pléomorphe, sous-type le plus fréquent (70-80% des cas), définis par l’association d’un contingent pseudo-sarcomateux montrant la présence de cellules fusiformes et/ou de cellules géantes devant représenter au moins 10% de la tumeur et d’un contingent épithélial comme par exemple un adénocarcinome, un carcinome épidermoïde, un carcinome adénosquameux, un carcinome à grandes cellules.
– des carcinosarcomes, entités rares, définis par l’association d’un contingent sarcomateux hétérologue vrai (chondrosarcome, ostéosarcome, rhabdomyosarcome, liposarcome) et d’un contingent épithélial.
– un peu à part et encore plus rare, le pneumoblastome associant un contingent d’adénocarcinome de type fœtal et une prolifération maligne mésenchymateuse.
Les formes pseudo-sarcomateuses pures composées : – des carcinomes à cellules fusiformes.
– des carcinomes à cellules géantes.
Le diagnostic pathologique peut être difficile. Il dépend de la qualité et de la taille des prélèvements tumoraux ainsi que du nombre de prélèvements réalisés par le pathologiste. Classiquement le diagnostic repose sur l’analyse des pièces opératoires. Cependant, le diagnostic est possible sur des biopsies de petite taille et peut être facilité par l’utilisation raisonnée de techniques complémentaires d’immuno-histochimie (IHC).
Plusieurs écueils diagnostiques existent. Le premier est de porter à tort le diagnostic d’authentique sarcome à la place d’une forme pure de carcinome à cellules fusiformes. Le synovialosarcome pulmonaire primitif touche préférentiellement l’adulte jeune et les enfants. Ces tumeurs sont périphériques et non encapsulées mais bien délimitées. Contrairement aux carcinomes sarcomatoïdes, le pléomorphisme cellulaire est moins marqué et la présentation
8 histologique existe sous deux formes principales : plus souvent monophasique à cellules fusiformes et rarement biphasique avec une composanteépithéliale, glandulaire et une composante fusiforme. L’étude en IHC est particulièrement utile dans les formes monophasiques en identifiant une positivité des cytokératines et/ou de l’EMA. Les anticorps BCL2 et CD99 sont fréquemment positifs mais non spécifiques. Le diagnostic de certitude est apporté par l’étude moléculaire par la mise en évidence de la translocation (X,18) (p11, q11) dans plus de 90 % des cas par FISH en paraffine ou PCR sur matériel congelé.
C’est la présence d’un contingent épithélial identifié en IHC qui permet de redresser le diagnostic de carcinome. La pancytokératine AE1AE3 semble être la plus sensible pour caractériser la présence d’un contingent épithélial, mais ne comporte pas la CK18 qui est parfois exprimée de façon isolée. D’autres marqueurs épithéliaux comme l’epithelial membran antigen (EMA), le CAM5.2 (pancytokératine comportant la CK 18) ou la P63-P40 peuvent aider à l’identification d’un contingent épithélial. En revanche, la vimentine, n’est pas un marqueur spécifique de tumeur mésenchymateuse, car nombre de carcinomes l’expriment et en particulier les adénocarcinomes bronchopulmonaires. Certains marqueurs tels que l’actine, la desmine, la myoglobine, et la protéine S100 permettent aussi de mieux caractériser le contingent pseudo-sarcomateux hétérologue des carcinosarcomes.
Il est parfois difficile de distinguer une lésion inflammatoire avec stroma-réaction fibreuse intense (par exemple pseudotumeur inflammatoire ou tumeur myofibroblastique inflammatoire) d’un carcinome sarcomatoïde à cellules fusiformes riche en cellules inflammatoires. Le profil d’expression des cytokératines oriente le diagnostic.
Il existe également des mésothéliomes de sous-type sarcomatoïdes particulièrement difficiles à distinguer des formes de carcinomes sarcomatoïdes à expression pleurale. Le
9 contexte clinique, radiologique et anatomopathologique (tumeur pulmonaire sous-jacente) orientera le diagnostic. En effet, l’expression de kératines est commune aux deux entités. Les marqueurs mésothéliaux habituels tels que la calrétinine et la CK5/6 sont parfois négatifs dans les formes sarcomatoïdes de mésothéliome. L’expression de Wilm's tumour 1 (WT1) et de la podoplanine (D240) peuvent être exprimés bien que moins fréquemment dans les carcinomes sarcomatoïdes que dans les mésothéliomes. Ainsi, le recours au réseau d’experts Mesopath est systématique en cas de doute quant à un mésothéliome.
Pour finir, il est parfois très difficile de trancher en particulier lorsqu’il existe des métastases de sites inhabituels quant à l’origine primitive du carcinome sarcomatoïde. En effet, il est décrit des carcinomes sarcomatoïdes de la sphère ORL, digestive, de la prostate, de la vessie, du rein, du sein. Le contexte clinique et radiologique orientera le diagnostic. Toute localisation viscérale en dehors des tissus mous d’une tumeur à cellules fusiformes fera évoquer le diagnostic de carcinome sarcomatoïde, rechercher des antécédents et justifier la comparaison morphologique et immuno-histochimique des lésions. L’expression de TTF1 oriente vers une origine primitive pulmonaire ; elle est positive jusqu’à 55 % des cas sur le contingent pseudo-sarcomateux et plus fréquemment sur le contingent épithélial, s’il est de type adénocarcinomateux. La cytokératine 20 est quasiment toujours négative sur les deux contingents. La cytokératine 7 (CK7) est positive dans 46 à 70 % des cas sur le contingent pseudo-sarcomateux et plus fréquemment sur le contingent épithélial, s’il est de type adénocarcinomateux. La CK7 peut être positive dans les tumeurs de la sphère digestive (estomac, pancréas), urothéliale et mammaire. Dans une étude comparant des carcinomes sarcomatoïdes d’organes différents, l’anticorps p63 semble être plus utile dans les carcinomes sarcomatoïdes ORL (63 % de positivité) que pulmonaires (50 %) et qu’urothéliaux (9 %). Les principales analyses immuno-histochimiques sont résumées dans le tableau 1.
10
Figure 2 : illustration des différents sous-types de carcinomes sarcomatoïdes
A
C
B
A Carcinome à cellules géantes avec empéripolèse (HES, grossissement x400) B Carcinome à cellules fusiformes (HES, grossissement x400)
11
Tableau I Caractéristiques immuno-histochimiques des carcinomes sarcomatoïdes (Vieira et al., 2012)
12 1.3.2 Caractéristiques cliniques
Les carcinomes sarcomatoïdes ne s’accompagnent pas de manifestations cliniques spécifiques. Les patients sont le plus souvent symptomatiques car la tumeur est volumineuse. Dans deux tiers des cas, la tumeur est plutôt de siège périphérique avec la possibilité d’un envahissement pleural et costal responsable de douleurs. Dans un tiers des cas, le siège est central avec une extension endobronchique, médiastinale ou vasculaire pouvant donner lieu à des hémoptysies. L’extension hématogène est fréquente avec des localisations métastatiques classiques mais aussi atypiques telles que péritonéales, retro péritonéales, gastriques, intestinales (Jia et al., 2010), pancréatiques, gingivales (Park et al., 2006), sous-cutanées, thyroïdiennes. En TEP-FDG, l’intensité maximale de fixation du FDG (SUVmax) au niveau du site primitif est importante (Kaira et al., 2011 ; Ito et al., 2010a). Ainsi, la présence d’une tumeur de grande taille, périphérique avec envahissement pariétal et un SUV max élevé peut faire évoquer ce diagnostic.
1.3.3 Caractéristiques moléculaires
Pour chaque anomalie génétique, nous rappellerons succinctement les données de la littérature concernant les carcinomes bronchiques non à petite cellules puis nous traiterons les données concernant les carcinomes sarcomatoïdes.
1.3.3.1 EGFR
L’epidermal growth factor receptor EGFR (ERBB1) est un récepteur membranaire à activité tyrosine kinase, dont le ligand est l’EGF. Après fixation du ligand, EGFR forme un hétéro-dimère, le plus souvent avec HER2 (ERBB2), et s’ensuit une activation du domaine tyrosine kinase intracellulaire avec activation des voies KRAS, PI3kinase (PI3K), STAT et PLCγ, qui
13 entraine la production de signaux de prolifération et de survie cellulaire au sein de la cellule tumorale. La présence d’une mutation somatique du gène de l’EGFR concerne 10 à 15% des CBNPC (Sharma et al., 2007). Elle prédomine dans les carcinomes non-épidermoïdes, et notamment dans les adénocarcinomes. Elle est plus souvent présente chez la femme, peu ou pas fumeuse et asiatique (Sharma et al., 2007). Ainsi, la fréquence des mutations de l’EGFR est de plus de 30% chez le non-fumeur (Barlesi et al., 2016a). Dans les CBNPC, les mutations d’EGFR touchent le domaine tyrosine kinase du récepteur, entrainant ainsi une auto-activation permanente de l’EGFR, plus ou moins indépendante de la fixation du ligand. Cette activation induit une activation, de qualité différente, des voies d’aval (Sharma et al., 2007). Les mutations d’EGFR les plus fréquentes sont présentes dans l’exon 19 (45% des cas), sous la forme d’une délétion, ou dans l’exon 21 (40 à 45% des cas), avec la substitution L858R. Toutes ces mutations sont associées à une sensibilité accrue aux ITK de l’EGFR, comme l’erlotinib, le gefitinib ou l’afatinib (Mok et al., 2009 ; Sharma et al., 2007). D’autres mutations sont possibles, comme les mutations des exons 18 ou 20 (avec notamment la substitution T790M), avec des réponses variables aux ITK de l’EGFR (sensibilité pour les mutations au sein de l’exon 18, résistance pour les mutations au sein de l’exon 20) (Beau-Faller et al., 2014). Il est actuellement admis que les mutations d’EGFR sont exclusives des mutations de KRAS et de la translocation ALK.
Concernant les carcinomes sarcomatoïdes, dans la série d’Italiano et al, l' EGFR était surexprimé chez tous les patients et amplifié chez 23%(>4 copies dans plus de4 0% des cellules) d’entre eux en FISH (Italiano et al., 2009). Des mutations d’EGFR ont été rapportés, en particulier dans les carcinomes sarcomatoïdes de type pléomorphe avec composante adénocarcinomateuse et plus fréquemment chez les asiatiques (Kaira et al., 2011)(Leone et al., 2011). Dans la série de Sequist et al, analysant 37 cas de CBNPC mutés pour EGFR et
re-14 biopsiés au moment de la progression sous inhibiteur de tyrosine kinase (ITK) de l’EGFR, trois cas avaient un phénotype de TEM dont un cas de carcinome sarcomatoïde. Celui-ci était associé à une perte de la mutation de l’EGFR (Sequist et al., 2011).
Ces données impliquent la recherche systématique de mutations d’EGFR dans les carcinomes sarcomatoïdes, en particulier pour les carcinomes pléomorphes avec composant adénocarcinomateux comme recommandée dans la classification OMS 2015.
1.3.3.2 KRAS
Les protéines RAS sont des protéines membranaires intervenant dans la transduction des signaux de croissance cellulaire reçus suite à l’activation des récepteurs tyrosines kinases membranaires (notamment EGFR, PDGFR, IGFR), et activant en aval la voie RAF – MEK – ERK, et la production de signaux cellulaires de prolifération. Les mutations de KRAS sont fréquentes (environ 25% des CBNPC) (Barlesi et al., 2016a). Elles concernent le codon 12 de
KRAS, ou plus rarement les codons 13 et 61. Elles entrainent une activation constitutive de
KRAS, indépendante des signaux d’amont. Il n’existe pas actuellement de thérapies ciblées en cas de mutation. La présence de mutations de KRAS est plus fréquente chez le patient fumeur (Figure 2B) (Barlesi et al., 2016a). Elle est également associée à une absence de réponse aux ITK de l’EGFR (Eberhard et al., 2005 ; Pao et al., 2005). Son caractère prédictif de résistance à la chimiothérapie cytotoxique est encore débattu (Loriot et al., 2009 ; Mellema et al., 2013). Il est actuellement admis que les mutations de KRAS sont exclusives des mutations d’EGFR et de la translocation ALK.
Les mutations de KRAS sont retrouvées jusqu’à 38% des cas, évaluées à partir de 7 séries de carcinomes sarcomatoïdes de 22 à 61 patients (Pelosi et al., 2004 ; Przygodzki et al.,
15 1996 ; Italiano et al., 2009 ; Leone et al., 2011 ; Chang et al., 2011 ; Jiang et al., 2011 ; Lee et al., 2011).
1.3.3.3 ALK
Anaplastic lymphoma kinase (ALK) est un récepteur membranaire tyrosine kinase de la superfamille des récepteurs à l'insuline. La translocation d’ALK est un événement rare (environ 5% des CBNPC) (Shaw et al., 2009 ; Kwak et al., 2010 ; Barlesi et al., 2016a). On la retrouve de manière préférentielle chez les patients jeunes, non-fumeurs et dans les adénocarcinomes (Shaw et al., 2009). Elle résulte le plus souvent d’une inversion au sein du chromosome 2, induisant alors la formation d’un gène de fusion et l’activation constitutive de la protéine ALK, avec stimulation des voies de signalisation d’aval (PI3K, STAT3, JAK2) (Soda et al., 2007). Les réarrangements de ALK impliquent l’exon 20 qui code pour la région tyrosine kinase d’ALK. Le gène partenaire du réarrangement est dans la très grande majorité des cas le gène EML4 (Echinoderm Microtubule associated protein Like 4). D’autres partenaires ont été décrits, comme les gènes TGF (TRK-fused gene), KIF5B (Kinesin Family member 5B) et KLC1 (Kinesin Light Chain 1). La présence d’une translocation d’ALK rend la tumeur particulièrement sensible à certaines thérapies ciblées, comme le crizotinib (Kwak et al., 2010). Il est actuellement admis que la translocation ALK est exclusive des mutations d’EGFR et des mutations de KRAS.
Dans les carcinomes sarcomatoïdes, de très rares réarrangements ALK ont été décrits (Alì et al., 2013 ; Kobayashi et al., 2013). En revanche, cinq amplifications de ALK sont décrits dans la série de 23 cas de Pelosi et al (Pelosi et al., 2016) dont l’impact thérapeutique n’est pas encore clairement défini.
16
1.3.3.4 MET
Le récepteur mesenchymal-epithelial transition factor (MET) est un récepteur membranaire tyrosine kinase. Son ligand est le facteur de croissance hépatocytaire (HGF). L’activation de MET entraine à la fois une activité mitogénique (prolifération cellulaire), motogénique (migration cellulaire), morphogénique (différenciation cellulaire) et antiapoptique. Ces différentes activités biologiques définissant la « croissance invasive », qui est quiescente chez les adultes. Elle peut être réactivée en cas d’agression tissulaire afin de restituer l’intégrité du tissu. Elle est également impliquée dans le développement et la progression tumorale et joue un rôle clé au cours de certains cancers, comme le cancer papillaire héréditaire du rein. Dans le cancer du rein, que ce soit dans les formes sporadiques ou héréditaires, des mutations ponctuelles au niveau du domaine tyrosine kinase de MET sont retrouvées. Ces mutations aboutissent à une activation constitutive du récepteur. Dans les CBNPC, différentes altérations oncogéniques de MET sont décrites : surexpression du récepteur, amplification génomique ou mutation.
Le gène MET est situé sur le bras long du chromosome 7 (7q31) et est constitué de 21 exons. Le récepteur MET fait partie de la famille des récepteurs tyrosine kinase. Il comprend une sous-unité ⍺ extra-cellulaire et une sous-unité β composée d’une partie extracellulaire avec une extrémité N-terminale contenant un domaine sémaphorine (SEMA), d’une partie transmembranaire et d’une partie intracellulaire comprenant un domaine juxtamembranaire et un domaine à activité tyrosine kinase.
Après liaison à son ligand HGF, MET forme des homodimères ou des hétérodimères avec d’autres récepteurs tyrosine kinase tels qu’EGFR, HER2 pouvant alors activer diverses voies de signalisation. La cascade RAS-MAP kinases permet de contrôler l’expression de
17 gènes impliqués dans la prolifération cellulaire, la migration cellulaire, l’invasion tissulaire et l’angiogenèse. La voie PI3K-AKT–mTOR favorise l’inhibition de l’apoptose et la motilité cellulaire (Ruppert et al., 2011).
Plusieurs mécanismes de dérégulation de la voie HGF–MET peuvent être impliqués dans le cancer bronchique : surexpression du récepteur, amplification génomique ou mutation. La surexpression de MET dans le cancer bronchique est fréquente. Elle est retrouvée davantage dans les adénocarcinomes (67 %), que dans les carcinomes indifférenciés à grandes cellules (57 %), les carcinomes épidermoïdes (57 %) et les cancers bronchiques à petites cellules (25 %) [15]. Dans la série de Tsuta et al, (Tsuta et al., 2012) la protéine MET et sa forme phosphorylée étaient surexprimés en immunohistochimie dans 80 et 90 % des 43 cas de carcinomes sarcomatoïdes analysés, respectivement.
La surexpression de MET a été l’objet d’un ciblage thérapeutique. L’onartuzumab (MetMab) est un anticorps monoclonal humanisé anti-MET. Sa fixation sur le récepteur MET est compétitive de celle du ligand de MET (HGF) et a pour conséquence le blocage du signal d’aval. Après avoir montré des résultats encourageants in vitro et dans les phases I, un essai de phase II randomisé (Spigel et al., 2013) incluait 128 patients en 2ième ou 3ième ligne de traitement dans 2 bras : erlotinib+onartuzumab vs. erlotinib + placebo. Les patients ayant une surexpression de MET en IHC voyaient leur survie sans progression doubler (médiane 1,5 versus 2,9 mois ; HR = 0,53 ; p = 0,04) et leur survie globale tripler (médiane 3,8 versus 12,6 mois ; HR = 0,37 ; p = 0,002) avec l’association erlotinib + MetMab par rapport au bras contrôle. Cette association était en revanche délétère chez les patients ne surexprimant pas MET. L’essai de phase III (Spigel et al., 2014), multicentrique, en double aveugle, incluait 499 patients avec un CBNPC sur-exprimant MET, randomisé de la même manière en 2ème ou
18 3ème ligne entre erlotinib + onartuzumab vs. erlotinib + placebo. Cependant l’étude était négative sur son critère de jugement principal, la survie globale. Il existait même une tendance négative (médiane 6,8 vs. 9,1 mois, p = 0,068) pour le bras expérimental. Il n’y avait aucune différence en termes de survie sans progression et de réponse. La surexpression de MET ne parait pas être une cible suffisante pour l’inhibition de ce dernier par des anticorps monoclonaux dans les CBNPC. L’association avec l’erlotinib n’était peut-être pas non plus un choix pertinent.
Le nombre de copies de MET peut être évalué par FISH ou par PCR en temps réel. L’amplification de MET est rare chez les patients atteints de CBNPC : elle est présente chez 2-6% des CBNPC dans plusieurs séries regroupant près de 600 patients (Bean et al., 2007 ; Cappuzzo et al., 2009 ; Chen et al., 2009 ; Kubo et al., 2009 ; Okuda et al., 2008 ; Katayama et al., 2009).L’amplification de MET joue un rôle dans la résistance acquise aux ITK d’EGFR chez les patients ayant un CBNPC muté EGFR (Bean et al., 2007 ; Engelman et al., 2007 ; Turke et al., 2010) et serait retrouvée dans 5 à 20% des cas (Arcila et al., 2011 ; Bean et al., 2007 ; Chen et al., 2009 ; Engelman et al., 2007 ; Turke et al., 2010 ; Sequist et al., 2011 ; Yu et al., 2013). La définition de l’amplification de MET reste débattue et plusieurs systèmes catégoriels ont été proposés (Tableau II, III, IV).
Tableau II : critères de F. Cappuzo
Score 1 Disomie équilibrée caractérisée par un maximum de 2 copies, dans plus de 90 % des cellules
Score 2 trisomie équilibrée de bas grade : ≤ 2 copies dans au moins 40 % des cellules tumorales et 3
copies dans 10 à 40 % et ≥ 4 copies dans moins de 10 %
Score 3 trisomie équilibrée haut grade : ≤ 2 copies dans au moins 40 % des cellules et 3 copies dans plus
de 40 % des cellules et ≥ 4 copies dans moins de 10 % des cellules
Score 4 polysomie équilibrée de bas grade : ≥ 4 copies dans 10 à 40 % des cellules
Score 5 polysomie équilibrée de haut niveau : ≥ 4 copies dans au moins 40 % des cellules
Score 6 amplification vraie définie par un ratio du nombre de copie du gène met sur le nombre de copies
du Chr 7 ≥ 2 avec présence de cluster ou un total de plus de 15 copies de met par cellule dans au moins 10 % des cellules tumorales analysées.
19
Tableau III : critère du programme AcSé (essai clinique unicancer, INCA)
Amplification Nombre de copies de MET >6 copies
Tableau IV : critères rapportés à l’ASCO par Camidge
amplification faible ≥1.8 et 1.8≤ Ratio Met/Ch7≤2.2,
amplification intermédiaire 2.2< Ratio Met/Ch7<5
amplification haute Ratio Met/Ch7≥5
L’augmentation du nombre de copies de MET n’est pas associée à un type histologique particulier. Deux séries rapportent les amplifications de MET dans les carcinomes sarcomatoïdes. Dans la série de Tsuta et al, il existait une amplification de MET dans 36% de 43 cas mis en évidence par technique BISH (bright-field in situ) (Tsuta et al., 2012). Lee et al retrouvaient une amplification de MET dans 18% des 61 cas des carcinomes sarcomatoïdes en FISH (Lee et al., 2011). Les amplifications de MET font l’objet d’un essai thérapeutique français « acse crizotinib ». En suivant une stratégie de diagnostic par immunohistochimie puis confirmation par FISH, 113/1550 patients atteints de cancer du poumon (7,3%) avaient une amplification de MET. Vingt-cinq ont été inclus dans cet essai. Le critère diagnostic utilisé était un nombre de copies de MET >6. La population incluse était pour trois quart des fumeurs sevrés ou actifs, et 36% avaient reçu un ITK d’EGFR. Le taux de réponse objective après deux cycles était de 32% (8/25) [9 ; 45] et le taux de contrôle de la maladie de 60% (15/25) [41 ; 71]. 32% des patients progressaient tout de même d’emblée sous traitement. La survie sans progression était estimée à 3,2 mois [IC95% : 1,9 ; 3,9] (Moro-Sibilot, oral 03.06, WCLC 2015). L’étude est toujours en cours, il est cependant important de comprendre pourquoi un tiers des patients progressent d’emblée. Les données ne sont pas encore matures.
20 En 2005 une équipe de Chicago s’est intéressée aux mutations ponctuelles de MET et a recherché ces altérations sur des lignées cellulaires de CBNPC et 127 prélèvements d’adénocarcinomes bronchiques réséqués (Patrick C Ma et al., 2005). Elles concernaient le domaine semaphorine, et le domaine juxta-membranaire. Les mutations étaient identifiées au niveau de séquences codantes du domaine juxta-membranaire. Elles sont responsables de l’épissage de l’exon 14. L’exon 14 code pour le site « Casitas B-lineage Lymphoma » (CBL) qui permet la liason de l’ubiquine ligase. L’ubiquination de MET participe à son renouvellement/degradation et donc à sa demi-vie. La perte de l’exon 14 abouti à un défaut d’ubiquination. Sur des modeles in vitro ces mutations sont oncogeniques et l’inhibition de MET est anti tumorale (Kong-Beltran et al., 2006a ; Ma et al., 2003). Ces mutations diffèrent de celles décrites dans le cancer papillaire rénal qui sont situées au niveau du domaine SEMA (site de fixation d’HGF) et du domaine tyrosine kinase (activation constitutive du récepteur).
Dans une série incluant 38 028 tumeurs dont 4402 adénocarcinome analysées par Next-Generation Sequencing (NGS), 131 (3%) présentaient des altérations de l’exon 14 (Frampton et al., 2015a). Dans ce travail, il est décrit 126 variants mutants : des insertions, des délétions, des substitutions dans les sites accepteur/donneur et dans l’intron adjacent au site accepteur/donneur ainsi que des délétions complètes de l’exon 14. Ces mutations étaient exclusives d’EGFR, KRAS, HER2, ALK, ROS1. En revanche des amplifications de MDM2 et
CDK4 pouvaient être associées.
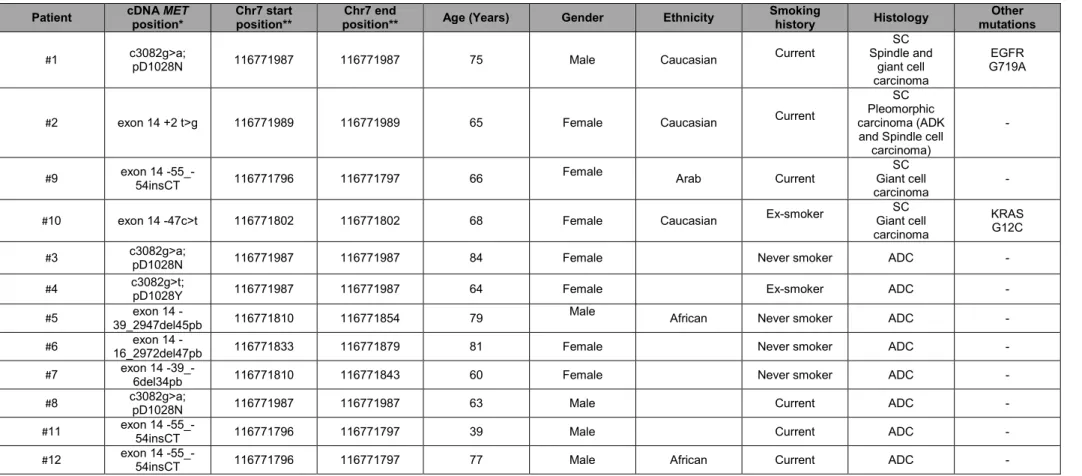
Dans une série récente étudiant les exomes de 10 cas de carcinomes sarcomatoïdes, deux mutations de MET responsables de l’épissage de l’exon 14 sont décrites (Liu et al., 2016a). L’étude s’est poursuivie avec l’analyse de 26 autres cas, avec une PCR recherchant spécifiquement ces anomalies. Au total, 8 patients avec un carcinome sarcomatoïde
21 présentaient une mutation responsable de l’épissage de l’exon 14 (22%). Ces 8 patients ne présentaient pas d’anomalies de KRAS, EGFR, BRAF, ALK. Un d’entre eux présentait une mutation associée de PIK3CA. Sept de ces 8 patients présentaient un contingent adénocarcinomateux au sein de leur tumeur sarcomatoïde. Une des patientes était traitée et présentait une réponse tumorale sous crizotinib (anti-ALK et anti-MET) après échec d’une chimiothérapie à base de platine. Chez cette patiente, la mutation était associée à une amplification de MET (> 9 copies).
Dans une cohorte de 1141 patients suivis pour CBNPC entre 2013 et 2015, 28 (3%) cas de mutations responsables de l’épissage de l’exon 14 de MET sont décrits. Dans cette population, il y avait 19 femmes (68%), un tiers de non-fumeur (n=10), tous caucasiens non hispaniques. Leur âge médian est de 72,5 ans. On y retrouve des stades I (n=13), II (n=2), III (n=4) et IV (n=9). Il s’agit principalement d’adénocarcinomes mais également des carcinomes sarcomatoïdes (4/15 soit 27%) avec un contingent adénocarcinomateux (Awad et al., 2016a).
Plus récemment, une analyse par NGS, incluant 11 205 patients, a mise en évidence un taux de 7.7% (n=8/104) de mutation de l’exon 14 parmi les carcinomes sarcomatoïdes (Schrock et al., 2016). Elle reste supérieure aux adénocarcinomes (3%). Une co-amplification de MET a été retrouvée (≥6 copies, ou ≥7 si tumeur triploïde; ≥8 si tumeur tétraploïde) dans 44/298 soit 14,8 % des patients dont 1/8 carcinome sarcomatoïde.
Ces mutations de MET semblent donc possiblement plus fréquentes dans le sous-type carcinome sarcomatoïde. Elles font l’objet d’une étude dans notre travail (cf. article 7).
22
1.3.3.5 Autres altérations moléculaires
Des altérations de P53 sont fréquemment décrites dans les carcinomes sarcomatoïdes, probablement du fait de la forte prévalence du tabagisme. Ces mutations de gènes suppresseurs de tumeurs ne sont pas mutuellement exclusives comme le sont les mutations des oncogènes. On retrouve ces mutations dans 30 à 60 % des cas des séries sarcomatoïdes publiés.
Enfin, une série asiatique de 61 patients atteints de carcinomes sarcomatoïdes a également rapportée une amplification du Fibroblast growth Factor Receptor 1 (FGFR1) dans 10% des cas (Lee et al., 2011).
Actuellement aucun réarrangement de RET ou ROS1 n’a été décrit dans cette population. 1.3.4 Immunité et cancer broncho-pulmonaire
1.3.4.1 Généralité
L’immunosurveillance est née avec Paul Ehrlich en 1909 qui emit l’hypothèse que les cellules du système immunitaire étaient capables de reconnaître et de détruire des cellules cancéreuses. Dans les années 80, furent mis en évidence pour la première fois les antigènes tumoraux, et les lymphocytes associés aux tumeurs. Dans les années 2000, Robert Schreiber émet l’hypothèse de « l’immunoediting » ou échappement immunitaire tumoral : les cellules cancéreuses vont être capables « d’épuiser » le système immunitaire. Les cellules cancéreuses sont capables de contourner les défenses anti-tumorales par divers mécanismes comme la perte de l’expression de molécules du complexe majeur d’histocompatibilité, une perte d’immunogénicité par mutations de certaines protéines ou antigènes, l’acquisition de
23 mécanismes immunosuppresseurs notamment au niveau des points de contrôle immunitaire appelés « checkpoint » en anglais.
Les points de contrôle immunitaire sont des molécules immuno-modulatrices c’est-à-dire permettant de moduler l’intensité de la réponse immunitaire adaptative quand elle est trop intense (dans le cas des rejets de greffe ou de maladies auto-immunes) ou trop faible (cancers, infections chroniques). Ces molécules agissent en tant que molécules de co-signalisation en complément de l’activation du T-cell receptor (TCR) ou du B-cell receptor (BCR) et modulent l’activité des lymphocytes. Ces molécules immuno-modulatrices peuvent être classées selon leur activité (inhibitrice ou stimulatrice des lymphocytes) ou leur structure (appartenance à la superfamille des immunoglobulines ou à la superfamille des récepteurs du TNF) (Page et al., 2014).
Parmi ces molécules immuno-modulatrices, sont particulièrement étudiées comme cibles thérapeutiques:
-CTLA4 (« cytotoxic T lymphocyte-associated antigen 4 ») : c’est un homologue de la molécule de co-stimulation CD28, qui provoque une inhibition de l’activité du lymphocyte qui l’exprime, une anergie (Krummel et Allison, 1995). Hautement exprimé par les Tregs, CTLA4 permet d’induire un processus de tolérance des cellules dendritiques par contact cellulaire direct.
-PD-1 (« programmed cell death 1 ») est une molécule dont l’expression est restreinte aux lymphocytes T activés permettant de réguler leur activation dans les tissus (Thompson, Allison, 1997) ; ses ligands, PD-L1 et PD-L2, sont exprimés par certaines tumeurs pour échapper à l’immuno-surveillance (Yao et al., 2013), et par ces cellules inflammatoires tels que les macrophages ou les lymphocytes (Herbst et al., 2014)
24
1.3.4.2 Essais cliniques dans les CBNPC
Le Nivolumab est un anticorps monoclonal anti PD-1 de type IgG4 et a obtenu son autorisation de mise sur le marché en deuxième ligne chez les patients atteints de carcinomes épidermoïdes métastatiques dans un premier temps puis à l’ensemble des CBNPC. L’essai de phase III Checkmate 017 (Brahmer et al., 2015) a randomisé 272 patients dans un bras Nivolumab (3 mg/kg toutes les 2 semaines) versus Docetaxel (75 mg/m2 toutes les 3
semaines) jusqu’à progression ou toxicité. Il s’agissait de patients ayant un carcinome épidermoïde de stade IIIB ou IV en progression après une première ligne comportant du platine. L’objectif principal était la survie globale, les objectifs secondaires la survie sans progression, la réponse objective, la qualité de vie, la tolérance et l’efficacité selon l’expression de PD-L1. L’objectif principal a été atteint avec une amélioration de la survie globale sous Nivolumab de plus de 3 mois (9.2 versus 6 mois) ce qui correspond à une diminution du risque de décès de 41% (HR 0.59 ; 95 CI, 0.44-0.79, p<0.001). Le taux de survie à 1 an était de 42% avec le Nivolumab versus 24% avec le Docetaxel. De même le taux de réponse objective était meilleur avec l’anti-PD1 : 20% versus 9% (p=0.008), la survie sans progression de 3,5 mois versus 2,8 mois (HR 0.62, 95% IC, 0.47-0.81, p<0.001).
L’étude checkmate 057 a le même schéma que la précédente (Nivolumab versus Docetaxel) mais concerne les non-épidermoïdes. Cette étude est positive sur son objectif principal qu’est la survie globale (Borghaei et al., 2015); néanmoins l’analyse des données met en évidence l’existence d’un sous-groupe de patients s’aggravant sous Nivolumab par rapport au Docetaxel et suggère une hétérogénéité des patients en termes de sensibilité au Nivolumab pour les non épidermoïdes.
25 Le tabagisme semble être associé une plus grande efficacité des anticorps anti PD-L1 ou PD-1. De même, que la présence des mutations de KRAS qui sont le plus souvent induites par le tabac, ainsi qu’une forte charge mutationnelle tumorale, témoignant probablement aussi d’une intoxication tababique. Une forte charge mutationnelle tumorale est associée à une forte présence de néoantigènes tumoraux et donc à une certaine immunogenicité suggerant une réponse immunitaire anti tumorale préexistante et réactivable par les anti-PD-L1 ou PD-1.
L’expression de PD-L1 par les cellules tumorales, et pour certains par les cellules immunitaires, par étude en IHC est un biomarqueur utilisé pour sélectionner les patients pour les essais de première ligne par anti-PD-L1 ou PD-1 versus chimiothérapie à base de platine. Dans l’étude de phase I de l’Atezolizumab (anti-PD-L1), l’expression stromale de PD-L1 était prédicteur de réponse (Herbst et al., 2014). Dans l’étude de phase II POPLAR incluant 287 CBNPC comparant Atezolizumab vs. Docetaxel, stratifiée sur le sous-type histologique (épidermoïde–non-épidermoïde), le niveau d’expression du PD-L1 (tumeur et stroma, 4 niveaux d’expressions) et le nombre de lignes préalables, la survie globale (critère principale) était supérieure dans le bras Atezolizumab (hazard ratio 0,73 [IC 95% 0,53-0,99]; p=0,04). Le Bénéfice sur la survie globale était d’autant plus grand que l’expression de PD-L1 est importante (Fehrenbacher et al., 2016). Dans l’étude de phase I utilisant le Pembrolizumab (anti-PD1), un haut niveau d’expression tumoral du PD-L1 (>50%) était associé à une meilleure réponse au traitement (Garon et al., 2015). Dans l’étude de phase II/III keynote-10 comparant en deuxième ligne Pembrolizumab versus Docetaxel, les patients (n=1034) étaient stratifiés sur le PS, la région d’origine, le score PD-L1 (1-49% / ≥50%). La survie globale (critère principale) était significativement supérieure pour le Pembrolizumab vs. Docetaxel (hazard ratio 0,71, IC95% 0,58-0,88; p=0,0008). Les patients avec un score PD-L1 ≥50% présentait une meilleure survie sans progression (Herbst et al., 2016, p. 10). L’étude de Phase
26 III keynote-024 qui inclut des CBNPC avec un score PD-L1 ≥50% compare en 1ère ligne le Pembrolizumab vs. un doublet à base de platine est annoncée positive par le laboratoire MSD à la fois en survie sans progression et en survie globale.
1.3.4.3 Données dans les carcinomes sarcomatoïdes
Les carcinomes sarcomatoïdes sont de potentiel bons «candidats » à l’immunothérapie. En effet ils sont associés à un fort tabagisme, une forte prévalence des mutations de KRAS, une réaction inflammatoire intense (classification OMS 2015). Cependant l’étude de l’immunité n’est que peu rapportée. Deux séries rapportent l’expression du PD-L1 dans les carcinomes sarcomatoïdes (Velcheti et al., 2013 ; Kim et al., 2015). Le taux d’expression de PD-L1 était respectivement de 69% et 90% (n=41). Dans le travail de Kim, ces derniers démontrent une corrélation entre une infiltration de lymphocytes CD8+ et l’expression du PD-L1.
1.3.5 Approches thérapeutiques actuelles
Actuellement, la prise en charge thérapeutique de ces cancers se base sur les principes généraux de celle des CBNPC que ce soit sur le plan chirurgical ou médical. Les données de survie proviennent de séries chirurgicales rétrospectives. La survie globale à 5 ans, tous stades confondus se situe entre 21 et 54 % selon les séries (Martin et al., 2007 ; Ito et al., 2010b ; Chen et al., 2012 ; Yuki et al., 2007). Certaines séries ont tenté d’apparier pour l’âge, le sexe et le stade, les patients ayant un carcinome sarcomatoïde aux patients ayant un autre type de CBNPC sur la même période d’étude. Dans ces séries chirurgicales, le pronostic des carcinomes sarcomatoïdes est moins bon que celui des autres types de CBNPC (Figure 3). La survie globale et sans récidive est plus courte pour les patients ayant un carcinome
27 sarcomatoïde de stade localement avancé. Les données sont moins claires pour les stades I et II. La survie globale ne varie pas en fonction du contingent épithélial ou pseudosarcomateux. En revanche, elle dépend du stade comprenant la taille tumorale (limite 6-7cm) et de l’extension ganglionnaire. L’importance de la nécrose est aussi un facteur pronostic péjoratif. En ce qui concerne la prise en charge des stades métastatiques, ces tumeurs sont réputées être chimio-résistantes. Cependant les séries médicales dédiées jusqu’à present sont peu nombreuses, rétrospectives et comprennent peu de patients. Les essais thérapeutiques n’individualisent pas et n’excluent pas toujours ces patients. Le traitement standard de première ligne est un sel de platine associé à une molécule de troisième génération. Les taux de progression sous première ligne à base de platine décrits dans ces petites séries sont très élevés allant de 60 à 85 % (Bae et al., 2007 ; Ito et al., 2010b ; Kaira et al., 2010).
28
Figure 3 : survie globale des carcinomes sarcomatoides comparée à celle des CBNPC (Martin et al., 2007)
SARC : sarcomatoide, CBNPC : carcinome bronchique non à petites cellules
Cette série de Martin et al inclut des malades diagnostiqués entre 1984 à 2003. Soixante-trois carcinomes sarcomatoïdes et 1 133 CBNPC d’autres sous-types sont comparés en survie globale tous stades confondus
29 1.3.1 Facteurs associés à la chimiorésistance
1.3.1.1 La transition épithélio-mésenchymateuse
La transition épithélio-mésenchymateuse (TEM) est un processus biologique qui permet aux cellules polarisées de prendre un phénotype mésenchymateux. Il s’agit d’un processus normal au cours de l’embryogénèse et observé lors des phénomènes de réparation. La réactivation aberrante de ce programme embryonnaire au cours de la tumorigenèse a été très largement associée à la dissémination métastatique. Les étapes de la dissémination métastatique impliquant la TEM sont multiples et concernent l’invasion des tissus adjacents, l’intravasation, la survie dans les flux et l’extravasation. Seule l’étape de colonisation semble davantage requérir le retour à un phénotype épithélial. Le potentiel métastatique d’une cellule cancéreuse dépend donc vraisemblablement de sa plasticité et de sa capacité à transiter d’un phénotype à l’autre.
Les inducteurs de TEM tel que TWIST, ZEB ET SNAIL, C-JUN sont fréquemment exprimés dans les tumeurs humaines et plus particulièrement dans les carcinomes. Ces facteurs de transcriptions sont capables de réprimer l’expression de l’E-cadhérine, facteur clé des jonctions adhérentes. En plus de contribuer à la perte des contacts intercellulaires et de la polarité apicobasale, la TEM est également associée à un gain de marqueurs mésenchymateux tels que la N-cadhérine, la fibronectine ou la vimentine. L’exposition de cellules épithéliales humaines à des cytokines telles que le TGFβ ou l’expression forcée de TWIST1, de SNAIL, de SLUG, de ZEB1 ou de ZEB2 favorisent la survenue d’une TEM, accompagnée de l’acquisition de propriétés migratoires et invasives, et de l’augmentation de leur potentiel métastatique.
30 La coexistence au sein des carcinomes sarcomatoïdes d’une prolifération de cellules d’aspect épithéliale et d’aspect mésenchymateux suggère cette plasticité tumorale et l’hypothèse d’un processus de transition épithélio-mésenchymateuse (TEM). Dans une série de 22 cas de carcinomes sarcomatoïdes (Blaukovitsch et al., 2006), l’E-cadhérine était exprimée de façon plus marquée par le contingent épithélial que par le contingent sarcomatoïde. Cette expression était associée négativement à celle de la vimentine et à l’activation de la voie de C-JUN, mais pas de celles de NOTCH, SNAIL ou du TGFβ1(Blaukovitsch et al., 2006).
Plusieurs travaux ont montré une association entre TEM et résistance à la chimiothérapie dans les CBNPC. Ainsi, l’inhibition de SNAIL sur une lignée d’adénocarcinome bronchique résistante au cisplatine, induit une sensibilisation au cisplatine, parallèlement à l’inhibition de la TEM, de la migration et de l’invasion cellulaire (Wang et al., 2014). Des résultats similaires ont été obtenus in vitro et in vivo sur un modèle d’adénocarcinome bronchique résistant au docetaxel, via l’inhibition de ZEB1 (Ren et al., 2013).
Parmi les mécanismes de chimiorésistance, l’induction d’une TEM peut conduire à un gain d’expression de divers transporteurs de la famille ABC. Ces protéines transmembranaires sont capables d’éluer différents substrats endogènes du cytoplasme vers le milieu extracellulaire tel que les drogues de chimiothérapies (Saxena et al., 2011). Par ailleurs les facteurs de transcriptions de la TEM apparaissent comme des régulateurs majeurs des voies oncosuppressives dépendantes des protéines p53 et Rb, et ont un effet tant sur leur activation et leur stabilisation que sur leur activité transcriptionnelle. Il a également été démontré qu’ils
31 étaient capables d’activer des voies de survie (AKT et NF-κB) et de moduler l’expression ou l’activité des membres de la famille BCL-2 en faveur des protéines antiapoptotiques.
Un nouveau champ d’investigation s’est récemment ouvert avec l’implication des micro-ARN dans la régulation de la TEM (miR-200, miR-448, miR-Let7c) Différents études menées sur des lignées tumorales mais aussi des échantillons tumoraux de patients ont montré une perte d’expression de ces divers micro-ARN après traitement par des molécules telles que les taxanes, le 5-FU et le cyclophosphamide. En cela, ces résultats démontrent non seulement l’implication des micro-ARN dans l’induction d’une TEM mais également leur implication dans une probable baisse, voire une perte, de sensibilité des tumeurs à plusieurs molécules thérapeutiques.
1.3.1.2 Un modèle de cellule souche ?
Les cellules souches cancéreuses (CSC) sont des cellules tumorales initiatrices du cancer et interviendraient dès les premières étapes de la carcinogenèse. Elles représentent une très faible proportion des cellules tumorales. Elles sont très indifférenciées, avec un fort potentiel de prolifération et de différenciation. Ces CSC partagent de nombreuses similitudes avec les cellules souches normales, en termes d’autorenouvèlement, de production de lignées cellulaires « filles », de phénotype membranaire et de voies de signalisation intracellulaires. On ignore si les CSC dérivent des cellules souches normales de l’organisme. Les CSC peuvent être définies par 3 propriétés essentielles : l’auto-renouvèlement, la production de lignées « filles » différenciées, et la capacité oncogénique in vivo, mesurée par la formation de xénogreffes. L’activation de plusieurs gènes spécifiques a été mise en évidence au sein des CSC, comme SOX2, NANOG ou OCT4/POU5F1. En 1982, Carney et al. ont montré à partir de tissus issus de mise en culture CBNPC et de carcinome bronchique à petites cellules que
32 certaines cellules, représentant moins de 2% de l’ensemble des cellules tumorales, étaient capables de former de nouvelles colonies de cellules tumorales. Ces colonies étaient capables de former des tumeurs in vivo, similaires à la tumeur initiale, après injection intracrânienne dans des souris immunodéprimées (Carney et al., 1982).
De nombreux travaux ont soulevé le rôle des CSC dans la chimiorésistance des CBNPC. La sélection in vitro des cellules tumorales sous chimiothérapie a été utilisée comme moyen de sélection des CSC (Li et al., 2008). L’expression de CD133, marqueur de cellule souche, dans le CBNPC était corrélée aux marqueurs de chimiorésistance, comme la glutathione S-transferase (Salnikov et al., 2010). L’administration de cisplatine, d’etoposide et de paclitaxel sur des CSC de CBNPC cultivées en sphères n’a aucune activité cytotoxique sur ces CSC (Eramo et al., 2008). De même, les cellules CD44+ isolées par tri cellulaire à partir de lignées de CBNPC (H1650 et H1299) sont beaucoup plus résistantes au cisplatine que les cellules CD44- ou les cellules non triées des mêmes lignées (Leung et al., 2010).
La voie Sonic Hedgehog (Shh) peut être activée au sein des CSC. Plusieurs études ont suggéré une relation entre l’activation de la voie Shh et la chimiorésistance. Le récepteur membranaire de Shh est Patched (Ptch1 et Ptch2). En l’absence de ligand Ptch inhibe la migration à la membrane de la protéine Smoothened (Smo). Lorsque Shh se fixe à son récepteur Ptch, il y a levée de l’inhibition de Smo qui va alors migrer à la membrane. Les facteurs de transcription Gli (Gli1, Gli2 et Gli3) se dissocient ensuite de SUFU pour migrer dans le noyau et activer la transcription des gènes cibles. Aucune différence en terme de gènes cibles n’a été mise en évidence entre Gli1, Gli2 et Gli3. Certains auteurs ont soulevé la possibilité d’une activation de Gli indépendante de la fixation de Shh sur son récepteur Ptch (activation « non-canonique » de Gli) (Lauth et al., 2007 ; Mimeault, Batra, 2010). Ainsi,
33 l’activation de KRAS, du TGFβ ou d’EGFR pourrait également activer Gli1 et Gli2 et la transcription des gènes cibles.
Il a ainsi été montré une corrélation entre l’activation de la voie Shh dans les cellules cancéreuses et l’expression de transporteurs ABC impliqués dans la chimiorésistance comme MDR1 et ABCG2, évaluée en Western Blot (Sims-Mourtada et al., 2007). De plus, Tian et al. (Tian et al., 2012) ont montré que le vismodegib, inhibiteur de SMO, inhibait de façon significative la prolifération tumorale in vitro des lignées cellulaires de CBNPC et de CBPC résistantes au cisplatine. L’inhibition de la voie Shh par vismodegib ou par siARN sensibilise également au traitement par ITK de l’EGFR. Ahmad et al. ont ainsi utilisé la lignée A549 rendue résistante au cisplatine et à l’erlotinib via un traitement par TGFβ qui permet l’acquisition d’un phénotype mésenchymateux (A549M) (Ahmad et al., 2013). Les IC50 du cisplatine et de l’erlotinib étaient significativement plus basses avec un pré-traitement par vismodegib ou siARN anti-Shh. Par ailleurs, l’administration de vismodegib entraînait la diminution de l’expression de marqueurs de CSC, comme Sox2 ou NANOG. Ces résultats soutiennent l’hypothèse actuellement admise que, in vivo, les CSC sont sélectionnées lors de l’administration d’une chimiothérapie cytotoxique, du fait de leur capacité de chimiorésistance. Elles seraient ainsi responsables de la progression tumorale ou de la rechute, car capables de recréer un clone tumoral.
2 Objectifs de thèse
Les carcinomes sarcomatoïdes par la présence de secteurs à la fois épithélial et mésenchymateux orientent vers une plasticité tumorale majeure. Ils semblent présenter des modifications génétiques communes aux CBNPC cependant ils se différencient par un
34 pronostic plus défavorable notamment au stade métastatique. Leur chimiorésistance est supputé sur la base d’un nombre restreint de tumeurs analysées. Elle pourrait impliquer la participation de mécanisme de transition épithélio-mesenchymateuse.
L’objectif du travail de thèse était de caractériser ces tumeurs et notamment de : – décrire les phénomènes de chimiorésistance
– décrire l’origine épithéliale de ces tumeurs – décrire le phénotype de TEM
– décrire les altérations moléculaires – décrire l’infiltration inflammatoire
– d’identifier des cibles thérapeutiques afin d’améliorer leur prise en charge thérapeutique
35
3 Résumés des articles et manuscrits
3.1 Détermination de la réponse à la chimiothérapie
3.1.1 Article 1 : étude des caractéristiques cliniques et moléculaires des
patients ayant un CBNPC de stade avancé et réfractaires à la chimiothérapie à base de platine
Clinical and molecular features in patients with advanced non-small cell lung carcinoma refractory to first-line platinum-based chemotherapy.
Giroux Leprieur E, Antoine M, Vieira T, Duruisseaux M, Poulot V, Rabbe N, Belmont L, Gounant V, Lavolé A, Milleron B, Lacave R, Cadranel J, Wislez M.
Lung Cancer, Volume 79, n°2, Février 2013, pages 167-172.
La majorité des patients ayant un CBNPC de stade avancé est traitée en première ligne par une chimiothérapie à base de platine. Environ un tiers à la moitié de ces patients présente une progression tumorale d’emblée sous ce traitement. Les patients sont alors considérés comme réfractaires à la chimiothérapie.
Une cohorte de 178 patients ayant un CBNPC de stade avancé a été traitée entre 2003 et 2006 dans le service de Pneumologie de l’Hôpital Tenon (AP-HP) par une chimiothérapie à base de platine, en première ligne. Nous avons comparé les caractéristiques cliniques, tumorales et moléculaires des patients réfractaires à celles des patients contrôlés. Les anomalies moléculaires étudiées étaient celles dépistées en routine en 2011 : mutations d’EGFR et de KRAS, translocation d’ALK. Le taux de patients réfractaires était de 25,6% (n=46).
36 La survie globale des patients réfractaires était de 5 mois (IQR 3,2-9,9) versus 15,4 mois (IQR 9,9-22,5) pour les patients contrôlés (n=132) p<0,001. Les caractéristiques cliniques étaient similaires entre les deux groupes. La seule différence concernait les sous types histologiques, avec 10,9% de carcinome sarcomatoïde (n=7) chez les patients réfractaires, versus 1,5% chez les patients contrôlés (p=0,057). Il n’y avait pas de différence en termes d’anomalies moléculaires entre les deux groupes. En analyse multivariée, seul le sous-type histologique sarcomatoïde était significativement associé au caractère réfractaire (OR=7,50, IC95% 1,02-55,45 ; p=0,048).
Discussion
Les techniques moléculaires pour la recherche de mutations (séquençage direct) d’EGFR et
KRAS étaient anciennes et moins sensibles et pourraient expliquer le taux de mutations plus
faibles dans cette série. La puissance statistique reste limitée au vu du petit effectif de carcinomes sarcomatoïdes. Elle donne un premier signal négatif quant au caractère chimiorésistant des carcinomes sarcomatoïdes. Elle déclenche le travail publié dans l’article 2.
43 3.1.2 Article 2 : étude de l’efficacité de la chimiothérapie dans le sous type
sarcomatoïde
Efficacy of first-line chemotherapy in patients with advanced lung sarcomatoid carcinoma. Vieira T, Girard N, Ung M, Monnet I, Cazes A, Bonnette P, Duruisseaux M, Mazieres J,
Antoine M, Cadranel J, Wislez M.
Journal of Thoracic Oncology. Volume 8, N°12, Décembre 2013, pages 1574-1577.
Nous avons repris les données cliniques et thérapeutiques d’une cohorte multicentrique française de 97 cas de carcinomes sarcomatoïdes de stade localement avancé ou métastatique. Nous avons étudié la réponse à la chimiothérapie selon les critères RECIST 1.1, la survie sans progression à la première ligne de traitement et la survie globale.
Les patients étaient souvent symptomatiques (80%). Les manifestations cliniques n’étaient pas différentes de celles des autres sous-types de CBNPC. Les hémoptysies y étaient peut-être plus fréquentes et peut être de plus grande abondance. Par ailleurs, l’extension hématogène était fréquente avec des localisations métastatiques classiques mais aussi atypiques telles que des localisations péritonéales, retro péritonéales, gastriques, intestinales, pancréatiques, gingivales, sous-cutanées, thyroïdiennes.
Dans cette cohorte, nous montrons que ces patients sont réfractaires à la chimiothérapie conventionnelle. En effet, soixante-neuf pourcent progressent d’emblée lors du premier bilan d’évaluation réalisé après deux cycles de chimiothérapie (figure 4). Enfin la survie sans progression en 1ère ligne était en médiane de 2 mois et la survie globale de 7 mois.
44 Ces données témoignent d’une grande agressivité et d’une résistance à la chimiothérapie conventionnelle. Ce travail reste néanmoins rétrospectif couvrant une longue période d’inclusion du fait de la rareté de ce sous type histologique avec des biais probables de sélection. Les chimiothérapies utilisées étaient parfois anciennes et n’associaient pas toujours un sel de platine et une molécule de troisième génération. Il était difficile de dégager un doublet à base de platine supérieur à un autre principalement du fait des effectifs restreints dans chaque association. Les thérapies s’approchant de celle des sarcomes ne semblaient pas efficace dans ces tumeurs.
Elle impose la nécessité d’explorer d’autres pistes thérapeutiques afin de comprendre la résistance aux chimiothérapies conventionnelles en particulier aux sels de platine.
Figure 4 : analyse de la réponse selon l’utilisation d’un sel de platine
20 11 69 4 27 69 0 10 20 30 40 50 60 70 80 90 réponse partielle stabilité progression (%)
sans sels de platine avec sels de platine
p=0,850
p=0,047
p=0,595
Evaluation de la réponse à la chimiothérapie selon les critères RECIST 1.1. et stratifiée en fonction de l’exposition à un sel de platine.
49 3.1.3 Article 3 : étude de la résistance à la chimiothérapie de lignées de
carcinomes sarcomatoïdes, implication de la voie SHH
Sonic Hedgehog Pathway Activation Is Associated With Resistance to Platinum-Based Chemotherapy in Advanced Non-Small-Cell Lung Carcinoma.
E. Giroux Leprieur*, T. Vieira*, M. Antoine, N. Rozensztajn, N. Rabbe, A.-M. Ruppert, A. Lavole, J. Cadranel, M. Wislez,
Clin. Lung Cancer, Juillet 2016, volume 17, N°4, pages 301-308 *Co-premier auteurs
Plusieurs études ont suggéré une relation entre l’activation de la voie Sonic Hedgehog (Shh), et la chimiorésistance. Par leur modèle de résistance clinique aux sels de platine, nous avons étudiés l’inhibition de la voie Shh dans les carcinomes sarcomatoïdes et d’autres sous types de CBNPC.
Etude de Gli1 et Gli2
Une cohorte de 178 patients ayant un CBNPC de stade avancé ayant été traitée entre 2003 et 2006 dans le service de Pneumologie de l’Hôpital Tenon (AP-HP) par une chimiothérapie à base de platine, en première ligne a été reprise (cohorte de l’article 1). Nous avons d’abord étudié l’activation de la voie Shh chez les patients ayant prélèvement chirurgical diagnostique (n=36), au travers de l’expression de Gli1 et Gli2 par immunohistochimie (anticorps sc-20687 (Gli1, Santa Cruz) et ab-26056 (Gli2, Abcam). Sur les 36 patients, 12 étaient réfractaires à la